

Abstract
Mutations of the human B-RAF gene are detected in ∼8% of cancer samples, primarily in cutaneous melanomas (70%). The most common mutation (90%) is a valine-to-glutamic acid mutation at residue 600 (V600E; formerly V599E according to previous nomenclature). Using a Cre/Lox approach, we have generated a conditional knock-in allele of V600EB-raf in mice. We show that widespread expression of V600EB-Raf cannot be tolerated in embryonic development, with embryos dying ∼7.5 dpc. Directed expression of mutant V600EB-Raf to somatic tissues using the IFN-inducible Mx1-Cre mouse strain induces a proliferative disorder and bone marrow failure with evidence of nonlymphoid neoplasia of the histiocytic type leading to death within 4 weeks of age. However, expression of mutant B-Raf does not alter the proliferation profile of all somatic tissues. In primary mouse embryonic fibroblasts, expression of endogenous V600EB-Raf induces morphologic transformation, increased cell proliferation, and loss of contact inhibition. Thus, V600EB-Raf is able to induce several hallmarks of transformation in some primary mouse cells without evidence for the involvement of a cooperating oncogene or tumor suppressor gene.
Introduction
Raf oncogenes were first identified as transforming genes in retroviruses that caused tumors in mice and chickens (1, 2). Oncogenic homologues of C-Raf and B-RAF were associated with the induction of carcinomas, sarcomas, and cancers of the hemopoietic lineage in these species (3, 4). It has since been discovered that oncogenic mutations of the B-RAF gene are present in human cancer samples (5). This study analyzed >900 cancer samples and human cancer cell lines and detected the presence of B-RAF missense mutations in ∼70% of human malignant melanomas. B-RAF mutations are also present at a high frequency in papillary thyroid cancer (36-53%; refs. 6-8), colorectal cancer (5-22%; ref. 9), and serous ovarian cancer (30%; ref. 10) and at a low frequency in a wide variety of other cancers (1-3%; refs. 5, 11, 12). Thus, in a similar way to RAS oncogenes that are detected in 15% to 30% of human cancers (13), B-RAF mutations are a critical event in the development of many human neoplasias.
Over 30 missense B-RAF mutations have now been identified in cancer samples (11, 14). The most common mutation, accounting for ∼90% of all those detected, is T1799A resulting in a glutamic acid-to-valine substitution at position 600 within the activation segment of the B-Raf kinase domain. V600EB-Raf induces 500-fold more active basal B-Raf kinase activity than basal WTB-RAF and exceeds G12VRAS-induced WTB-RAF activity (14). Recent structural studies have shown that this is due to the ability of the V600E mutation to disrupt the normal hydrophobic interaction between the activation segment and P-loop that maintains basal B-Raf in an inactive conformation (14). In WTB-RAF, phosphorylation of residues Thr599 and Ser602 normally disrupt this interaction, allowing activation of B-RAF. The increased activity of V600EB-RAF has the effect of stimulating endogenous, basal mitogen-activated protein/extracellular signal-regulated kinase (MAP/ERK) kinase (MEK) and ERK1/2 activation, leading to increased cell proliferation and survival (14-17). The effect of V600EB-RAF on proliferation in mouse melanocytes and melanomas can be reversed by treatment of these cells with MEK inhibitors or with small interfering RNA (siRNA) to B-RAF. Thus, this mutant is thought to transform and mediate its tumorigenic effects in a similar way to oncogenic RAS. This is reinforced by the fact that B-RAF and RAS mutations are rarely present in the same cancer samples, indicating that they may have overlapping functions in tumorigenesis (5, 11, 12).
Although V600EB-RAF has the ability to induce G0-G1-S progression in tissue culture systems, it is not clear whether this is the case in tumor development in vivo. Oncogenic B-RAF mutations have been detected in benign nevi and premalignant colon polyps, suggesting that additional mutations in other key oncogenes/tumor suppressor genes are required to unleash the tumorigenic effects of B-RAF (9, 18). Candidate genes for such additional mutation are INK4A and PTEN that are frequently found mutated in melanomas (19, 20). Work in tissue culture systems has also shown that the ability of the RAF/MEK/ERK cascade to induce G0-G1-S cell cycle progression is dependent on the cooperative activation of the phosphatidylinositol 3-kinase (PI3K)/PDK1/AKT pathway (21) or inactivation of cyclin-dependent kinase inhibitors (22, 23), including p16INK4A (24, 25).
To investigate how B-RAF is involved in tumorigenesis, and whether oncogene/tumor suppressor gene cooperation has a key function in development of tumors with B-RAF mutations, we have produced a tractable mouse model that mimics the somatic expression of V600EB-Raf in human cancers. These transgenic mice contain a heterozygous knock-in mutation of V600EB-raf whose expression is determined by the presence or absence of a Lox-STOP-Lox (LSL) cassette. We show that following deletion of LSL by the Cre recombinase, expression of V600EB-Raf in somatic tissues leads to development of some hallmarks of cancer in mice.
