

Abstract
Homogeneous N-glycoproteins carrying defined natural N-glycans are essential for detailed structural and functional studies. The transglycosylation activity of the endo-β-N-acetylglucosaminidases from Arthrobacter protophormiae (Endo-A) and Mucor hiemalis (Endo-M) holds a great potential for glycoprotein synthesis, but the wild type enzymes are not practical for making glycoproteins carrying native N-glycans because of their predominant activity for product hydrolysis. We report in this article the studies on two endoglycosidase-based glycosynthases, EndoM-N175A and EndoA-N171A, and their usefulness for constructing homogeneous N-glycoproteins carrying natural N-glycans. Oligosaccharide oxazoline corresponding to the bi-antennary complex type N-glycan was synthesized and tested with the two glycosynthases. The EndoM-N175A mutant was able to efficiently transfer the complex type glycan oxazoline to a GlcNAc-peptide and GlcNAc-containing ribonuclease to form the corresponding homogeneous glycopeptide/glycoprotein. The EndoA-N171A did not recognize complex type N-glycan oxazoline but could efficient use the high-mannose type glycan oxazoline for transglycosylation. These mutants possess the transglycosylation activity but lack the hydrolytic activity toward the product. Kinetic studies revealed that the dramatically enhanced synthetic efficiency of the EndoA-N171A mutant was due to the significantly reduced hydrolytic activity toward both the Man9GlcNAc oxazoline and the product, as well as its enhanced activity for transglycosylation. Thus, the two mutants described here represent the first endoglycosidase-based glycosynthases enabling a high efficient synthesis of homogeneous natural N-glycoproteins.
Keywords: Glycosynthase, Oligosaccharide oxazoline, Transglycosylation, Homogeneous N-glycoprotein, Endoglycosidase
Introduction
Homogeneous glycopeptides and glycoproteins carrying defined glycan structures are indispensible tools for structural and functional investigation of glycoproteins.1 Since natural glycoproteins are usually present as heterogeneous mixtures of glycoforms, from which pure glycoforms are difficult to isolate with current techniques, synthesis has become an essential source to obtain homogeneous materials. A number of elegant synthetic methods have been developed, as discussed in recent reviews.1-4 For example, mild chemo-selective ligation methods, including native chemical ligation and auxiliary-assisted ligation, have been designed to overcome a series of technical difficulties in synthesizing homogeneous glycopeptides and glycoproteins.5 Meanwhile, the chemoenzymatic approach, based on enzymatic elaboration of sugar chains on free monosaccharide-tagged polypeptide or protein, represents another major focus of research in this field.3, 4 Both glycosyltransferases and endoglycosidases have been used to elaborate sugar chains for glycopeptide/glycoprotein synthesis. The chemoenzymatic synthesis using the transglycosylation activity of endo-β-N-acetylglucosaminidase (ENGase) constitutes a highly convergent approach, as the endoglycosidase is able to attach a large intact oligosaccharide moiety to a pre-assembled GlcNAc-polypeptide or GlcNAc-protein in a single step and in a regio- and stereo-specific manner, without the need for any protecting groups.4, 6 So far, two ENGases have been frequently used for the chemoenzymatic synthesis, including the Endo-A from Arthrobactor protophormiae7-12 that is specific for high-mannose type N-glycans, and the Endo-M from Mucor hiemalis13, 14 that can act on both complex and high-mannose type N-glycans. However, a major barrier that limits the use of this enzymatic method for synthetic purpose is the low efficiency caused by the inherently low transglycosylation yield with natural donor substrates (natural N-glycans or N-glycopeptides) as well as the rapid product hydrolysis. For example, in vitro remodeling of a glycoprotein using a natural complex type N-glycopeptide as the donor substrate and wild type Endo-M as the enzyme resulted in only a low yield (<5%) of the desired glycoprotein even when a large excess (over 100-fold) of the donor substrate was used.15 To circumvent this problem, several research groups, including ours, have recently explored synthetic sugar oxazolines, a highly activated species mimicking the presumed oxazolinium ion intermediate generated by a substrate-assisted mechanism of catalysis, as donor substrates for glycopeptide synthesis.16-19 Extension of this approach to the synthesis of homogeneous glycoproteins carrying a defined oligosaccharide moiety was also achieved.19 It was demonstrated that the activated oligosaccharide oxazoline corresponding to the Man3GlcNAc tetrasaccharide core, and some of its structurally modified forms, were able to serve as donor substrates for transglycosylation. Notably, the resulting glycopeptides that carry the modified N-glycans, regarded as “ground-state”, became poor substrates for hydrolysis because of the slight structural modifications. Thus a big difference in reactivity between the highly activated oxazoline and the ground-state product enables an efficient synthesis of corresponding glycopeptides or glycoproteins with truncated or modified N-glycans. Recently, we have successfully applied this chemoenzymatic approach for the glycoengineering of human IgG1-Fc and for the synthesis of an array of homogeneous glycoforms of ribonuclease B.20, 21 However, when it was applied to glycoproteins carrying natural, full-size N-glycans, a rapid product hydrolysis by the wild-type enzymes would be difficult to avoid, as the natural glycoforms, once formed, would become excellent substrates for the respective ENGases.
One solution to this problem is to create novel glycosynthases, via glycosidase mutagenesis, that lack activity to hydrolyze the product but are able to take a highly activated species, such as glycosyl fluoride in the opposite anomeric configuration, as the donor substrate.22 A general approach to creating glycosynthase from glycosidase is to mutate the essential nucleophilic residue to abolish its function for catalytic hydrolysis. The ENGases of the family 85 are a class of endoglycosidases that catalyze the reaction by a presumed substrate-assisted mechanism in which the 2-acetamido group in the substrate acts as the nucleophile. Thus the common approach to generating glycosynthase by “knocking out” the nucleophilic residue could not apply to ENGases. To address this issue, we have recently performed site-directed mutagenesis in the putative catalytic region of Endo-M and identified a novel Endo-M mutant, N175A, which became the first glycosynthase derived from the family 85 endoglycosidases.23 It was demonstrated that this mutant was able to use a full-size, high-mannose type glycan oxazoline (Man9GlcNAc oxazoline) as a donor substrate for transglycosylation, but lacked the activity to hydrolyze the resulting natural high-mannose type N-glycopeptide product, implicating a great potential for this type of mutant. However, a number of questions remain to be answered: Is this Endo-M mutant applicable for synthesizing glycopeptides carrying natural, full-size complex type N-glycans without hydrolysis? Can this glycosynthase approach be efficiently extended to the synthesis of intact glycoproteins carrying native N-glycans? Does the corresponding Endo-A mutant behave similarly? Does the mutation change the substrate specificity (high-mannose type vs. complex type)? What's the kinetic feature of the mutant-catalyzed reactions? To answer these questions, we describe in this paper the synthesis of a key oligosaccharide oxazoline corresponding to the natural, bi-antennary complex-type N-glycan. The corresponding Endo-A mutant, EndoA-N171A, was generated. The substrate specificity and kinetics of the two mutant enzymes were evaluated. Our experiments have revealed that the EndoM-N175A mutant could take both the high-mannose type and complex-type N-glycan oxazolines as substrates for transglycosylation without product hydrolysis, but the EndoA-N171A mutant recognizes only the high-mannose type glycan oxazoline as the substrate for transglycosylation. Moreover, we have demonstrated that the glycosynthase approach can be efficiently applied for the synthesis of homogeneous N-glycoproteins carrying intact natural N-glycans.
Results and Discussion
Semi-synthesis of oligosaccharide oxazoline corresponding to the complex type N-glycan
A semi-synthesis of the complex type N-glycan oxazoline (CT-oxazoline) was achieved using the biantennary sialylglycopeptide (SGP) as the starting material, which was readily isolated from hen's egg yolk on large scale according to previously reported procedures.11, 24 To simplify the synthesis, the sialic acid residues in SGP were removed by sialidase treatment, followed by Endo-M catalyzed hydrolysis, to give the octa-saccharide (1) in 65% yield in two steps. O-Acetylation of the free oligosaccharide (1) gave the per-acetate 2. Oxazoline ring formation was achieved by treatment of 2 with TMSBr and boron trifluoride in the presence of collidine to obtain the oxazoline derivative 3 in 75% yield. Finally, global de-O-acetylation with a catalytic amount of MeONa in MeOH afforded the complex type N-glycan oxazoline (4) in quantitative yield (Scheme 1).
