

SUMMARY
The CRAF protein kinase regulates proliferative, differentiation, and survival signals from activated RAS proteins to downstream effectors, most often by inducing MEK/ERK activation. A well-established model of CRAF regulation involves RAS-mediated translocation of CRAF to the plasma membrane, where it is activated by a series of events including phosphor-ylation. Here we have discovered a new mode of regulation that occurs prior to this step. By creating a kinase-defective version of CRAF in mice or by use of the RAF inhibitor sorafenib, we show that CRAF must first undergo autophosphorylation of serine 621 (S621). Autophosphorylation occurs in cis, does not involve MEK/ERK activation, and is essential to ensure the correct folding and stability of the protein. In the absence of S621 phosphorylation, CRAF is degraded by the proteasome by mechanisms that do not uniquely rely on the E3 ubiquitin ligase CHIP.
INTRODUCTION
The RAS/RAF/MEK/ERK cascade is a conserved intracellular signaling pathway that plays a crucial role in controlling the ability of cells to respond to their environment (Avruch et al., 2001; Marais and Marshall, 1996). ERKs are the ultimate effectors of the pathway that can phosphorylate and activate numerous nuclear and cytoplasmic substrates involved in mediating the appropriate cellular response (Yoon and Seger, 2006). A major point of control of ERK occurs at the level of RAF, of which there are three family members in mammals: ARAF, BRAF, and CRAF (Mercer and Pritchard, 2003; Wellbrock et al., 2004). Of these three, BRAF has by far the strongest ability to activate ERK (Huser et al., 2001; Pritchard et al., 1995) and is a key mediator of ERK activation in several physiological settings (Rushworth et al., 2006). The predominant role of BRAF is underpinned by the discovery of somatic mutations of BRAF in human cancer samples but the rarity of CRAF mutations and the absence of ARAF mutations (Davies et al., 2002; Garnett and Marais, 2004). The most common BRAF mutation, a valine to glutamic acid change at residue 600 (V600EBRAF), is activated by more than 500-fold and promotes tumor progression by inducing constitutive ERK activation (Garnett and Marais, 2004; Mercer and Pritchard, 2003; Wan et al., 2004).
Although recent emphasis has been placed on BRAF, much of our understanding of the control of RAF activity is based on CRAF (Wellbrock et al., 2004). CRAF normally resides in the cytoplasm as an inactive kinase bound to 14-3-3 adaptor/scaffold proteins (Kolch, 2000). Conversion of RAS.GDP to RAS.GTP leads to the displacement of 14-3-3, dephosphorylation of serine 259, and the recruitment of CRAF to the plasma membrane (Leevers et al., 1994; Marais et al., 1997; Stokoe et al., 1994). Here, CRAF undergoes regulatory phosphorylation events and can bind to lipids and other proteins that together convert CRAF to an active kinase. Five phosphorylation sites within or flanking the kinase domain are involved in this. Binding of 14-3-3 to the C terminus of CRAF is mediated by phosphorylation of serine 621 (S621) (Muslin et al., 1996; Wellbrock et al., 2004). Phosphorylation of serine 338 (S338) and tyrosine 341 (Y341) within the N region (Marais et al., 1995; Mason et al., 1999) and threonine 491 (T491)/serine 494 (S494) in the activation segment of the kinase domain (Chong et al., 2001) occur following CRAF membrane localization. The kinases involved in these essential steps have not all been elucidated, although S621 is known to be a site of CRAF autophosphorylation (Hekman et al., 2004; Mischak et al., 1996; Thorson et al., 1998), and it has been shown that casein kinase 2 (CK2) can phosphorylate S338 (Ritt et al., 2007).
Apart from its ability to activate MEK/ERK, CRAF is known to have other targets in the cell. Ablation of craf in mice causes widespread apoptosis and embryonic lethality without alterations in MEK/ERK activity, and the creation of a MEK kinase-inactive version of CRaf with the YY340/341FF mutations (CrafFF) confirmed that this phenotype is MEK/ERK independent (Huser et al., 2001; Mikula et al., 2001). Biochemical evidence has since shown that CRaf suppresses apoptosis by binding to and inhibiting the activity of two proapoptotic kinases: apoptosis-signal-regulating kinase 1 (ASK1) (Chen et al., 2001) and mammalian sterile 20-like kinase 2 (MST2) (O'Neill et al., 2004). It can also inhibit the ability of Rokα to control Fas clustering and internalization at the cell membrane in a MEK kinase-independent manner (Piazzolla et al., 2005). The fact that knockout mutation of ask1 rescues cardiomyocyte apoptosis induced by ablation of craf is supportive of a role of ASK1 as an apoptotic effector of CRAF (Yamaguchi et al., 2004).