Materials and Methods
Generation of LSL-B-rafV600E mice
The LSL targeting vector was assembled by cloning left, middle, and right arms in the ploxP-neo-loxP vector. The right arm fragment contains exon 15 with the T1799A mutation. The middle arm contains a LSL cassette with a cDNA encoding exons 15 to 18 of mouse B-raf. This was PCR amplified from a mouse B-raf cDNA clone. The forward primer for this PCR reaction had the sequence 5′-TACTAACCATGCCTTCTTCTTTTTCCTACAGATATATTTCTTCATGAAGACCTCACGGT-3′and contained 31 nucleotides corresponding to a splice acceptor sequence from the α-globin gene followed by 28 nucleotides corresponding to exon 15 of the mouse B-raf gene. The reverse primer had the sequence 5′-CGAGTGAGAGACACAAAAAATTCCAACACACTATTGTTGCTACCTTACTTTATTGTCTCACTC-3′and contained 35 nucleotides corresponding to the α-globin pause site and 28 nucleotides corresponding to the 3′-untranslated region of the mouse B-raf gene. The vector was electroporated into E14.1 embryonic stem cells, and homologous recombination events were identified. Positive embryonic stem clones were injected into mouse C57BL6 blastocysts and chimeric mice derived using standard procedures. Inheritance of the targeted allele was assessed by PCR genotyping (26, 27) of mouse tail DNA using primers A (5′-GCCCAGGCTCTTTATGAGAA-3′) and B (5′-GCTTGGCTGGACGTAAACTC-3′) for the LSL-B-rafV600E–targeted allele combined with primers A and C (5′-AGTCAATCATCCACAGAGACCT-3′) for the wild-type allele (Fig. 1). A breeding colony of LSL-B-rafV600E mice were established by backcrossing on the C57BL6 background. Genotyping of offspring from LSL-B-rafV600E × Cre crosses was done by using a multiplex PCR reaction using primers A to C as well as primers D (5′-GTTCGCAAGAACCTGATGGACA-3′) and E (5′-CTAGAGCCTGTTTTGCACGTTC-3′) for identifying the Cre gene. To detect the Lox-B-RAFV600E allele, primers A and C were used. All PCRs were done using Reddymix PCR reagent (ABgene, Surrey, United Kingdom) with 35 cycles of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds.
Figure 1.

A, generation of LSL-B-rafV600E and Lox-B-rafV600E alleles. The targeting vector contains left and right arms with the right arm containing exon 15 with the T1799A mutation (*). These two arms are separated by the LSL cassette. This cassette contains three LoxP sequences (black arrows), a minigene (MG) encoding exons 15 to 18 of wild-type B-Raf with a splice acceptor (SA) sequence at the 5′end. Two STOP sequences, represented by polyadenylation (PA) sequences, are located at the 3′end of the minigene and at the 3′end of the neoR cassette. Homologous recombination between the targeting vector and the wild-type B-raf gene in embryonic stem cells generated the LSL-B-rafV600E allele. Expression of the Cre recombinase allows deletion of the LSL cassette and generation of the Lox-B-rafV600E allele. Location of primers used for PCR genotyping (arrows and A-C). B, PCR genotyping to detect LSL-B-rafV600E, WT B-raf, and Cre alleles. Primers A-E were used in combination on tail DNA samples from intercrosses between heterozygous LSL-B-rafV600E mice and Cre mice. In this example, the WT B-raf allele (466 bp), Cre allele (350 bp), and LSL-B-rafV600E allele (140 bp) are analyzed in tail DNAs from heterozygous LSL-B-rafV600E mice without Cre (lane 1), wild-type mice with Cre (lane 2), and heterozygous LSL-B-rafV600E mice with Cre (lane 3). C, PCR genotyping to detect Lox-B-rafV600E and WT B-raf alleles. Primers A and C were used on the same DNA samples described above in (B). A 518-bp product indicates the presence of the Lox-B-rafV600E, and a 466-bp product indicates the presence of the WT B-raf allele. D, photographs of E7.5 embryos resulting from LSL-B-rafV600E × CMV-Cre intercrosses showing LSL-B-rafV600E − CMV-Cre embryo (left) and LSL-B-rafV600E + CMV-Cre embryo (right). Bar, 250 μm.
Histology, immunohistochemistry, and proliferation assays
Mouse tissues were processed for histology by previously described methods (26). Sections were either stained with H&E or used for immunohistochemistry with antibodies for Ki67 (Novocastra Laboratories Ltd., Newcastle upon Tyne, United Kingdom), Mac-2 (Cedarlane Laboratories Ltd., Hornby, Ontario, Canada), and phospho-ERK (Cell Signaling Technology, Beverly, MA) or used for terminal deoxynucleotidyl transferase–mediated nick-end labeling (TUNEL) analysis as described previously (26, 27). To assess proliferation, mice were injected with 10 μg/mL bromodeoxyuridine (BrdUrd), and tissues were either analyzed by immunohistochemistry with an α-BrdUrd antibody or, in the case of the spleen, cells were dissociated by passing through a mesh, counted, and then stained with α-BrdUrd-FITC and analyzed by fluorescence-activated cell sorting (FACS) using the kit and method provided by BD Biosciences (San Jose, CA). Bone marrow was collected from the femurs of freshly culled mice, and cells were quantitated using a Coulter counter (Becton Dickinson, Mountain View, CA).
Northern blot analysis
Mouse spleens were snap-frozen in liquid nitrogen and ground to a powder. Total cellular RNA was isolated using the RNAzol-B technique (Biogenesis Ltd., Poole, United Kingdom). RNA was resolved by denaturing 1.2% agarose/18% (v/v) formaldehyde gel electrophoresis and transferred to GeneScreen (Dupont, Boston, MA), before UV cross-linking. cDNA inserts were labeled (Random primers DNA labeling system, Amersham Biosciences, Buckinghamshire, United Kingdom) using [α-32P]dATP (Amersham; 3,000 Ci/mmol). Filters were hybridized in a solution containing 7% (w/v) SDS, 1% (w/v) bovine serum albumin, 0.25 mol/L NaCl, and 0.5 mol/L Na2HPO4 buffer (pH 7.2) at 65°C for 16 hours. They were washed sequentially at 65°C in 4× SSC, 0.5× SSC, and 0.2× SSC, each containing 0.1% SDS, and resolved by autoradiography.