Scheme 1.

Semi-synthesis of oligosaccharide oxazoline corresponding to complex type N-glycan
To evaluate whether EndoM-N175A mutant can take oxazoline 4 as a donor substrate for transglycosylation, we used a GlcNAc-containing pentapeptide (5)18 as the acceptor. The enzymatic reaction was performed in a phosphate buffer (pH 6.8) at 23 °C (molar ratio of donor to acceptor, 3:1). It was found that the enzymatic transglycosylation with oxazoline 4 proceeded smoothly with a steady formation of the glycopeptide product (6) (Scheme 2). After 4 h, the yield of the product reached 85% as monitored by reverse-phase HPLC. Incubation of EndoM-N175A with the isolated glycopeptide product (6) under the enzymatic reaction condition indicated that the EndoM-N175A mutant did not hydrolyze the natural complex type N-glycopeptide. These experimental data suggest that the EndoM-N175A mutant is a typical glycosynthase capable of using a highly activated species as the substrate for glycosylation, but lacks the activity to hydrolyze the product. The identity of the transglycosylation product, glycopeptide 6, was characterized by a combination of several analytic means. Deconvolution of the ESI-MS of 6 revealed a molecular mass of m/z 2197.32, which is consistent with molecular mass of the expected adductive product (calculated, M = 2197.11). Further characterization of the product was performed by pronase digestion of 6 and subsequent analysis of the resulting Asn-linked oligosaccharide. Thus, treatment of glycopeptide 6 with pronase gave an Asn-linked oligosaccharide, which showed no difference with the authentic Asn-linked N-glycan (Galβ1,4GlcNAcβ1,2Manα1,6)[Galβ1,4GlcNAcβ1,2Manα1,3]-Manβ1,4-GlcNAcβ1,4GlcNAcβ1-Asn, prepared from the standard desialylated glycopeptide of hen's egg yolk,25 by ESI-MS and HPAEC-PED analysis (Figure S4, Supporting Information). In addition, the 1H-NMR spectrum of the Asn-linked oligosaccharide obtained from pronase digestion of glycopeptide 6 was essentially identical to that of the authentic Asn-linked glycan (Figure S5, Supporting Information). The anomeric proton H-1b (i.e., H-1') of the second GlcNAc moiety appeared as a doublet at 4.58 ppm with a relatively large coupling constant (J1',2' = 7.5 Hz), clearly indicating that the newly formed glycosidic bond was in the expected β-stereochemistry. To further confirm the linkage type, we performed a detailed 2D NMR (1H-13C HSQC and 1H-1H NOESY) analysis of the Asn-linked oligosaccharide obtained from glycopeptide 6 (Figure S6, Supporting Information). A clear NOE correlation between the H-1b (the anomeric proton of the second GlcNAc) and the H-4a (the H-4 of the first GlcNAc) suggested that the newly generated glycosidic bond was in a β-1,4 linkage type. Taken together, these experimental data have unambiguously confirmed that the mutant N175A-catalyzed transglycosylation proceeded in a regio- and stereo-specific manner to form the desired N-glycopeptide, in which the newly generated glycosidic bond was in the natural, β-1,4-glycosidic linkage.
Scheme 2.

Transglycosylation with complex type glycan oxazoline by glycosynthase EndoM-N175A
We also examined the transglycosylation activity of wild type Endo-M (WT-EndoM) and the mutant Y217F 23 with the newly synthesized oxazoline (4). Both the WT-EndoM and the mutant Y217F were able to use oxazoline 4 as donor substrate for transglycosylation, and the initial transglycosylation rate for mutant Y217F was much higher than that of the WT-EndoM and EndoM-N175A mutant (Figure 1). However, in contrast to N175A that lacked the hydrolytic activity, both WT-EndoM and EndoM-Y217F gradually hydrolyzed the natural N-glycopeptide 6 thus produced and, after 2 h, all of glycopeptide product was essentially hydrolyzed (Figure 1).
Figure 1.
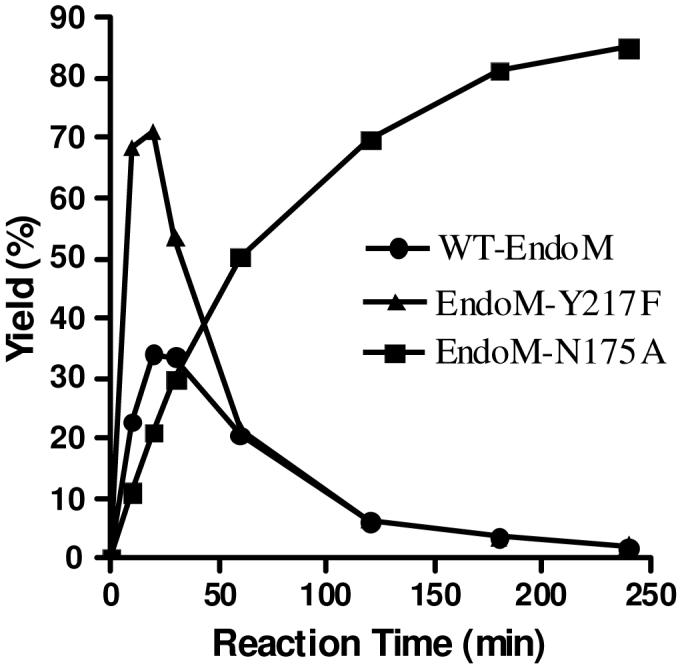
Transglycosylation with the glycan oxazoline (4) by Endo-M and its mutants.
Synthesis of a HIV-1 gp41 C34-glycopeptide carrying a natural complex-type N-glycan
To demonstrate the usefulness of the glycosynthase for assembling large, natural complex type N-glycopeptides, we have chosen a HIV-1 gp41 glycopeptide C34 as a target, in which a complex type N-glycan was attached at the conserved glycosylation site Asn-637 (HIV-1 HXB2 numbering) (Scheme 3). We have previously synthesized a high-mannose type C34-glycopeptide using natural Man9GlcNAc2Asn as the donor substrate and wild type Endo-A as the enzyme, which gave the product in only 12% yield under an optimal transglycosylation condition.12 Incubation of the complex-type oxazoline 4 and the pre-synthesized GlcNAc-C34 (7)12 (molar ratio of donor to acceptor, 5:1) with EndoM-N175A for 4 h in a phosphate buffer (pH 7.0) containing 20% DMSO (used to increase the solubility of GlcNAc-C34 in aqueous solution) gave the desired glycopeptide (8), which was readily isolated by RP-HPLC in 71% yield: ESI-MS of 8, calculated, M = 5912.62; found, 1479.62 [M + 4H]4+, 1183.88 [M + 5H]5+. This result indicates that the EndoM-N175A mutant is efficient for the synthesis of large complex-type N-glycopeptides.
Scheme 3.