CRAF is also part of a multiprotein complex containing a number of chaperone proteins including heat shock protein 90 (HSP90), p50/cdc37, and HSP70 (Kolch, 2000; Wartmann and Davis, 1994). Through an ATP-driven process, HSP90 is able to ensure the correct folding of the protein and disruption of the CRAF-HSP90 interaction with geldanamycin leads to CRAF misfolding and its consequent degradation via the proteasome (Demand et al., 2001; Schulte et al., 1997; Schulte et al., 1995). The degradation of misfolded CRAF is thought to occur by the recruitment of the E3 ubiquitin ligase CHIP (C terminus of Hsp70-interacting protein) to the complex. CHIP interacts with HSP70 via tandem tetratricopeptide (TPR) repeats and also has a RING-finger-like domain that facilitates the ubiquitylation of chaperone client proteins like CRAF (Connell et al., 2001; Demand et al., 2001). The degradation of CRAF is facilitated by the ubiquitin-binding protein BAG-1, which targets ubiquitylated proteins to the proteolytic complex (Demand et al., 2001; Song et al., 2001).
In this study, we have further investigated CRAF with the aim of clarifying its kinase-dependent and -independent functions. This involved the creation of mice expressing a kinase-inactive version of CRAF in which aspartic acid residue 486 within the DFG motif of the activation segment was converted to alanine (D486ACRaf). Homozygous D486Acraf mice have increased levels of apoptosis in a similar way to mice with a craf knockout mutation. Here we show that this phenotype can be attributed to the fact that CRAF kinase activity is required for stabilizing its own protein expression by a mechanism involving autophosphorylation of S621 via an intramolecular reaction. In the absence of this phosphorylation event, CRAF is misfolded and targeted to the proteasome, although we show that this process does not uniquely rely on CHIP or BAG-1. Thus we have identified a new mode of regulation of CRAF that is absolutely essential to fulfil its role in signaling.
RESULTS
Generation and Analysis of Kinase-Dead CRAF in Mice
We generated homozygous mice expressing the D486Acraf mutation by using a gene-targeting knockin approach (Huser et al., 2001; Figure 1A). The mutation creates a kinase-inactive protein as measured using the immunoprecipitation MEK-ERK kinase cascade assay (Marais et al., 1997) (Figure 1B). Unexpectedly, we found that CRaf protein levels were reduced by >50-fold in crafDA/DA embryos and mouse embryonic fibroblasts (MEFs) compared to craf+/+ embryos and MEFs (Figures 1C and 1D). The expression of other components of the RAF/MEK/ERK pathway was not affected (Figure 1D), and total protein synthesis was not reduced in crafDA/DA cells (Figure 1E), suggesting that the effect was specific for the CRaf protein. Craf mRNA levels (Figure 1F) and splicing (see Figure S1 available online) were not altered, excluding the possibility that the targeting event disrupted craf mRNA production or processing. Thus, CRaf kinase activity is required for controlling its own expression at the protein level.
Figure 1. Generation and Analysis of D486Acraf Mice.

(A) Gene targeting in mice was used to create the D486Acraf allele. Black arrows represent loxP sequences. To delete the neoR cassette, craf+/DA mice were crossed to CMV-Cre mice. The lower figure shows PCR genotyping of a craf+/DA intercross. Primers A and B were used in PCR reaction to distinguish craf+/+ (lanes 2 and 3), craf+/DA (lanes 1, 4, and 6), and crafDA/DA (lanes 5 and 7) alleles.
(B) CRaf kinase assays. craf+/+ and crafDA/DA MEFs were serum starved and then either untreated or treated with 10% (v/v) FCS for 10 min. Protein lysates were subjected to a Raf assay. Values represent mean of three different experiments, and error bars represent standard error.
(C) Western blot analysis of CRaf expression in embryos resulting from craf+/DA intercross.
(D) Western blot analysis of RAF/MEK/ERK component proteins in crafDA/DA MEFs compared to craf+/+ cells. Quantitation of each protein by NIH ImageJ software is shown in the lower bar chart. The mean of three independent experiments for three different MEFs of each genotype is shown, and error bars represent standard error.