Western blots and kinase assays
Mouse tissues were snap-frozen in liquid nitrogen and homogenized in NP40 lysis buffer [50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 0.5% NP40, 5 mmol/L NaF] with 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L aprotinin, 1 mmol/L leupeptin, and 1 mmol/L sodium orthovanadate. Western blots were carried out as described previously (28). B-Raf kinase assays were done using the immunoprecipitation kinase cascade assay as previously reported (29) using the B-Raf antibody provided by Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Primary antibodies were a 1:1,000 dilution of a mouse monoclonal antibody against Thr202/Tyr204 phospho-p44/42 ERK1/2 (Cell Signaling Technology), a 1:1,000 dilution of a rabbit polyclonal antibody for ERK2 (Zymed Laboratories, Inc., South San Francisco, CA), a 1:1,000 dilution of a rabbit polyclonal antibody against Ser217/Ser221 phospho-MEK1/2 (Cell Signaling Technology), a 1:1,000 dilution of a cyclin D1 antibody (Cell Signaling Technology), a 1:1,000 dilution of an antibody for actin (Sigma, St. Louis, MO), and a 1:1,000 dilution of an antibody for p21CIP1 (Cell Signaling Technology).
Mouse embryonic fibroblast analysis
Mouse embryonic fibroblasts (MEF) were isolated and cultured from homogenized E14.5 embryos arising from timed matings between LSL-B-rafV600E animals and wild-type C57BL6 animals using standard methods (26). MEFs were transfected with the pCrePac plasmid using a Nucleofector under the MEF conditions recommended by the manufacturer (Amaxa Biosystems, Koln, Germany). Forty-eight hours following transfection, cells were photographed and processed for immunofluorescence analysis of the actin cytoskeleton by staining with FITC-phalloidin as described previously (30). For proliferation, mock-transfected and Cre-transfected cells, 24 hours after transfection, were placed in serum-free media for 20 hours and then either left unstimulated or stimulated with 10% (v/v) FCS for 16 hours. BrdUrd incorporation assays were done by using the BrdUrd labeling and detection kit (Roche, Indianapolis, IN). The percentage of BrdUrd-positive cells was visualized by fluorescence microscopy using a Zeiss Axiophot microscope. For protein analysis, 24 hours following transfection, cells were serum starved for 20 hours and then stimulated with FCS for 10 minutes or for 16 hours. Protein lysates were generated, and Western blots were carried out as described above. Focus-forming assays were done as described elsewhere (31), and agar assays were done as described previously (32).
Results
Numbering of the amino acids in B-Raf
The valine residue that is most commonly mutated in human cancer was originally reported as being at position 599 of the human B-Raf protein. However, as highlighted by Wellbrock et al. (33), more recent sequencing data have shown that there was a previous error in the sequencing of the B-RAF 5′coding region, such that this valine is now reclassified as being at residue 600. In the mouse, the 5′coding region has not yet been fully sequenced and thus the residue number of the equivalent valine is not known. For the present purposes, this residue will also be referred to as Val600.
Generation of LSL-B-rafV600E mice
We used the Cre-LoxP system to generate transgenic animals containing a conditional knock-in allele that expresses V600EB-Raf only after Cre-mediated recombination (Fig. 1A). The targeting vector contained a left arm spanning exon 14 and a right arm containing exon 15 with the B-rafV600E mutation, representing a T1799A nucleotide change. These two arms were separated from each other by the neoR gene and a minigene cDNA encoding wild-type exons 15 to 18. Three LoxP sequences were present in the vector and two polyadenylation STOP sequences. In this arrangement, wild-type B-Raf is expressed from the targeted gene with exons 15 to 18 being encoded by the minigene. Introduction of the Cre recombinase allows deletion of the LSL cassette and expression of V600EB-Raf. The vector was introduced into embryonic stem cells, and homologous recombination events were identified. Mice were generated from the positive clones, and a breeding colony was established on the C57BL6 background. PCR genotyping confirmed the inheritance of the LSL-B-rafV600E allele (Fig. 1B).
Expression of V600EB-Raf in embryogenesis
We generated mice that permanently express V599EBRaf in all tissues. This was achieved by crossing the LSL-B-rafV600E mice to heterozygous CMV-Cre mice that express the Cre transgene in germ cells as well as in a mosaic manner during development (34). Multiplex PCR genotyping was used to identify LSL-B-rafV600E + CMV-Cre animals (Fig. 1B). These were then analyzed by PCR for the presence of the Cre-deleted Lox-B-rafV600E allele expressing V600EB-Raf (Fig. 1C). Genotyping of the surviving offspring revealed no viable LSL-B-rafV600E + CMV-Cre animals (Table 1). The LSL-B-rafV600E and CMV-Cre mice were thus mated at timed intervals, and embryos were harvested at various ages of gestation. LSL-B-rafV600E + CMV-Cre embryos were obtained at the expected frequency at E7.5, but lethality was observed from this point onwards. At E7.5, all embryos with the LSL-B-rafV600E allele were abnormal and in the process of being reabsorbed (Fig. 1D). Only those embryos that were mosaic for the Lox-B-rafV600E and LSL-B-rafV600E alleles survived to later ages of gestation (Table 1), but even these all died before birth. This indicates that the expression of V600EB-Raf in gestation is lethal to the animal, although the embryos are able to implant.
Table 1.
Genotypes of embryos arising from intercrosses between LSL-B-raf V600E and CMV-Cre heterozygous animals
| Age | B-raf +/+ | B-raf +/+ + CMV-Cre | LSL-B-raf V600E − CMV-Cre | LSL-B-raf V600E + CMV-Cre | Untyped |
|---|---|---|---|---|---|
| E7.5 | 7 | 6 | 4 | 10 | 0 |
| E8.5 | 5 | 4 | 5 | 5 | 1 |
| E9.5 | 0 | 4 | 2 | 1* | 0 |
| E10.5 | 3 | 6 | 3 | 2* | 5 |
| E12.5 | 2 | 5 | 5 | 1* | 9 |
| Birth (P1) | 13 | 12 | 13 | 0 | 0 |
NOTE: Analysis of embryos at E7.5 and E8.5 showed that LSL-B-rafV600E + CMV-Cre embryos were obtained at the expected frequency at these developmental ages but were obtained at reduced frequencies at later developmental ages, and none survived to birth. At E7.5 and E8.5, all LSL-B-rafV600E + CMV-Cre embryos containing the Lox-B-rafV600E allele were abnormal and in the process of being reabsorbed (see Fig. 1D).