Synthesis of a complex type glycopeptide drived from HIV-1 gp41
A correspondent Endo-A mutant is a glycosynthase specific for high-mannose type oxazoline substrate
Encouraged by the promising glycosynthase activity of EndoM-N175A, we next turned our attention to Endo-A, another endo-β-N-acetylglucosaminidase that possesses transglycosylation and hydrolytic activity for high-mannose type N-glycans.7, 8 Based on sequence alignment, we created an EndoA-N171A mutant by site-directed mutagenesis, which can be regarded as the Endo-A equivalent of the EndoM-N175A. A test of the EndoA-N171A with oxazoline 4 indicated that the EndoA-N171A mutant could not recognize the complex type sugar oxazoline 4 as substrate for transglycosylation. It was found that the wild type Endo-A could not take the complex type glycan oxazoline, either. Despite the fact that the sugar oxazoline 4 was a highly activated species, and the fact that Endo-A could use Man3GlcNAc oxazolines and some structurally modified oxazolines with substituents at the 4 and 6-hydroxyl groups of the outer mannose residues of the Man3GlcNAc core,18, 19 no cross-reactivity was observed for Endo-A or its N171A mutant toward the complex type oxazoline. These results implicate that the attachment of β-1,2-linked GlcNAc residues at the outer mannose residues of the Man3GlcNAc core, as in the case of the complex type N-glycan oxazoline 4, rendered the substrate inactive to Endo-A enzymes. As wild type Endo-A can quickly hydrolyze natural high-mannose type N-glycans, we then tested how the EndoA-N171A acted on natural high-mannose type N-glycans and its oxazolines. It was found that EndoA-N171A was unable to hydrolyze the high-mannose type N-glycan Man9GlcNAc2Asn, but it could take the synthetic Man9GlcNAc oxazoline (9)23 for transglycosylation to form the corresponding natural glycopeptide (10) with high efficiency (Scheme 4). Like EndoM-N175A, the EndoA-N171A mutant lacked the activity to hydrolyze the natural glycopeptide product thus formed. Therefore, EndoA-N171A represents another endoglycosidase-based glycosynthase that is valuable for the construction of large natural N-glycopeptides of high-mannose type.
Scheme 4.

Transglycosylation with high-complex type glycan oxazoline by glycosynthase EndoA-N171A
While EndoM-N175A can use both complex and high-mannose type N-glycan oxazolines as substrates for transglycosylation, the EndoA-N171A mutant only uses the oxazoline corresponding to high-mannose type N-glycan, indicating a more strict substrate specificity for the Endo-A mutant. During the preparation of this manuscript, Fairbanks and co-workers reported two different Endo-A mutants, E173H and E173Q, that demonstrated further decreased or aborted hydrolytic activity on the Man3GlcNAc2-peptide and related products thus formed, although their specific activity for transglycosylation with corresponding sugar oxazolines was also significantly reduced in comparison with the wild type Endo-A.26 We are aware that in this reported study, a relatively large amount of enzymes as well as a prolonged reaction time was employed to demonstrate the difference in the transglycosylation efficiency/product hydrolysis between the wild type and the mutant enzymes. Previously, it has been shown that Endo-A could use Man3GlcNAc-oxazoline and its modified forms for transglycosylation to efficiently produce homogeneous N-glycopeptides and glycoproteins, but the enzymatic hydrolysis of the glycopeptide products that carry the Man3GlcNAc2 core pentasaccharide and its modified forms was only marginal, when a catalytic amount of the wild type Endo-A was used for the reactions.17-21 It is to be demonstrated how the two E173H and E173Q mutants act on sugar oxazolines corresponding to natural, full-size high-mannose-type N-glycans.
Kinetic studies with the glycosynthases
It is known that wild type Endo-A can use natural high-mannose type N-glycans such as Man9GlcNAc2Asn as donor substrate for transglycosylation to form natural glycopeptide, despite the relatively low yield of synthesis due to Endo-A catalyzed hydrolysis of both the starting N-glycan and the product.7, 8, 11, 12 Therefore, it would be interesting to see how wild type Endo-A acts on the Man9GlcNAc oxazoline (9). To our surprise, incubation of the Man9GlcNAc oxazoline (9) and the GlcNAc-pentapeptide (5) (an acceptor) in the presence of wild type Endo-A gave no trace of transglycosylation product, except for the hydrolysis of the Man9GlcNAc oxazoline (9). This result implicates that the wild type Endo-A might hydrolyze the Man9GlcNAc oxazoline much faster than its transglycosylation reaction. To clarify this point, we performed kinetic studies on the hydrolysis and transglycosylation of Man9GlcNAc oxazoline (9) by wild type Endo-A and the N171A mutant. Dionex HPAEC-PAD was used to monitor and quantify the formation of the hydrolytic product Man9GlcNAc.27 It was found that the Km of EndoA-N171A mutant (2.4 mM) is about 6-fold higher than that of the wild type Endo-A (Km = 0.43 mM) for the substrate Man9GlcNAc-oxazoline. More significantly, the catalytic constant (kcat) of N171A mutant (kcat = 0.44 min-1) is 300-fold less than that of the wild type (kcat = 130 min-1). The specificity constant (kcat/Km) of N171A (0.18 min-1mM-1) for Man9GlcNAc oxazoline hydrolysis is thus about 1600-fold less than that of the wild type Endo-A (kcat/Km = 290 min-1mM-1) (Table 1). Taken together, these data indicate that the N to A mutation at position 171 dramatically diminished the ability of the enzyme to hydrolyze the Man9GlcNAc oxazoline substrate, thus eliminating a key detrimental property of the wild type Endo-A in terms of its synthetic potential.
Table 1.
Kinetic parameters of the hydrolysis of the Man9GlcNAc oxazoline by wild type Endo-A and the EndoA-N171A mutant
| Man9GlcNAc oxazoline (9) |
|||
|---|---|---|---|
| kcat (min-1) | Km(mM) | kcat/Km (min-1 mM-1) | |
| WT-EndoA | 130 | 0.43 | 290 |
| EndoA- N171A | 0.44 | 2.4 | 0.18 |
We next sought to determine the kinetic parameters for the mutant N171A-catalyzed transglycosylation with oxazoline (9) and the acceptor GlcNAc-pentapeptide (5) (Table 2). Interestingly, while the Km of Man9GlcNAc oxazoline (9) was found about the same (2.7 mM) for the hydrolysis and transglycosylation reactions, the kcat for the oxazoline transglycosylation (3.4 min-1) was found to be about 8-fold higher than that of the oxazoline hydrolysis (kcat = 0.44 min-1). These data suggested that while the affinity of N171A to the oxazoline substrate was independent of the presence or absence of the acceptor (5), the presence of a suitable acceptor such as the GlcNAc-pentapeptide (5) significantly promoted the reaction of the oxazoline to form the transglycosylation product. It is likely that the N171A mutation resulted in a diminished affinity of the enzyme for the nucleophilic water molecule at the catalytic site (as reflected by the diminished hydrolytic activity of the N171A mutant toward the oxazoline) and meanwhile led to an enhanced affinity of the enzyme for the GlcNAc-peptide acceptor for transglycosylation. Unfortunately, we could not accurately measure the kinetic parameters of the transglycosylation by the wild type Endo-A for comparison, because of the rapid hydrolysis of the donor substrate oxazoline (9), as well as the presumed in situ rapid hydrolysis of the glycopeptide product once formed. The kinetic parameters of the corresponding Endo-M mutant, EndoM-N175A, for the transglycosylation between the oxazoline 9 and the acceptor 5 were also measured and compared with those of the EndoA-N171A mutant (Table 2).
Table 2.
Kinetic parameters of the transglycosylation activity of EndoA-N171A and EndoM-N175A
| Man9GlcNAc oxazoline (9) |
GlcNAc-pentapeptide (5) |
|||||
|---|---|---|---|---|---|---|
| kcat | Km | kcat/Km | kcat | Km | kcat/Km | |
| (min-1) | (mM) | (min-1 mM-1) | (min-1) | (mM) | (min-1 mM-1) | |
| EndoA-N171A | 3.4 | 2.7 | 1.3 | 2.1 | 5.0 | 0.40 |
| EndoM-N175A | 4.5 | 3.1 | 1.4 | 4.0 | 6.3 | 0.63 |
The two mutants were very similar in both Km (ca. 3 mM for the oxazoline 9 and ca. 5 mM for the the acceptor 5) and kcat (ca. 4 min-1 for the oxazoline 9 and ca. 3 min-1 for the acceptor 5). These data suggest that the two mutants have comparable affinity for the high-mannose type sugar oxazoline and the acceptor substrates. They also have similar catalytic efficiency for the transglycosylation reactions (Table 2). As to the EndoM-N175A catalyzed reaction between the complex type glycan oxazoline 4 and the acceptor 5, accurate kinetic parameters were not obtained because of the difficulty to reach a saturated substrate concentration of the complex-type oxazoline substrate. The Km for the oxazoline 4 was estimated to be larger than 10 mM. In comparison, the Km of N175A for the high-mannose type oxazoline 9 (3 mM) was much smaller than that of the complex-type oxazoline 4 (Km > 10 mM). Therefore, the Endo-M mutant appeared to have a higher affinity for the high-mannose type glycan oxazoline than the complex-type glycan oxazoline.