(E) Total protein synthesis in crafDA/DA MEFs using 35S labeling. The mean level of incorporation of 35S for three MEFs of each genotype is shown, and error bars represent standard error.
(F) Abundance of craf mRNA in crafDA/DA cells compared to craf+/+ cells using qRT-PCR. Relative abundance was determined by calculating the expression ratio (2−ΔΔCT value) after normalizing each CT value for craf to the CT value for gapdh. The mean of three different MEF samples of each genotype performed in triplicate is shown, and error bars represent standard error.
A consequence of the low level of expression of kinase-dead CRaf is that crafDA/DA embryos have a phenotype similar to the craf−/− null phenotype (Figure 2) (Huser et al., 2001; Mikula et al., 2001). Like the craf−/− embryos, the crafDA/DA embryos are developmentally retarded and have widespread apoptosis (Figure 2A), dying at E10.5–E12.5. crafDA/DA MEFs have a reduced capacity to grow (Figure S2) with increased levels of apoptosis in response to serum withdrawal, etoposide, and α-CD95 antibody (Figure 2B). Consistent with observations in craf−/− cells (Huser et al., 2001; Mikula et al., 2001), ERK phosphorylation and activation is not disrupted in crafDA/DA cells in response to stimulation of cells with exogenous growth factors or apoptosis inducers (Figures 2C–2E and Figure S2). Indeed, we have consistently observed increased levels of active ERK in both crafDA/DA and craf−/− cells in response to a variety of extracellular stimuli. These results confirm that the MEK kinase activity of CRAF is not essential in suppressing apoptosis and also suggest that the MEK/ERK pathway is not involved in mediating the effect of CRAF in regulating its own expression.
Figure 2. crafDA/DA Mice and MEFs Show a Similar Phenotype to the craf−/− Phenotype.
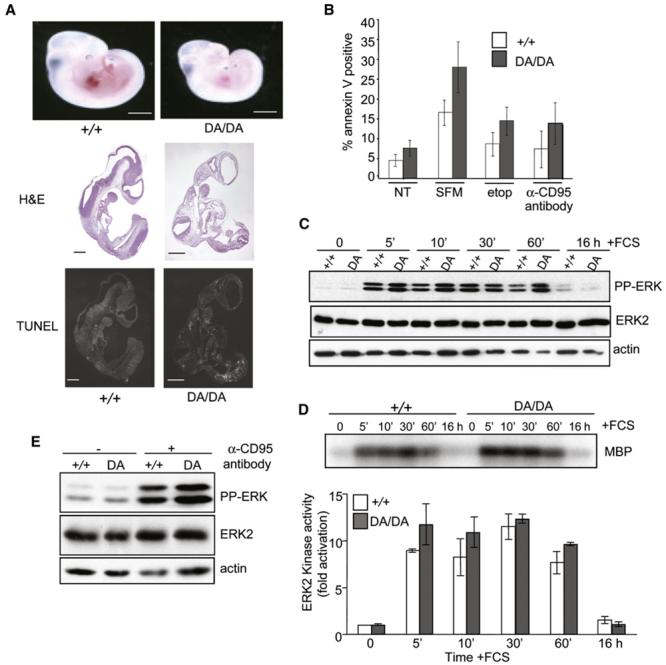
(A) Embryos. Whole-mount photographs of craf+/+ and crafDA/DA E10.5 embryos are shown in the top panels. These embryos were also subjected to H&E (middle panels) and TUNEL (bottom panels) analysis. Scale bars, 250 μm.
(B) MEF apoptosis analysis. MEFs were either not treated (NT) or treated with serum-free media (SFM), etoposide, or α-CD95 antibody, and apoptosis was quantitated by annexin V staining. The mean of three independent experiments for three different MEFs of each genotype is shown, and error bars represent standard error.
(C) ERK phosphorylation following FCS stimulation. MEFs were serum starved for 24 hr and then stimulated with 10% (v/v) FCS over a time course of up to 16 hr; protein lysates were harvested and analyzed with the antibodies indicated.
(D) ERK2 activation. Protein lysates from the same time course as (C) were subjected to an ERK2 kinase assay using the immunocomplex MBP kinase assay. Levels of MBP phosphorylation at each time point were quantitated. Data presented are the mean of three different experiments, and error bars represent standard deviation.