These later surviving embryos were all mosaic for the LSL-B-rafV600E and Lox-B-rafV600E alleles.
Somatic expression of V600EB-Raf
To examine the effect of the oncogenic V600EB-raf mutation on somatic tissues, we crossed the LSL-B-rafV600E mice to Mx1-Cre mice and examined the offspring. The Mx1-Cre mouse strain expresses the Cre recombinase from an IFN-inducible promoter and can be induced to express the Cre recombinase either by injection of IFN or polyriboinosic/polyribocytidylic acid colpolymer (pI-pC; ref. 35). Consistent with previous observations with the Mx1-Cre mice, we found that the LSL-B-rafV600E + Mx1-Cre animals showed a phenotype even without injection of pI-pC (Fig. 2A; refs. 36, 37). This is likely because of endogenous IFN production in response to a subclinical infection or other stimulus. All of the LSL-B-rafV600E + Mx1-Cre animals died before 4 weeks of age, whereas the LSL-B-rafV600E − Mx1-Cre animals survived the normal life span of a mouse with no symptoms (Fig. 2A). Wild-type mice with the Mx1-Cre allele also showed no phenotype (data not shown). PCR analysis of DNA from various tissues of LSL-B-rafV600E + Mx1-Cre animals indicated the presence of the LSL-B-rafV600E allele in many tissues, including the spleen, liver, lung, and heart (Fig. 2B). However, this allele was present at considerably lower levels in some tissues, particularly, the kidney, testis, gut, and thymus.
Figure 2.
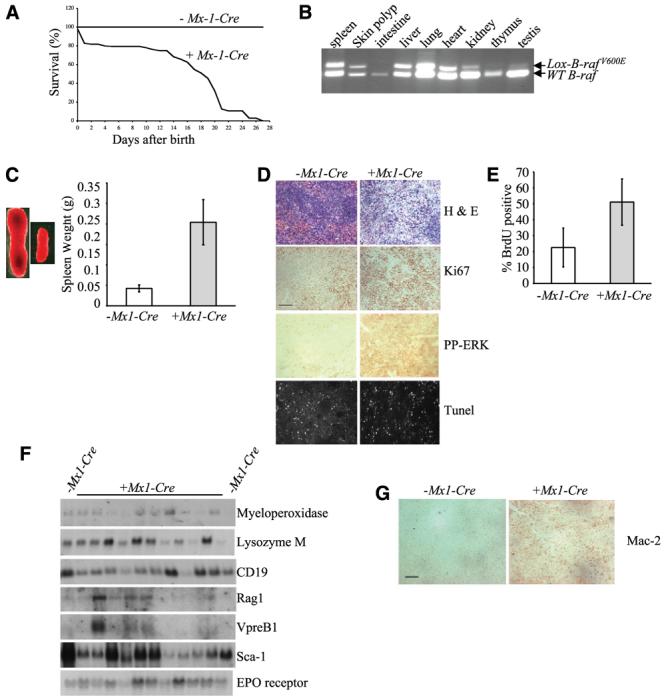
Analysis of mice resulting from Lox-B-rafV600E × Mx1-Cre intercrosses. A, % survival of LSL-B-rafV600E + Mx1-Cre animals versus LSL-B-rafV600E − Mx1-Cre animals at various ages after birth. B, PCR genotyping to detect Lox-B-rafV600E allele in DNA from various tissues derived from LSL-B-rafV600E + Mx1-Cre mice. C, splenomegaly in LSL-B-rafV600E + Mx1-Cre mice. Left, increase in size of the spleen of P21 animals. Right, pooled weight data from four spleens of LSL-B-rafV600E ± Mx1-Cre mice at P21. LSL-B-rafV600E + Mx1-Cre mice have a mean spleen weight of 0.2 g compared with 0.04 g for the control LSL-B-rafV600E − Mx1-Cre mice. Columns, means; bars, SD. D, histologic analysis of spleen sections from LSL-B-rafV600E ± Mx1-Cre mice. Spleen sections were either stained with H&E, with antibodies for Ki67 or phospho-ERK, or processed for TUNEL analysis. Bar, 50 μm (the same for all sections). E, proliferation analysis of spleens from LSL-B-rafV600E ± Mx1-Cre mice. Mice were injected with BrdUrd, splenocytes were isolated, and the % incorporation of BrdUrd was assessed by FACS analysis. Columns, means; bars, SD. F, Northern blot analysis of spleen RNA isolated from LSL-B-rafV600E ± Mx1-Cre mice. Northern blots were probed with the markers indicated. G, histologic analysis of spleen sections from LSL-B-rafV600E ± Mx1-Cre mice with a histiocyte marker. Spleen sections were stained with a Mac-2 antibody to detect histiocyte amplification. Bar, 50 μm.
Tissues with the LSL-B-rafV600E allele were subjected to further investigation. No alterations were detected in the morphology or pathology of the lung, heart, kidney, gut, or testis (data not shown). However, the spleen and liver were abnormal. Splenomegaly was observed in all LSL-B-rafV600E +Mx1-Cre animals (Fig. 2C). 2C). Histologic analysis of spleen sections showed the infiltration of a proliferating, pleomorphic population of cells with abundant eosinophilic cytoplasm and irregular nuclei as well as extramedullary sites of hemopoiesis (Fig. 2D). The spleens of the LSL-B-rafV600E + Mx1-Cre mice also stained far more strongly with antibodies for the S-phase marker Ki67 as well as phospho-ERK than LSL-B-rafV600E − Mx1-Cre spleens, but there were no noticeable changes in apoptosis, as measured by TUNEL analysis (Fig. 2D). To quantitate proliferation in vivo, mice were injected with BrdUrd, and incorporation into splenocytes was measured by FACS analysis (Fig. 2E). The splenocytes of LSL-B-rafV600E + Mx1-Cre Cre mice (mean = 51.1%, n = 6) proliferated 2-3× more than splenocytes from LSL-B-rafV600E − Mx1-Cre mice (mean = 22.6%, n = 6, P = 0.0015). To identify which cell types were amplifying in the spleen, Northern blot analysis was done on spleen RNA with probes for genes expressed in the myeloid (myeloperoxidase and lysozyme M), lymphoid (CD19, rag-1, and VpreB1), stem cell (Sca1), and erythroid (erythropoietin receptor) lineages. No consistent increase in expression of any of these genes was observed in LSL-B-rafV600E + Mx1-Cre spleens compared with LSL-B-rafV600E − Mx1-Cre spleens (Fig. 2F). However, the spleens from LSL-B-rafV600E + Mx1-Cre mice stained strongly for Mac2 (Fig. 2G), indicating the presence of an amplified population of histiocytes.