EndoA-N171A is more thermo-stable than EndoM-N175A
Originally, Endo-A was a bacterial enzyme isolated from Arthrobactor protophormiae7, 8, and Endo-M was a fungus enzyme isolated from Mucor hiemalis.13 For the present study, the Endo-A and Endo-M mutants were successfully overproduced in E. coli. As Endo-M appeared to be much more sensitive than Endo-A during our transglycosylation reactions, we examined and compared the thermo-stability of the two mutants. Thus, the two mutant enzymes were incubated in a phosphate buffer (50 mM, pH 6.8) at different temperatures (30-50 °C) for 10 min and then their transglycosylation activities were measured using the Man9GlcNAc oxazoline (9) as the donor and the GlcNAc-pentapeptide (5) as the acceptor substrate. As shown in Figure 2, pre-incubation of the EndoM-N175A at 45 °C for 10 min resulted in complete loss of its transglycosylation activity, while the EndoA-N171A mutant retained its full activity after the same treatment (Figure 2). Therefore, the EndoA-171A mutant was much more thermo-stable than the EndoM-N175A mutant.
Figure 2.

The thermo-stability of EndoA-N171A and EndoM-N175A
Glycosynthase-catalyzed synthesis of homogeneous glycoproteins carrying full-size natural N-glycans
We have demonstrated that the two endoglycosidase mutants, EndoM-N175A and EndoA-N171A, were able to take the N-glycan oxazolines as donor substrates for transglycosylation to form corresponding natural N-glycopeptides without the hydrolysis of the product. These results prompted us to examine the efficiency of the two glycosynthases for synthesizing full-size homogeneous glycoproteins carrying natural N-glycans. Again, we chose bovine ribonuclease B, a natural glycoprotein consisting of 124 amino acid residues, as a model system to test the two glycosynthases. Natural bovine ribonuclease B (RNase B) carries a heterogeneous high-mannose type N-glycan (Man5-9GlcNAc2) at Asn-34. It was previously used by us and others for showcasing new chemoenzymatic methods for glycoprotein synthesis and glycosylation remodeling.7, 15, 19, 21, 28 Thus, the heterogeneous N-glycan in the natural RNase B was removed by Endo-H treatment, leaving only the inner most N-acetylglucosamine (GlcNAc) at the Asn-34 site to give the homogeneous GlcNAc-RNase B, which was used as the acceptor substrate for the enzymatic transglycosylation. It was found that incubation of the complex-type glycan oxazoline (4) and GlcNAc-RNase B (molar ratio of donor to acceptor, 10:1) with the mutant EndoM-N175A in a phosphate buffer (pH 7.0) at 23 °C resulted in the formation of the transglycosylation product (11). After 8 h, ca. 80% of the GlcNAc-RNase B was converted into the glycoprotein product (11), which was eluted slightly earlier than GlcNAc-RNase B in RP-HPLC. The product could be easily isolated by HPLC and was characterized as the RNase B glycoform carrying a complex type N-glycan. Deconvolution of the ESI-MS of 11 gave a molecular mass of 15308 Da, which is in good agreement with the calculated molecular mass (15305 Da) of the expected RNase B glycoform carrying the complex-type N-glycan. For further characterization, the attached N-glycan in glycoprotein 11 was released by PNGase F treatment. MALDI-TOF MS of the released N-glycan gave a single m/z species of 1664.53 [M + Na]+, which is consistent with the bi-antennary complex type N-glycan (Galβ1,4GlcNAcβ1,2Manα1,6)-[Galβ1,4GlcNAcβ1,2Manα1,3]-Manβ1,4GlcNAcβ1,4GlcNAc (calculated, M = 1641.49). It was found that the transglycosylation with the GlcNAc-RNase B could be driven to completion to form the glycoprotein product when additional CT-oxazoline (4) was added (Scheme 5). It was also observed that a relatively large amount of enzyme was required for an efficient transglycosylation, particularly when the substrate concentration was low. Therefore, future research should be directed to the improvement of the specific activity of the mutants for transglycosylation. This might be achieved by mutagenesis and screening of mutant library based on the EndoM-N175A mutant.
Scheme 5.

Chemoenzymatic synthesis of homogeneous glycoforms of RNase B
For the synthesis of the RNase B glycoform carrying a full-size high-mannose type N-glycan, GlcNAc-RNase B was incubated with the Man9GlcNAc oxazoline (9) (molar ratio of donor to acceptor, 8:1) in a phosphate buffer in the presence of the mutant EndoA-N171A at 23°C. Under this condition, the transglycosylation was found to take place smoothly to form the desired glycoprotein product (12), which was isolated by HPLC in 82% yield (Scheme 5). Again, the obtained glycoprotein product was confirmed to be the desired Man9GlcNAc2-RNase B (12) by ESI-MS analysis of the glycoprotein and the MALDI-TOF MS analysis of the N-glycan released by PNGase F treatment. The deconvoluted ESI-MS of 12 (found, M = 15549) matches well with the calculated molecular mass of glycoprotein 12 (calculated, M = 15547). The ESI-MS profiles of the synthetic glycoproteins 11 and 12 were shown in Figure 3.
Figure 3.

Deconvoluted ESI-MS of the synthetic glycoforms of RNase B. panel A, RNase B carrying the bi-antennary complex-type N-glycan; panel B, RNase B carrying the Man9GlcNAc2 glycan.
These experimental data clearly indicate that the two endoglycosynthase mutants, EndoM-N175A and EndoA-N171A, when coupled with the use of an appropriate N-glycan oxazoline as the donor substrate, enable an efficient synthesis of homogeneous N-glycoproteins carrying either a natural complex-type or a natural high-mannose type N-glycan, without product hydrolysis. It should be pointed out that the application of the glycosynthases for glycoprotein synthesis would rely on the access of a GlcNAc-protein as the precursor. Fortunately, a given GlcNAc-protein could be potentially prepared by several approaches, including: a) overproduction of the protein in a high-yield yeast or insect expression system coupled with Endo-H de-glycosylation; (b) total protein synthesis via native chemical ligation or expressed protein ligation with incorporation of a GlcNAc moiety at a pre-determined site during the synthesis;29 and c) overproduction of a GlcNAc-containing protein in E. coli through the novel nonsense codon suppression technology.30
Conclusion
The properties and synthetic potential of two novel endoglycosidase-based glycosynthases, EndoM-N175A and EndoA-N171A, were described. The two glycosynthases were able to take an appropriate highly-activated glycan oxazoline as donor substrate for transglycosylation to form homogeneous natural N-glycoproteins without the hydrolysis of the product. Thus, the creation of these glycosynthases provides a beautiful solution to the problem of product hydrolysis associated with the wild type enzymes. The EndoM-N175A was found to be capable of transferring complex type N-glycan oxazoline to form homogeneous, complex-type N-glycoproteins, while the EndoA-N171A mutant is a more convenient glycosynthase for the synthesis of high-mannose type N-glycoproteins. Therefore, the novel glycosynthases described here provides a timely and potentially powerful tool for the synthesis of various homogeneous N-glycoproteins carrying defined natural N-glycans, which are essential for detailed structural and functional studies.
Experimental
Materials
The GlcNAc-pentapeptide (1), derived from erythropoietin (amino acid sequence 37-41), was synthesized as described in the literature.18 The GlcNAc-C34 (7) a 34-mer peptide derived from HIV-1 gp41, was prepared by automatic solid-phase peptide synthesis using the Fmoc approach, according to our previously reported method.12 Man9GlcNAc oxazoline (9) was synthesized from Man9GlcNAc2Asn according to our previously reported method.23 The bi-antennary complex-type sialylglycopeptide (SGP), Lys-Val-Ala-Asn[(NeuAc-Gal-GlcNAc-Man)2Man-GlcNAc2)]-Lys-Thr, was prepared from hen egg yolks following the reported procedure.11, 24 The recombinant wild type Endo-A was overproduced in E. coli and purified by affinity chromatography according to the literature.10 The pGEX-2T/Endo-A plasmid used for the expression was kindly provided by Prof. Takegawa. The recombinant wild type Endo-M and the two Endo-M mutants, EndoM-N175A and EndoM-Y217F, were overproduced according to our previously reported method.23 Neuraminidase (Vibrio cholerae) and peptide N-glycosidase F (PNGase F) were purchased from New England Biolabs Inc. All other reagents were purchased from Sigma/Aldrich and used as received.