(E) ERK phosphorylation following α-CD95 antibody treatment. MEFs were either untreated or treated with α-CD95 antibody plus cycloheximide for 10 min, and protein lysates were harvested and analyzed with the antibodies indicated.
Kinase-Inactive CRaf Is Unstable and Degraded by the Proteasome
To determine whether the kinase activity of CRAF is required for stabilizing the protein, the half-lives of wild-type CRAF and kinase-inactive CRAF were compared. In initial experiments, the t1/2 of endogenous CRaf in craf+/+ cells was examined by pulse labeling and found to be ~7 hr (Figure S3). Although several attempts were made to measure the t1/2 of endogenous D486ACRaf in crafDA/DA cells, it proved impossible to pulse label this protein due to its low expression level. Therefore, experiments were performed by transfection of myc-tagged vectors expressing WTCRAF and two different kinase inactive versions of CRAF: K375MCRAF and D486ACRAF. Transfections were performed into craf−/− cells in order to prevent any effects of endogenous CRaf in cross-stabilizing the transfected proteins. The t1/2 values were as follows: WTCRAF, ~80 min; K375MCRAF, ~45 min; and D486ACRAF, ~50 min, showing that kinase-inactive CRAF is ~40%–60% less stable than WTCRAF and that both mutants are alike in this respect (Figure 3A). Transfected WTCRAF is also considerably less stable than endogenous WTCRAF, possibly because proteins involved in binding and stabilizing CRAF are titrated out as a consequence of the overexpression.
Figure 3. Kinase-Inactive CRaf Is Unstable, Misfolded, Ubiquitylated, and Targeted by the Proteasome.

(A) Kinase-inactive CRaf is unstable. Myc-tagged CRaf expression vectors were transfected into craf−/− cells. Twenty-four hours later, cells were pulse labeled and chased over a time course of up to 200 min. Protein lysates were harvested and immunoprecipitated with the α-myc antibody, and immunoprecipitated CRaf was quantitated. A typical example of data obtained from four independent experiments is presented.
(B) CRaf expression is rescued by proteasomal inhibition. crafDA/DA cells were either not treated (NT) or treated with proteasomal inhibitors lactacystin (L), epoxomicin (E), or MG132 (M). Protein lysates were prepared from soluble and insoluble Triton X-100 fractions and CRaf expression levels assessed.
(C) D486ACRaf is misfolded. Protein lysates were prepared from craf+/+ and crafDA/DA cells, and CRaf (C) or BRaf (B) proteins were immunoprecipitated. Immunoprecipitated proteins as well as total protein lysate (L) were analyzed with an antibody for HSP90. The strong interaction of D486ACRaf with HSP90 is marked by an asterisk. A longer exposure of the X-ray film is shown in Figure S4A in order to visualize the coimmunoprecipitation of WTCRAF and HSP90.
(D) CRaf is ubiquitylated. Myc-tagged expression vectors were either transfected alone or together with an expression vector for HA-ubiquitin into craf−/− cells. Cells were treated for 5 hr with lactacystin and protein lysates harvested and immunoprecipitated using the α-myc antibody. Immunoprecipitated material was analyzed with the antibodies indicated.
(E) D486ACRaf degradation by the proteasome does not rely on CHIP. crafDA/DA cells were either mock transfected or transfected with 100 μm CHIP siRNA or control siRNA and CRaf expression levels analyzed.
(F) D486ACRaf degradation by the proteasome does not rely on Bag-1. crafDA/DA cells were either mock transfected or transfected with 75 nm Bag-1 siRNA or control siRNA and CRaf expression levels analyzed.
To examine the role of the proteasome in regulating CRAF stability, crafDA/DA MEFs were treated with a range of proteasomal inhibitors and CRaf expression levels analyzed in Triton X-100 soluble and insoluble fractions. All treatments rescued the expression of CRaf to levels similar to that in craf+/+ cells, suggesting that proteasomal degradation is the cause of the reduced CRaf expression (Figure 3B). With epoxomicin and MG132, in particular, a significant proportion of D486ACRaf was in the insoluble fraction and showed a laddering pattern typically observed in proteins that are ubiquitylated.