Histologic analysis of the liver also indicated the presence of extramedullary sites of hemopoiesis. Ki67 staining indicated increased proliferation in the liver of LSL-B-rafV600E + Mx1-Cre mice, associated with increased phospho-ERK staining (Fig. 3A). However, in addition to this, the liver of these mice was also in the process of undergoing apoptosis as indicated by increased TUNEL staining (Fig. 3A). The Lox-BrafV600E allele was strongly detected in the bone marrow of LSL-B-rafV600E + Mx1-Cre animals. This was associated with the virtual absence of RBC in the bone marrow and a reduction in the production of WBC by at least 50% (Fig. 3B). Circulating RBC and WBC were also significantly reduced (data not shown). Additionally, LSL-B-rafV600E + Mx1-Cre animals developed LSL-B-rafV600E–containing histiocytes (Fig. 2B) at extranodal sites that were predominantly observed on the skin (Fig. 3C), although occasionally on the gut wall. These were detectable at birth and contained proliferating, phospho-ERK positive cells with a typical histiocyte morphology that stained strongly for Mac2 (Fig. 3D). The appearance of splenomegaly and Mac2-positive staining cells in the spleen and at extranodal sites is consistent with a diagnosis of nonlymphoid leukemia of the histiocytic type (according to Bethesda classification of hemopoietic neoplasms.4
Figure 3.
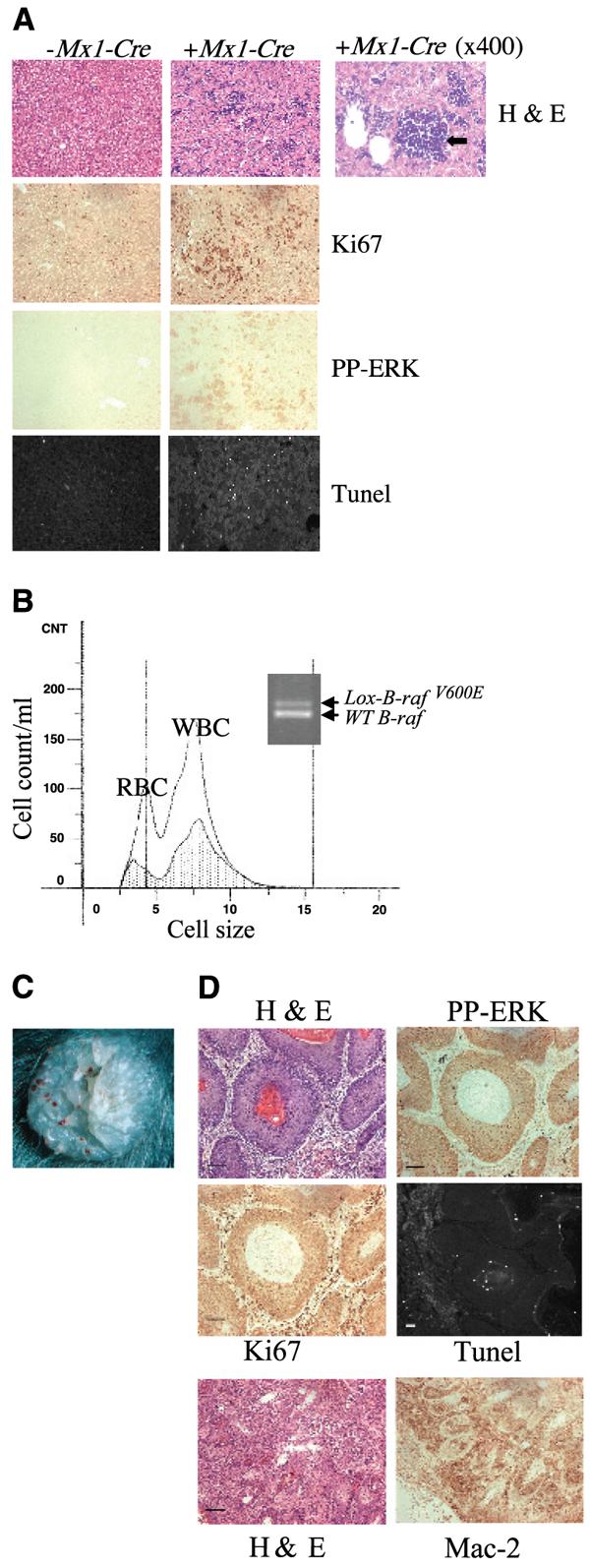
Analysis of tissues from mice resulting from LSL-B-rafV600E × Mx1-Cre intercrosses. A, histologic analysis of liver sections from LSL-B-rafV600E ± Mx1-Cre mice. Liver sections were either stained with H&E, with a Ki67 antibody, with a phospho-ERK antibody or processed for TUNEL analysis. Right, H&E section was photographed at higher magnification (×400) to indicate site of hemopoiesis (black arrow) in the mutant liver. Bar, 50 μm (the same for all sections) except for 25 μm (enlarged H&E section, right). B, analysis of bone marrow from LSL-B-rafV600E ± Mx1-Cre mice. Bone marrow cells were isolated from the femurs of LSL-B-rafV600E ± Mx1-Cre mice and analyzed by the Coulter counter. There are far fewer RBC and WBC in the bone marrow of LSL-B-rafV600E + Mx1-Cre mice (shaded area) than LSL-B-rafV600E − Mx1-Cre mice (clear area). PCR analysis of DNA from bone marrow of LSL-B-rafV600E + Mx1-Cre animal, indicating the presence of the Lox-B-rafV600E allele. C, photograph of histiocyte arising on skin of LSL-B-rafV600E + Mx1-Cre animal. D, histologic analysis of histiocyte sections from LSL-B-rafV600E ± Mx1-Cre mice. Sections were either stained with H&E, with antibodies for Ki67, phospho-ERK, or Mac-2, or processed for TUNEL analysis. Bar, 50 μm.