Methods
Analytical RP-HPLC was performed on a Waters 626 HPLC instrument with a Symmetry300™ C18 column (5.0 μm, 4.6 × 250 mm) at 40°C. Depending on the properties of the compounds to be separated, the column was eluted with a linear gradient of MeCN at a flow rate of 1 mL/min with one of the following four gradients: Method A, 0-20% MeCN containing 0.1% trifluoroacetic acid (TFA) for 20 min; Method B, 0-30% MeCN containing 0.1% TFA for 18 min; Method C, 0-90% MeCN containing 0.1% TFA for 30 min; Method D, 23-29% MeCN containing 0.1% TFA for 30 min. Preparative HPLC was performed on a Waters 600 HPLC instrument with a preparative C18 column (Symmetry300, 19 × 300 mm). The column was eluted with a suitable gradient of water-acetonitrile containing 0.1% TFA. NMR spectra were measured with JEOL ECX 400 MHz and/or Inova 500 MHz NMR machines. The chemical shifts were assigned in ppm. ESI-MS Spectra were measured on a micromass ZQ-4000 single quadruple mass spectrometer. MALDI-TOF MS measurement was performed on an Autoflex II MALDI-TOF mass spectrometer (Bruker Daltonics). The instrument was calibrated by using ProteoMass Peptide MALDI-MS calibration kit (MSCAL2, Sigma/Aldirich). The matrix used for glycans is 2,5-dihydroxybenzoic acid (DHB) (10 mg/mL in 50% acetonitrile containing 0.1% trifluoroacetic acid). The parameters for measurement are: 337 nm nitrogen laser with 100 μJ output; laser frequency 50.0 Hz; laser power 35%; linear mode; positive polarity; detection range 1000-3000; pulsed ion extraction: 70ns; high voltage: on; realtime smooth: high; shots: 500-2000.
Creation and expression of EndoA-N171A mutant
Mutation of Asn171 to Ala was performed using Quick-change mutagenesis Kit following the manufacturer's instruction (Stratagene, La Jolla, CA). The oligonucleotide primers were list here with the mutated site underlined: Forward: 5' - GAC GGC TGG TTT ATT GCC CAA GAA ACA GAA GGG - 3'; Reverse: 5' - CCC TTC TGT TTC TTG GGC AAT AAA CCA GCC GTC - 3'. Mutations were verified by DNA sequencing. The recombinant EndoA-N171A was over-expressed and finally purified by affinity chromatography following our previously described procedures.23
Synthesis of β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→2)-α-D-mannopyranosyl-(1→3)-[β-D-Galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→2)-α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-2-acetamido-2-deoxy-D-glucopyranose (1)
Sialylglycopeptide (SGP) (70 mg, 24.4 μmol) was incubated with neuraminidase (Vibrio cholerae) (5 U) in a phosphate buffer (200 μL, 50 mM, pH 6.0) at 30 °C. The de-sialylation reaction was monitored by analytic HPLC (method A, tR = 10.5 min). The reaction was complete after 12 h. The product was purified by preparative HPLC to give the asialo-SGP (55 mg, 98%). 1H NMR (D2O, 400 MHz): δ 5.01 (s, 1H, H-1 of Man4), 4.92 (d, 1H, J = 8.0 Hz, H-1 of GlcNAc1), 4.81 (s, 1H, H-1 of Man4'), 4.68 (s, 1H, H-1 of Man3), 4.50-4.44 (m, 3H, H-1 of GlcNAc2, GlcNAc5 and GlcNAc5'), 4.35 (m, 2H, H-1 of Gal6 and Gal6'), 1.96 (s, 3H, Ac), 1.92 (s, 3H, Ac), 1.91 (s, 3H, Ac), 1.88 (s, 3H, Ac), 1.24 (d, 3H, J = 7.6 Hz, Thr-CH3), 1.09 (d, 3H, J = 7.6 Hz, Ala-CH3), 0.82 (d, 6H, J = 7.6 Hz, Val-CH3); ESI-MS: calculated for C90H155N13O54, M = 2283.24; found, 1141.16 [M + 2H]2+, 761.82 [M + 3H]3+.
The asialo-SGP (53 mg) thus obtained was incubated with Mucor hiemalis Endo-β-N-acetylglucosaminidase (Endo-M) (50 mU) in a phosphate buffer (50 mM, pH 6.5, 300 μL) at 30 °C for 16 h. The residue was treated with ion-exchange resin DOWEX 50WX2-400 and DOWEX 1×2-100 to completely remove cation and anion species and the solution was filtered and concentrated. The residue that contains the neutral oligosaccharide was dissolved in water and was loaded to a column (1.5×85 cm) of Sephadex G-25. The material was eluted with 0.1M acetic acid. The fractions containing the oligosaccharide were combined and lyophilized to give 1 (23 mg, 66%). 1H NMR (D2O, 400 MHz): δ 5.10 (d, 1H, J = 3.6 Hz, H-1 of GlcNAc1), 5.01 (s, 1H, H-1 of Man3), 4.82 (s, 1H, H-1 of Man3'), 4.65 (s, 1H, H-1 of Man2), 4.46 (d, 2H, J = 8.0 Hz, H-1 of GlcNAc4 and GlcNAc4'), 4.34 (d, 2H, J = 8.0 Hz, H-1 of Gal5 and Gal5'), 4.15 (m, 1H), 4.08 (m, 1H), 4.01 (m, 1H), 1.95 (s, 3H, Ac), 1.94 (s, 3H, Ac), 1.93 (s, 3H, Ac); ESI-MS: calculated for C54H91N3O41, M = 1437.51; found, 1438.49 [M + H]+, 719.93 [M + 2H]2+.
Synthesis of 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→2)-3,4,6-tri-O-acetyl-α-D-mannopyranosyl-(1→3)-[2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→2)-3,4,6-tri-O-acetyl-α-D-mannopyranosyl-(1→6)]-2,4-di-O-acetyl-β-D-mannopyranosyl-(1→4)-1,3,6-tri-O-acetyl-2-acetamido-2-deoxy-D-glucopyranose (2)
Compound 1 (23 mg, 16 μmol) was dissolved in pyridine (2 mL) and acetic anhydride (2 mL) and the solution was stirred at rt for 20 h. The reaction mixture was concentrated under vacuum to dryness, and the residue was subject to column chromatography on silica gel using CH2Cl2/MeOH (20:1) as the eluent to afford the fully acetylated compound 2 (26 mg, 68%). 1H NMR (CDCl3, 500 MHz): δ 5.42 (m, 1H, H-1 of GlcNAc1), 5.03 (s, 1H, H-1 of Man3), 4.82 (s, 1H, H-1 of Man3), 4.64 (s, 1H, H-1 of Man2), 4.51 (m, 2H, H-1 of GlcNAc4 and GlcNAc4'), 4.36 (m, 2H, H-1 of Gal5 and Gal5'), 2.19-1.95 (m, 78H, 26 × Ac); ESI-MS: calculated for C100H137N3O64, M = 2403.75; found, 2404.85 [M + H]+, 1203.31 [M + 2H]2+.