CRaf is a HSP90 client protein, and this interaction is necessary for establishing and stabilizing CRaf tertiary structure (Kolch, 2000; Wartmann and Davis, 1994). It has also been demonstrated that the CHIP E3-ubiquitin ligase stimulates the degradation of chaperone substrates such as CRAF by a process involving the chaperone cofactor BAG1 (Demand et al., 2001). To examine whether this pathway is involved in the increased turnover of kinase-inactive CRaf, we first examined the interaction of D486ACRaf with chaperone proteins. D486ACRaf had a significantly increased ability to bind HSP90 compared to the wild-type protein (Figure 3C), indicating that the D486Acraf mutation generates a misfolded protein, although binding to HSP70 was not noticeably altered (Figure S4B). To examine CRaf ubiquitylation, vectors expressing myc-tagged kinase-inactive K375MCRAF and WTCRAF as well as the S621ACRAF mutant were cotransfected into cells with HA-tagged ubiquitin. Following treatment with lactacystin, CRAF immunoprecipitates were analyzed with an antibody for HA. All forms of CRaf showed evidence of mono- and/or polyubiquitylation (Figure 3D). siRNA was used to knock down CHIP in crafDA/DA cells, but this did not lead to alterations in the level of expression of D486ACRaf (Figure 3E), suggesting that CRAF is not uniquely ubiquitylated by CHIP. These data were supported by the fact that the expression of K375MCRaf is not stabilized in CHIP−/− cells (Figure S5). In addition, knockdown of the ubiquitin-binding protein BAG1 also does not resurrect D486ACRaf levels (Figure 3F). Thus, although kinase-inactive CRaf is misfolded and degraded by the proteasome, this process is not uniquely modulated by the CHIP E3 ubiquitin ligase/chaperone.
Phosphorylation of S621 Is Required to Stabilize CRaf
Data provided above (Figure 2) rule out a role of the best-characterized effector pathway of CRaf—the MEK/ERK pathway—in mediating the effect of CRaf in stabilizing itself. However, in our analysis, we consistently observed that D486ACRaf migrated faster in SDS-PAGE than WTCRaf (Figures 1C, 1D, and 3B). Since phosphorylation of CRaf is known to play a role in this, we analyzed phosphorylation of kinase-inactive CRaf. Phosphorylation of S621 was found to be virtually absent in the D486ACRaf protein expressed in crafDA/DA cells (Figure 4A) as well as in K375MCRAF and D486ACRAF ectopically expressed in craf−/− cells (Figure 4B). Interestingly, although the majority of WTCRaf was detected in the soluble fraction and was phosphorylated on S621, a significant portion of WTCRaf was in the insoluble fraction, but this was not phosphorylated on S621 (Figure 4A). Other CRaf phosphorylation sites including S259 and S338 were not affected by the kinase-inactivating mutations (Figures 4B). Although it was not possible to examine the phosphorylation of Y341, T491, or S494 due to the lack of good phosphoantibodies for these sites, we examined whether mutation of these sites affected the stability of the protein. Following transfection into craf−/− cells, the activation segment mutant T491A/S494ACRAF was expressed at similar levels as WTCRAF (Figure 4C). In addition, CRaf expression level was not altered in MEFs expressing CRafFF (Figure 4D). These results suggest that phosphorylation of Y341, T491, and S494 does not play a role in stabilizing CRaf.
Figure 4. CRaf Kinase Activity Is Required for S621 Phosphorylation.

(A) D486ACRaf is not phosphorylated on S621. Soluble (S) and insoluble (I) proteins were collected from craf+/+ and crafDA/DA MEFs, immunoprecipitated with a CRaf antibody, and analyzed with P-S621 and CRaf antibodies. The Ig light chain confirms equal loading of the immunoprecipitates.
(B) K375MCRAF and D486ACRAF are not phosphorylated on S621. craf−/− cells were transfected with myc-tagged expression vectors, and CRaf was immunoprecipitated and analyzed with the antibodies indicated. Due to the reduced stability of the mutants, protein loading was adjusted in order to compare equivalent levels of CRaf in all cases.
(C) CRaf expression level is not affected by the T491A/S494A mutations. Protein lysates were prepared from craf−/− MEFs transfected with WTCRAF or AACRAF expression vectors and analyzed with antibodies for CRaf and actin.
(D) CRaf expression level is not affected by the YY340/341FF mutations. Protein lysates were prepared from embryos resulting from a craf+/FF intercross and analyzed with antibodies for CRaf and actin.