Expression of V600EB-Raf induces activation of the B-RAF/mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase cascade
We did B-Raf kinase assays on protein lysates generated from spleen, liver, and lung of LSL-B-rafV600E ± Mx1-Cre animals. B-Raf kinase activity was elevated by ∼11-fold in the spleen and by ∼3-fold in the liver and lung of LSL-B-rafV600E + Mx1-Cre animals compared with controls (Fig. 4A). Western blot analysis also showed that levels of phospho-MEK and phospho-ERK were significantly increased in tissues affected by the expression of V600E B-Raf (i.e., the spleen and liver). With regard to cyclin D1, levels of expression were increased in the spleen of LSL-B-rafV600E + Mx1-Cre mice but not the liver, probably because this tissue is also undergoing apoptosis (Fig. 3A). However, tissues that were not morphologically affected by the mutation (lung or thymus) showed an increase in levels of phospho-MEK but not an increase in phospho-ERK or cyclin D1 levels (Fig. 4B).
Figure 4.

Analysis of B-RAF/MEK/ERK activation in tissues derived from LSL-B-rafV600E ± Mx1-Cre mice. Protein lysates were harvested from various tissues and subjected to (A) B-Raf kinase assays and (B) Western blot analysis with antibodies for phospho-ERK, phospho-MEK, and cyclin D1. Immunoblotting with a total ERK antibody was used to confirm protein loading.
Expression of V600EB-Raf induces transformation of primary mouse embryonic fibroblasts
To further investigate the transforming effects of endogenous V600EB-Raf, we derived primary MEFs from LSL-B-rafV600E mice and transfected these cells with a plasmid expressing the Cre recombinase. LSL-B-rafV600E MEFs without Cre showed a typical nontransformed morphology (Fig. 5A). However, transfection with the Cre plasmid induced morphologic transformation and collapse of the actin cytoskeleton (Fig. 5A). In addition, LSL-B-rafV600E MEFs with the Cre plasmid showed a significant increase in proliferation (Fig. 5B). When serum starved, 6.75% of mock-transfected cells entered S phase, whereas in the presence of Cre 9.57% of cells entered S phase (P < 0.002). In the presence of FCS, 11.5% of cells without Cre entered S phase, whereas in the presence of Cre, 26.2% of cells entered S phase (P < 10−6). Thus, serum-starved, Cre-containing LSL-B-rafV600E MEFs undergo proliferation at a similar rate to serum-stimulated, mock-transfected LSL-B-rafV600E MEFs, although proliferation of the Cre-containing LSL-B-rafV600E MEFs was further stimulated by the presence of growth factors. Concomitant with the increase in proliferation in serum-starved, Cre-containing cells, levels of phospho-ERK (Fig. 5C) and cyclin D1 (Fig. 5D) were increased in these cells compared with nontransfected controls, but p21CIP1 levels were drastically reduced (Fig. 5D). LSL-B-rafV600E primary MEFs transfected with the Cre plasmid also acquired the ability to grow to a very high density, with loss of contact inhibition and focus formation after 3 weeks in culture (Fig. 5E), and were able to form very small colonies when grown in soft agar (Fig. 5F).
Figure 5.

Analysis of primary MEFs derived from LSL-B-rafV600E mice. A, morphologic transformation. Primary LSL-B-rafV600E MEFs were either mock-transfected (−Cre) or transfected with the pCrePac plasmid (+Cre). After 48 hours, the cells were either photographed (top) or subjected to immunofluorescence analysis with FITC-phalloidin (bottom). B, proliferation analysis. Mock- and Cre-transfected cells were serum starved for 20 hours (open columns) and then stimulated with serum for 16 hours (gray columns). % BrdUrd incorporation was assessed under each condition. Columns, means; bars, SD. The cells transfected with Cre undergo a high level of proliferation even in the absence of serum compared with mock-transfected controls. C, Western blot analysis of lysates prepared from Cre- and mock-transfected MEFs with an antibody for phospho-ERK. Immunoblotting with a total ERK antibody was used to confirm protein loading. D, Western blot analysis of lysates prepared from Cre- and mock-transfected MEFs with antibodies for cyclin D1 and p21CIP1. Immunoblotting with an antibody for actin was used to confirm protein loading. E, focus formation assay of Cre- and mock-transfected LSL-B-rafV600E primary MEFs. MEFs were grown to confluency and the media was changed every 2 to 3 days until foci formed for 18 days. F, soft agar assays of Cre- and mock-transfected LSL-B-rafV600E primary MEFs.
Discussion
We report the generation of mice with a LSL conditional knock-in mutation of V600EBraf, whereby the expression of endogenous V600EB-Raf is dependent on delivery of the Cre recombinase and deletion of the LSL cassette. We show that somatic tissues expressing endogenous V600EB-Raf have elevated B-Raf kinase activity, elevated levels of phospho-MEK, phospho-ERK, and cyclin D1 expression, and increased proliferation. In addition, expression of V600EB-Raf in primary MEFs induces morphologic transformation as well as hyperproliferation, the loss of contact inhibition, and a weak ability to grow in an anchorage-independent manner. These results show that at least in some primary mouse cells, V600EB-Raf is able to induce several hallmarks of transformation without evidence for the involvement of a second cooperating oncogene or loss of a key tumor suppressor gene.