Synthesis of 2-methyl-[2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→2)-3,4,6-tri-O-acetyl-α-D-mannopyranosyl-(1→3)-[2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranosyl-(1→2)-3,4,6-tri-O-acetyl-α-D-mannopyranosyl-(1→6)]-2,4-di-O-acetyl-β-D-mannopyranosyl-(1→4)-3,6-di-O-acetyl-1,2-dideoxy-α-D-glucopyranose]-[2,1-d]-oxazoline (3)
Compound 2 (26 mg, 10.8 μmol) was dissolved in anhydrous 1,2-dichloroethane (2 mL), and then TMS-Br (20 μL, 0.17 mmol), BF3·Et2O (20 μL, 0.17 mmol) and 2,4,6-collidine (23 μL, 0.17 mmol) were added sequentially under an argon atmosphere. The mixture was stirred at rt for 12 h and then diluted with chloroform (20 mL) and washed with saturated sodium bicarbonate solution and brine. The organic layer was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated and the residue was purified by column chromatography on silica gel using CH2Cl2/MeOH (20:1) as the eluent to give a crude product as a yellow solid. The crude product was further purified by gel filtration (Sephadex LH20, eluted with MeOH) to afford compound 3 as a pale yellow foam (19 mg, 75%). 1H NMR (CDCl3, 500 MHz): δ 5.99 (d, 1H, J = 7.0 Hz, H-1 of oxazoline), 5.16 (s, 1H, H-1 of Man3), 4.94 (s, 1H, H-1 of Man3), 4.72 (s, 1H, H-1 of Man2), 4.52 (m, 2H, H-1 of GlcNAc4 and GlcNAc4'), 4.34 (m, 2H, H-1 of Gal5 and Gal5'), 2.19-1.85 (m, 75H, 25 × Ac); ESI-MS: calculated for C98H133N3O62, M = 2343.73; found, 2344.96 [M + H]+, 1173.42 [M + 2H]2+.
Synthesis of 2-methyl-[β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→2)-α-D-mannopyranosyl-(1→3)-[β-D-galactopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→2)-α-D-mannopyranosyl-(1→6)]-β-D-mannopyranosyl-(1→4)-1,2-dideoxy-α-D-glucopyranose]-[2,1-d]-oxazoline (4)
Compound 3 (19 mg, 8.1 μmol) was dissolved in anhydrous methanol (1 mL) and was treated with MeONa (0.5 M in MeOH, 2 μL, 1 μmol), and the de-O-acetylation was monitored by ESI-MS. After 22 h, the solvent was removed by evaporation, and the residue was subject to gel filtration (Sephadex G-10, eluted by water containing 0.03% Et3N). The fractions containing the product were combined and lyophilized to give the oxazoline 4 as a pale yellow solid (11mg, quantitative yield). 1H NMR (D2O, 500 MHz): δ 6.05 (d, 1H, J = 7.0 Hz, H-1 of oxazoline), 5.12 (s, 1H, H-1 of Man3), 5.07 (s, 1H, H-1 of Man3), 4.89 (s, 1H, H-1 of Man2), 4.57(m, 2H, H-1 of GlcNAc4 and GlcNAc4',), 4.42 (m, 2H, H-1 of Gal5 and Gal5'), 2.02 (s, 3H, Ac), 2.01 (s, 3H, Ac), 1.84 (s, 3H, CH3 of oxazoline); ESI-MS: calculated for C54H89N3O40, M = 1419.50; found, 1420.36 [M + H]+.
EndoM-N175A catalyzed transglycosylation with complex type glycan oxazoline (4). Synthesis of Glu-Asn[(Gal-GlcNAc-Man)2Man-GlcNAc2]-Ile-Thr-Val (6)
CT-oxazoline 4 (1 mg, 0.7 μmol) and GlcNAc-pentapeptide 5 (110 μg, 0.14 μmol) in phosphate buffer (50 mM, pH 7.0, 35 μL) was incubated with EndoM-N175A (7 μg) at 23 °C for 4 h. The glycopeptide 6 was obtained via preparative HPLC purification (260 μg, 85%). tR = 14.2 min (analytic HPLC, method B). ESI-MS: calculated for C86H145N11O54, M = 2197.11; found, 1099.78 [M + 2H]2+, 916.75 [M - GalGlcNAc + 2H]2+, 733.46 [M + 3H]3+. For comparative study, the same reaction was carried out with wild type Endo-M and mutant Y217F, and aliquots were taken at intervals for HPLC analysis (method B) to monitor the product formation (The results were shown in Figure 1).
EndoM-175A catalyzed transfer of complex N-glycan to GlcNAc-C34. Synthesis of (Gal-GlcNAc-Man)2Man-GlcNAc2-C34 (8)
A solution of CT-oxazoline 4 (1 mg, 0.7 μmol) and GlcNAc-C34 (7) (630 μg, 0.14 μmol) in a phosphate buffer (50 mM, pH 7.0, 50 μL) containing 20% DMSO was incubated with EndoM-N175A (10 μg) at 23 °C for 4 h. The glycopeptide 8 was obtained via preparative HPLC purification (590 μg, 71%). tR = 19.4 min (analytic HPLC, method C). ESI-MS: calculated for C248H388N55O109S, M = 5912.62; found, 1479.62 [M + 4H]4+, 1388.22 [M - GalGlcNAc + 4H]4+, 1183.88 [M + 5H]5+, 1110.81 [M - GalGlcNAc + 5H]5+.
EndoA-N171A catalyzed transglycosylation with the Man9GlcNAc oxazoline. Synthesis of Glu-Asn(Man9GlcNAc2)-Ile-Thr-Val (10)
A solution of Man9GlcNAc oxazoline (9) (1 mg, 0.6 μmol) and GlcNAc-pentapeptide (5) (100 μg, 0.128 μmol) in a phosphate buffer (50 mM, pH 7.0, 30 μL) was incubated with EndoA-N171A (6 μg) at 23 °C for 4 h. The transglycosylation product was then purified by preparative HPLC to give glycopeptide 10 (250 μg, 78%), tR = 13.8 min (analytic HPLC, method B). ESI-MS: calculated for C94H159N9O54, M = 2437.94; found, 1220.78 [M+2H]2+, 1139.66 [MMan+2H]2+.
EndoM-N175A catalyzed transfer of complex type N-glycan to GlcNAc-RNase B. Synthesis of RNase B glycoform (11) carrying a complex type N-glycan
A solution of CT-oxazoline 4 (500 μg, 0.35 μmol) and GlcNAc-RNase B (500 μg, 0.036 μmol) in a phosphate buffer (50 mM, pH 7.0, 5 μL) was incubated with EndoM-N175A (200 μg) at 23 °C for 8 h. The transglycosylation product was isolated by preparative HPLC to afford the glycoprotein product 11 (415 μg, 75%). tR = 21.3 min (analytic HPLC, method D). ESI-MS: calculated, M = 15305; found, 15307.7 (deconvolution data) Further structural characterization of the glycoprotein was performed by treatment of the glycoprotein with PNGase F. The N-glycan released from the glycoprotein was confirmed to be the complex type N-glycan, (Galβ1,4GlcNAcβ1,2Manα1,6)[Galβ1,4GlcNAcβ1,2Manα1,3]Manβ1,4GlcNAcβ1,4GlcNAc. MALDI-TOF MS of the released N-glycan, calculated for C62H104N4O46, M = 1641.49; Found, 1664.53 [M + Na]+.
EndoA-N171A catalyzed transfer of high-mannose type glycan to GlcNAc-RNase B. Synthesis of Man9GlcNAc2-RNase B (12)
A solution of Man9GlcNAc oxazoline (9) (500 μg, 0.30 μmol) and GlcNAc-RNase B19 (500 μg, 0.036 μmol) in a phosphate buffer (50 mM, pH 7.0, 10 μL) was incubated with EndoA-N171A (200 μg) at 23 °C for 8 h. The reaction was monitored by analytic HPLC and the glycoprotein product was isolated by preparative HPLC to give the Man9GlcNAc2-RNase B (12) as a white foam after lyophilization (460 μg, 82%). tR = 21.2 min (analytic HPLC, method D). ESI-MS: calculated, M = 15547; found, 15548.9 (deconvolution data). Further structural characterization of the glycoprotein product was performed by treatment of the glycoprotein with PNGase F. The glycan released from the glycoprotein was confirmed to be Man9GlcNAc2. MALDI-TOF of the released N-glycan: calculated for C70H118N2O56, M = 1882.64; found, 1905.99 [M + Na]+, 1944.32 [M - mannose + Na]+.