Classic studies have previously established a requirement for oncogene cooperation in transformation of primary cells (38). RAS and RAF oncogenes overexpressed in primary cells have been shown to induce cell senescence unless accompanied by mutations in the p19ARF/p53 pathway (23-25). However, it has recently been shown that the expression of lower, physiologic levels of G12DK-ras in mice promotes proliferation and partial transformation in primary cells, without a cooperating event (39). These authors were able to indicate clear differences in the biological effects of overexpressed oncogenic K-ras, which induces senescence, versus oncogenic expression of endogenous K-ras at physiologic levels that induces proliferation. We have found that like G12DK-Ras, V600EB-Raf expressed at physiologic levels in mice can promote proliferation and partial transformation of some primary cell types. This is perhaps a more surprising result than that with oncogenic RAS as RAS has multiple effector pathways, notably, the RAF/MEK/ERK and PI3K/PDK1/PKB pathways, both of which can impinge on the cell cycle, whereas oncogenic RAF is only known to activate the MEK/ERK pathway. However, our observations are consistent with previous data showing that the effects of V600EB-Raf on proliferation of mouse melanocytes and human melanoma cells containing the V600EB-Raf mutation can be reversed by siRNA to B-RAF (15-17, 40). In the development of human malignant cancers in vivo, the situation may well be more complex as V600EB-Raf mutations are detected in a high proportion of benign melanocytic lesions (nevi) and premalignant colon polyps in which the cells are thought to be senescent (9, 18, 41, 42). This has been taken as evidence that B-RAF activation itself is not sufficient to induce all hallmarks of cancer in vivo, at least in melanocytes and colonic epithelial cells.
We expressed V600EB-Raf ubiquitously, by crossing the LSL-B-rafV600E mice to CMV-Cre mice, and show that this cannot be tolerated in embryonic development with embryos harboring the Lox-B-rafV600E allele embryos dying before E7.5. This result is similar to that obtained with the ubiquitous expression of endogenous G12DK-ras, which also induces early embryonic lethality (39). Using the Mx1-Cre strain, we found that the expression of V600EB-Raf in adult somatic tissues induces hyperproliferation as well as bone marrow failure. The effects on proliferation are highly tissue dependent. Pathologic effects on proliferation were observed in the Lox-B-rafV600E–containing spleen and liver, and the histiocytes were observed on the skin but not in the Lox-B-rafV600E–containing lung, kidney, or thymus, which showed no evidence for the presence of histiocytes. Despite this, similar levels of B-Raf kinase activity were observed in the Lox-B-rafV600E–containing lungs of LSL-B-rafV600E + Mx1-Cre mice compared with the Lox-B-rafV600E–containing livers. This observation may be related to the fact that there is no noticeable increase in levels of phospho-ERK in the lung of LSL-B-rafV600E + Mx1-Cre animals, although phospho-MEK levels are increased, which would suggest an uncoupling between B-RAF/MEK and ERK in the lung, possibly due to the action of tissue-specific MAP kinase phosphatases. The liver is affected in a complex way by the expression of V600EB-Raf. Higher levels of apoptosis are observed as well as increased proliferation. The increase in apoptosis may be an indirect consequence of the bone marrow failure and reduction in circulating RBC rather than a direct consequence of V600EB-Raf expression in this tissue.
Expression of V600EB-Raf using the Mx1-Cre strain leads to the development of a form of hemopoietic dysplasia with some of the characteristics of nonlymphoid leukemia of the histiocyte type, in which there is an amplification of circulating cells of the histiocyte/macrophage lineage. At present, it is not clear why these cells in particular are susceptible to the proliferative effects of V600EB-Raf. Histiocytes have been observed in other mouse tumor models for cancer, particularly those on the C57BL6 strain background, including Bax/ARF double null mice (43) as well as mice expressing endogenous G12VK-ras (44). However, histiocytes are rare in humans and have not thus far been associated with the presence of B-RAF mutations. Indeed, despite the high level of expression of B-Raf in hemopoietic cells, B-RAF mutations are extremely rare in hemopoietic neoplasms, and none of those reported thus far represent V600EB-RAF mutations (45-47). By contrast, NRAS and KRAS mutations occur in ∼30% of acute myeloid leukemias and myeloproliferative disorders (13), and somatic activation of G12DK-ras in mice using the Mx1-Cre strain induces a similar myeloproliferative disorder (37, 48), indicating that oncogenic B-RAF and RAS target different populations of Mx1-Cre–expressing cells.
Studies with Ras oncomice have clearly illustrated the importance of expression level in mediating the effects of oncogenic Ras on tumor development (39). We have developed a tractable strain of transgenic mouse that allows the somatic expression of V600EB-Raf at physiological levels. The next important step is to deliver the Cre recombinase to these mice in such a way that it allows the somatic expression of V600EB-Raf in cell types most affected by B-RAF mutations in the development of human tumors, most notably melanocytes, papillary thyrocytes, and colonic and ovarian epithelial cells, as well as cancer stem cells.
Acknowledgments
Grant support: Cancer Research UK project grant #C1362/A2402 (C. Pritchard and R. Marais) and core funding grant #C107/A3096 (R. Marais).
We thank Peter Greaves (University of Leicester) for providing histopathological advice; Misha Roodbari (University of Leicester) for helping with the MEF proliferation assays; staff in Biomedical Services at Leicester for helping with mouse breeding colonies; and Graham Freeth (Animal Resource Centre, Western Australia) for providing CMV-Cre and Mx1-Cre mice.
Footnotes
D. Lloyd is currently at the Genomics Institute of the Novartis Foundation, 10675 John Jay Hopkins Drive, San Diego, CA 92121.
Publisher's Disclaimer: The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Kan NC, Flordellis CS, Garon CF, Duesberg PH, Papas TS. Avian carcinoma virus MH2 contains a transformation-specific sequence, mht, and shares the myc sequence with MC29, CMII, OK10 viruses. Proc Natl Acad Sci U S A. 1983;80:6566–70. doi: 10.1073/pnas.80.21.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rapp UR, Goldsborough MD, Mark GE, et al. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci U S A. 1983;80:4218–22. doi: 10.1073/pnas.80.14.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen HW, Lurz R, Bister K, Bonner TI, Mark GE, Rapp UR. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature. 1984;307:281–4. doi: 10.1038/307281a0. [DOI] [PubMed] [Google Scholar]
- 4.Storm SM, Brennscheidt U, Sithanandam G, Rapp UR. raf oncogenes in carcinogenesis. Crit Rev Oncog. 1990;2:1–8. [PubMed] [Google Scholar]
- 5.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 6.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–7. [PubMed] [Google Scholar]
- 7.Soares P, Trovisco V, Rocha AS, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22:4578–80. doi: 10.1038/sj.onc.1206706. [DOI] [PubMed] [Google Scholar]
- 8.Xu X, Quiros RM, Gattuso P, Ain KB, Prinz RA. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003;63:4561–7. [PubMed] [Google Scholar]
- 9.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 10.Singer G, Oldt R, III, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–6. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 11.Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6:313–9. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 12.Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 13.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9. [PubMed] [Google Scholar]
- 14.Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 15.Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–202. [PubMed] [Google Scholar]
- 16.Karasarides M, Chiloeches A, Hayward R, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;21:21. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 17.Wellbrock C, Ogilvie L, Hedley D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–42. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 18.Yazdi AS, Palmedo G, Flaig MJ, et al. Mutations of the BRAF gene in benign and malignant melanocytic lesions. J Invest Dermatol. 2003;121:1160–2. doi: 10.1046/j.1523-1747.2003.12559.x. [DOI] [PubMed] [Google Scholar]
- 19.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–77. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 20.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–41. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirza AM, Gysin S, Malek N, Nakayama K, Roberts JM, McMahon M. Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Mol Cell Biol. 2004;24:10868–81. doi: 10.1128/MCB.24.24.10868-10881.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5588–97. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–19. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMahon M, Woods D. Regulation of the p53 pathway by Ras, the plot thickens. Biochim Biophys Acta. 2001;1471:M63–71. doi: 10.1016/s0304-419x(00)00027-5. [DOI] [PubMed] [Google Scholar]
- 26.Huser M, Luckett J, Chiloeches A, et al. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001;20:1940–51. doi: 10.1093/emboj/20.8.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercer K, Giblett S, Oakden A, Brown J, Marais R, Pritchard C. A-Raf and Raf-1 work together to influence transient ERK phosphorylation and G1/S cell cycle progression. Oncogene. 2005;25:25. doi: 10.1038/sj.onc.1208707. [DOI] [PubMed] [Google Scholar]
- 28.Luckett JC, Huser MB, Giagtzoglou N, Brown JE, Pritchard CA. Expression of the A-raf proto-oncogene in the normal adult and embryonic mouse. Cell Growth Differ. 2000;11:163–71. [PubMed] [Google Scholar]
- 29.Marais R, Light Y, Paterson HF, Mason CS, Marshall CJ. Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J Biol Chem. 1997;272:4378–83. doi: 10.1074/jbc.272.7.4378. [DOI] [PubMed] [Google Scholar]
- 30.Pritchard CA, Hayes L, Wojnowski L, Zimmer A, Marais RM, Norman JC. B-Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol Cell Biol. 2004;24:5937–52. doi: 10.1128/MCB.24.13.5937-5952.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sage J, Mulligan GJ, Attardi LD, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–50. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mercer K, Chiloeches A, Huser M, Kiernan M, Marais R, Pritchard C. ERK signalling and oncogene transformation are not impaired in cells lacking A-Raf. Oncogene. 2002;21:347–55. doi: 10.1038/sj.onc.1205101. [DOI] [PubMed] [Google Scholar]
- 33.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 34.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–1. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–9. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 36.Le DT, Kong N, Zhu Y, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103:4243–50. doi: 10.1182/blood-2003-08-2650. Epub 2004 Feb 24. [DOI] [PubMed] [Google Scholar]
- 37.Chan IT, Kutok JL, Williams IR, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–38. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 39.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 40.Sumimoto H, Miyagishi M, Miyoshi H, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;23:6031–9. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 41.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 42.Yuen ST, Davies H, Chan TL, et al. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002;62:6451–5. [PubMed] [Google Scholar]
- 43.Eischen CM, Rehg JE, Korsmeyer SJ, Cleveland JL. Loss of Bax alters tumor spectrum and tumor numbers in ARF-deficient mice. Cancer Res. 2002;62:2184–91. [PubMed] [Google Scholar]
- 44.Guerra C, Mijimolle N, Dhawahir A, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–20. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 45.Lee JW, Yoo NJ, Soung YH, et al. BRAF mutations in non-Hodgkin's lymphoma. Br J Cancer. 2003;89:1958–60. doi: 10.1038/sj.bjc.6601371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith ML, Snaddon J, Neat M, et al. Mutation of BRAF is uncommon in AML FAB type M1 and M2. Leukemia. 2003;17:274–5. doi: 10.1038/sj.leu.2402787. [DOI] [PubMed] [Google Scholar]
- 47.Lee JW, Soung YH, Park WS, et al. BRAF mutations in acute leukemias. Leukemia. 2004;18:170–2. doi: 10.1038/sj.leu.2403201. [DOI] [PubMed] [Google Scholar]
- 48.Braun BS, Tuveson DA, Kong N, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004;101:597–602. doi: 10.1073/pnas.0307203101. Epub 2003 Dec 29. [DOI] [PMC free article] [PubMed] [Google Scholar]