Kinetic studies
For the measurement of the kinetic parameters of Man9GlcNAc oxazoline hydrolysis by Endo-A and its mutant, a solution of Man9GlcNAc oxazoline (9) at various concentrations (0.3125, 0.625, 1.25, 2.5, 5.0 and 10 mM, respectively) in a Tris-HCl buffer (200 mM, pH 8.0, total volume, 5 μl) was incubated with 10 ng of EndoA or 3 μg of N171A at 30 °C. The reaction was terminated after 30 min by the addition of an equal volume of 20% tetrahydrofuran (THF) in aqueous sodium hydroxide (0.1 M). The aliquots were then analyzed by high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) and the hydrolytic product Man9GlcNAc was quantified according to our previously described method.27 For the measurement of the kinetic parameters for the transglycosylation by EndoA-N171A and EndoM-N175A with oxazoline 9 and GlcNAc-pentapeptide 5, the enzymatic reactions were performed at 30 °C for 30 min in a Tris-HCl buffer (200 mM, pH 8.0, total volume, 5 μl). To determine the Km value for the Man9GlcNAc oxazoline, the oxazoline at various concentrations (0.3125, 0.625, 1.25, 2.5, 5.0 and 10 mM, respectively) was incubated with 6 μg of EndoA-N171A or EndoM-N175A and a fixed concentration (20 mM) of GlcNAc-pentapeptide 5 in the buffer. To determine the apparent Km value of GlcNAc-pentapeptide (5), the oxazoline concentration was fixed at 10 mM, and the concentrations of 5 were varied as follows: (0.3125, 0.625, 1.25, 2.5, 5.0, 10, and 20 mM, respectively. The reaction was monitored by analytic HPLC. The parameters Km and Vmax were obtained by fitting the experimental data into the Michaelis-Menten kinetics model in GraphPad Prism.
Thermo-stability of EndoA and EndoM mutants
To determine the thermo-stability of mutants, 5 μg of EndoA-N171A or EndoM-N175A was incubated at various temperatures (30, 35, 40, 45, and 50 °C) for 10 min. Then the enzymes were cooled on ice for 5 min and their activity was assayed as follows: an aliquot of the treated enzyme was incubated with the oxazoline (9) (10 mM) and the GlcNAc-pentapeptide 5 (20 mM) in a Tris-HCl buffer (200 mM, pH 8.0, total volume: 5 μL) at 30 °C for 1 h. The reaction was then terminated by 10% TFA and the transglycosylation product 10 was quantified by HPLC analysis.
Supplementary Material
Acknowledgment
This work was supported by the National Institutes of Health (GM080374 and AI067111).
Footnotes
Supporting Information available: The 1H-NMR spectra of the asialo-glycopeptide from hen's egg yolk, the complex-type oligosaccharide (1), and the complex-type glycan oxazoline (4); The 2D NMR (1H-13C HSQC and 1H-1H NOESY) spectra of the Asn-linked N-glycan obtained from pronase digestion of glycopeptide (6). This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).(a) Grogan MJ, Pratt MR, Marcaurelle LA, Bertozzi CR. Annu. Rev. Biochem. 2002;71:593–634. doi: 10.1146/annurev.biochem.71.110601.135334. [DOI] [PubMed] [Google Scholar]; (b) Davis BG. Chem. Rev. 2002;102:579–601. doi: 10.1021/cr0004310. [DOI] [PubMed] [Google Scholar]; (c) Buskas T, Ingale S, Boons GJ. Glycobiology. 2006;16:113R–136R. doi: 10.1093/glycob/cwj125. [DOI] [PubMed] [Google Scholar]
- (2).(a) Seitz O. ChemBioChem. 2000;1:214–246. doi: 10.1002/1439-7633(20001117)1:4<214::AID-CBIC214>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]; (b) Herzner H, Reipen T, Schultz M, Kunz H. Chem. Rev. 2000;100:4495–4538. doi: 10.1021/cr990308c. [DOI] [PubMed] [Google Scholar]; (c) Wong CH. J. Org. Chem. 2005;70:4219–4225. doi: 10.1021/jo050278f. [DOI] [PubMed] [Google Scholar]; (d) Pratt MR, Bertozzi CR. Chem. Soc. Rev. 2005;34:58–68. doi: 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]; (e) Liu L, Bennett CS, Wong CH. Chem. Commun. (Camb) 2006:21–33. doi: 10.1039/b513165k. [DOI] [PubMed] [Google Scholar]; (f) Brik A, Ficht S, Wong CH. Curr. Opin. Chem. Biol. 2006;10:638–644. doi: 10.1016/j.cbpa.2006.10.003. [DOI] [PubMed] [Google Scholar]
- (3).Bennett CS, Wong CH. Chem. Soc. Rev. 2007;36:1227–1238. doi: 10.1039/b617709c. [DOI] [PubMed] [Google Scholar]
- (4).Wang LX. Carbohydr. Res. 2008;343:1509–1522. doi: 10.1016/j.carres.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:6576–6578. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]; (b) Macmillan D, Bertozzi CR. Angew. Chem. Int. Ed. 2004;43:1355–1359. doi: 10.1002/anie.200352673. [DOI] [PubMed] [Google Scholar]; (c) Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew. Chem. Int. Ed. 2006;45:4116–4125. doi: 10.1002/anie.200600538. [DOI] [PubMed] [Google Scholar]; (d) Brik A, Ficht S, Yang YY, Bennett CS, Wong CH. J. Am. Chem. Soc. 2006;128:15026–15033. doi: 10.1021/ja065601q. [DOI] [PubMed] [Google Scholar]; (e) Yang M, Davies GJ, Davis BG. Angew. Chem. Int. Ed. 2007;46:3885–3888. doi: 10.1002/anie.200604177. [DOI] [PubMed] [Google Scholar]; (f) Wan Q, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]; (g) Chen J, Wan Q, Yuan Y, Zhu J, Danishefsky SJ. Angew. Chem. Int. Ed. 2008;47:8521–8524. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. J. Am. Chem. Soc. 2008;130:501–510. doi: 10.1021/ja072543f. [DOI] [PubMed] [Google Scholar]; (i) Bennett CS, Dean SM, Payne RJ, Ficht S, Brik A, Wong CH. J. Am. Chem. Soc. 2008;130:11945–11952. doi: 10.1021/ja8010513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yamamoto K. J. Biosci. Bioeng. 2001;92:493–501. doi: 10.1263/jbb.92.493. [DOI] [PubMed] [Google Scholar]
- (7).Takegawa K, Tabuchi M, Yamaguchi S, Kondo A, Kato I, Iwahara S. J. Biol. Chem. 1995;270:3094–3099. doi: 10.1074/jbc.270.7.3094. [DOI] [PubMed] [Google Scholar]
- (8).Wang LX, Tang M, Suzuki T, Kitajima K, Inoue Y, Inoue S, Fan JQ, Lee YC. J. Am. Chem. Soc. 1997;119:11137–11146. [Google Scholar]
- (9).(a) Deras IL, Takegawa K, Kondo A, Kato I, Lee YC. Bioorg. Med. Chem. Lett. 1998;8:1763–1766. doi: 10.1016/s0960-894x(98)00306-0. [DOI] [PubMed] [Google Scholar]; (b) Singh S, Ni J, Wang LX. Bioorg. Med. Chem. Lett. 2003;13:327–330. doi: 10.1016/s0960-894x(02)01025-9. [DOI] [PubMed] [Google Scholar]
- (10).Fujita K, Tanaka N, Sano M, Kato I, Asada Y, Takegawa K. Biochem. Biophys. Res. Commun. 2000;267:134–138. doi: 10.1006/bbrc.1999.1963. [DOI] [PubMed] [Google Scholar]
- (11).Li H, Singh S, Zeng Y, Song H, Wang LX. Bioorg. Med. Chem. Lett. 2005;15:895–898. doi: 10.1016/j.bmcl.2004.12.066. [DOI] [PubMed] [Google Scholar]
- (12).Wang LX, Song H, Liu S, Lu H, Jiang S, Ni J, Li H. ChemBioChem. 2005;6:1068–1074. doi: 10.1002/cbic.200400440. [DOI] [PubMed] [Google Scholar]
- (13).(a) Yamamoto K, Kadowaki S, Watanabe J, Kumagai H. Biochem. Biophys. Res. Commun. 1994;203:244–252. doi: 10.1006/bbrc.1994.2174. [DOI] [PubMed] [Google Scholar]; (b) Haneda K, Inazu T, Yamamoto K, Kumagai H, Nakahara Y, Kobata A. Carbohydr. Res. 1996;292:61–70. doi: 10.1016/s0008-6215(96)91025-3. [DOI] [PubMed] [Google Scholar]; (c) Mizuno M, Haneda K, Iguchi R, Muramoto I, Kawakami T, Aimoto S, Yamamoto K, Inazu T. J. Am. Chem. Soc. 1999;121:284–290. [Google Scholar]
- (14).(a) O'Connor SE, Pohlmann J, Imperiali B, Saskiawan I, Yamamoto K. J. Am. Chem. Soc. 2001;123:6187–6188. doi: 10.1021/ja010094s. [DOI] [PubMed] [Google Scholar]; (b) Saskiawan I, Mizuno M, Inazu T, Haneda K, Harashima S, Kumagai H, Yamamoto K. Arch. Biochem. Biophys. 2002;406:127–134. doi: 10.1016/s0003-9861(02)00416-2. [DOI] [PubMed] [Google Scholar]; (c) Akaike E, Tsutsumida M, Osumi K, Fujita M, Yamanoi T, Yamamoto K, Fujita K. Carbohydr. Res. 2004;339:719–722. doi: 10.1016/j.carres.2003.12.007. [DOI] [PubMed] [Google Scholar]; (d) Osumi K, Makino Y, Akaike E, Yamanoi T, Mizuno M, Noguchi M, Inazu T, Yamamoto K, Fujita K. Carbohydr. Res. 2004;339:2633–2635. doi: 10.1016/j.carres.2004.08.014. [DOI] [PubMed] [Google Scholar]; (e) Haneda K, Tagashira M, Yoshino E, Takeuchi M, Inazu T, Toma K, Iijima H, Isogai Y, Hori M, Takamatsu S, Fujibayashi Y, Kobayashi K, Yamamoto K. Glycoconj. J. 2004;21:377–386. doi: 10.1023/B:GLYC.0000046277.92806.74. [DOI] [PubMed] [Google Scholar]; (f) Haneda K, Takeuchi M, Tagashira M, Inazu T, Toma K, Isogai Y, Hori M, Kobayashi K, Takegawa K, Yamamoto K. Carbohydr. Res. 2006;341:181–190. doi: 10.1016/j.carres.2005.11.015. [DOI] [PubMed] [Google Scholar]
- (15).Fujita K, Yamamoto K. Biochim. Biophys. Acta. 2006;1760:1631–1635. doi: 10.1016/j.bbagen.2006.09.003. [DOI] [PubMed] [Google Scholar]
- (16).(a) Fujita M, Shoda S, Haneda K, Inazu T, Takegawa K, Yamamoto K. Biochim. Biophys. Acta. 2001;1528:9–14. doi: 10.1016/s0304-4165(01)00164-7. [DOI] [PubMed] [Google Scholar]; (b) Li H, Li B, Song H, Breydo L, Baskakov IV, Wang LX. J. Org. Chem. 2005;70:9990–9996. doi: 10.1021/jo051729z. [DOI] [PubMed] [Google Scholar]; (c) Rising TW, Claridge TD, Moir JW, Fairbanks AJ. ChemBioChem. 2006;7:1177–1180. doi: 10.1002/cbic.200600183. [DOI] [PubMed] [Google Scholar]; (d) Rising TW, Claridge TD, Davies N, Gamblin DP, Moir JW, Fairbanks AJ. Carbohydr. Res. 2006;341:1574–1596. doi: 10.1016/j.carres.2006.03.007. [DOI] [PubMed] [Google Scholar]; (e) Rising TW, Heidecke CD, Moir JW, Ling Z, Fairbanks AJ. Chem. Eur. J. 2008;14:6444–6464. doi: 10.1002/chem.200800365. [DOI] [PubMed] [Google Scholar]
- (17).Li B, Zeng Y, Hauser S, Song H, Wang LX. J. Am. Chem. Soc. 2005;127:9692–9693. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]
- (18).Zeng Y, Wang J, Li B, Hauser S, Li H, Wang LX. Chem. Eur. J. 2006;12:3355–3364. doi: 10.1002/chem.200501196. [DOI] [PubMed] [Google Scholar]
- (19).Li B, Song H, Hauser S, Wang LX. Org. Lett. 2006;8:3081–3084. doi: 10.1021/ol061056m. [DOI] [PubMed] [Google Scholar]
- (20).Wei Y, Li C, Huang W, Li B, Strome S, Wang LX. Biochemistry. 2008;47:10294–10304. doi: 10.1021/bi800874y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ochiai H, Huang W, Wang LX. J. Am. Chem. Soc. 2008;130:13790–13803. doi: 10.1021/ja805044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Mackenzie LF, Wang QP, Warren RAJ, Withers SG. J. Am. Chem. Soc. 1998;120:5583–5584. [Google Scholar]; (b) Malet C, Planas A. FEBS Lett. 1998;440:208–212. doi: 10.1016/s0014-5793(98)01448-3. [DOI] [PubMed] [Google Scholar]; (c) Perugino G, Trincone A, Rossi M, Moracci M. Trends Biotechnol. 2004;22:31–37. doi: 10.1016/j.tibtech.2003.10.008. [DOI] [PubMed] [Google Scholar]; (d) Hancock SM, Vaughan MD, Withers SG. Curr. Opin. Chem. Biol. 2006;10:509–519. doi: 10.1016/j.cbpa.2006.07.015. [DOI] [PubMed] [Google Scholar]; (e) Faijes M, Planas A. Carbohydr. Res. 2007;342:1581–1594. doi: 10.1016/j.carres.2007.06.015. [DOI] [PubMed] [Google Scholar]
- (23).Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang LX, Yamamoto K. J. Biol. Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]
- (24).Seko A, Koketsu M, Nishizono M, Enoki Y, Ibrahim HR, Juneja LR, Kim M, Yamamoto T. Biochim. Biophys. Acta. 1997;1335:23–32. doi: 10.1016/s0304-4165(96)00118-3. [DOI] [PubMed] [Google Scholar]
- (25).Kajihara Y, Suzuki Y, Yamamoto N, Sasaki K, Sakakibara T, Juneja LR. Chem. Eur. J. 2004;10:971–985. doi: 10.1002/chem.200305115. [DOI] [PubMed] [Google Scholar]
- (26).Heidecke CD, Ling Z, Bruce NC, Moir JW, Parsons TB, Fairbanks AJ. ChemBioChem. 2008;9:2045–2051. doi: 10.1002/cbic.200800214. [DOI] [PubMed] [Google Scholar]
- (27).Wang LX, Ni J, Singh S, Li H. Chem. Biol. 2004;11:127–134. doi: 10.1016/j.chembiol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- (28).Witte K, Sears P, Martin R, Wong CH. J. Am. Chem. Soc. 1997;119:2114–2118. [Google Scholar]
- (29).(a) Dawson PE, Kent SB. Annu. Rev. Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923. [DOI] [PubMed] [Google Scholar]; (b) Schwarzer D, Cole PA. Curr. Opin. Chem. Biol. 2005;9:561–569. doi: 10.1016/j.cbpa.2005.09.018. [DOI] [PubMed] [Google Scholar]
- (30).(a) Wang L, Schultz PG. Angew. Chem. Int. Ed. 2004;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z, Gildersleeve J, Yang YY, Xu R, Loo JA, Uryu S, Wong CH, Schultz PG. Science. 2004;303:371–373. doi: 10.1126/science.1089509. [DOI] [PubMed] [Google Scholar]; (c) Fahmi NE, Dedkova L, Wang B, Golovine S, Hecht SM. J. Am. Chem. Soc. 2007;129:3586–3597. doi: 10.1021/ja067466n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.