(E) Sorafenib disrupts S621 phosphorylation and destabilizes CRaf. craf−/− cells were transfected with a vector expressing WTCRAF and the cells treated with 0–20 μm sorafenib for 2 hr. In the upper panels, immunoprecipitated CRaf was analyzed with antibodies for P-S621 and CRaf. In the lower panels, total protein lysates were analyzed with antibodies for CRaf and actin.
(F) Mutation of S621 creates an unstable protein. craf−/− cells were transfected with vectors expressing either S621ACRAF or WTCRAF, after which a pulse-chase analysis was performed as described above. A typical example of data obtained from four independent experiments is presented.
To confirm that disruption of RAF activity per se is responsible for this effect, treatment of cells with the RAF kinase inhibitor sorafenib was assessed and led to a significant disruption of S621 phosphorylation and decreased stability of the protein (Figure 4E). To confirm a role of S621 phosphorylation in stabilizing CRaf, the stability of the nonphosphorylatable S621ACRAF mutant was examined by pulse-chase labeling following transfection into craf−/− cells. In a similar way to kinase-inactive CRaf, this mutant was found to be ~50% less stable than WTCRAF, with a t1/2 of ~40 min (Figure 4F). These results suggest that the kinase activity of CRaf is required for S621 phosphorylation and that in its absence an unstable protein is formed.
CRaf Autophosphorylation Occurs In cis
Although we have shown that the phosphorylation of S621 of CRaf requires its own kinase activity, it is conceivable that an intermediary kinase is involved whose activity is dependent on CRaf kinase activity. Given the known heterodimerization of RAF kinases (Rushworth et al., 2006; Wan et al., 2004), we investigated whether ARaf or BRaf may play a role in phosphorylating and stabilizing CRaf. However, we found that neither the expression level of CRaf nor S621 phosphorylation was altered in MEFs with knockout mutations of araf or braf (Figure 5A). In addition, AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA) have both been previously suggested to be S621 kinases (Mischak et al., 1996; Sprenkle et al., 1997). However, we found that neither the expression level of CRaf nor S621 phosphorylation was altered following treatment of crafDA/DA cells with agonists known to stimulate these kinases (Figures 5B and 5C).
Figure 5. CRaf Autophosphorylation Occurs In cis.

(A) CRaf expression and S621 phosphorylation are not dependent on ARaf or BRaf. Protein lysates were prepared from wild-type, ARaf, or BRaf knockout MEFs and analyzed with the antibodies indicated.
(B) CRaf expression and S621 phosphorylation are not dependent on AMPK. In the top four panels, crafDA/DA or craf+/+ cells were treated with 0–300 μm of the AMPK agonist A-769662 for 1 hr and protein lysates generated and analyzed with the antibodies indicated. Phosphorylation of acetyl-CoA carboxylase (P-ACC) provides a readout of AMPK activation and confirms activation of AMPK following treatment with 100 and 300 μm A-769662. D486ACRaf expression levels are not raised under these conditions. In the lower two panels, CRaf was immunoprecipitated from either craf+/+ cells or crafDA/DA cells that were treated with 0 or 300 μM A-769662, and protein lysates were analyzed with antibodies for CRaf or phosphoser621. Due to the reduced stability of D486ACRaf, protein loading was adjusted to obtain equivalent levels of immunoprecipitated CRaf between craf+/+ and crafDA/DA samples.
(C) CRaf expression and S621 phosphorylation are not dependent on PKA. craf+/+ and crafDA/DA cells were serum-starved for 24 hr and then either left untreated or treated with the PKA agonist forskolin/IBMX (F/I) for 20 min. In the top two panels, protein lysates were analyzed for expression of CRaf, showing that F/I does not raise the expression of D486ACRaf. In the lower three panels, protein lysates were immunoprecipitated for CRaf and analyzed with antibodies for phosphoser621 or phosphoser43. We have previously shown that PKA stimulates CRaf serine 43 phosphorylation (Dumaz et al., 2002), and the increase in serine 43 phosphorylation by F/I shown here confirms activation of PKA in both cell types. Again, protein loading of the immunoprecipitates was adjusted to obtain equivalent levels of immunoprecipitated CRaf between cell lines.
(D) Transfected WTCRAF cannot transphosphorylate transfected KDCRAF on S621. A vector expressing HA-tagged WTCRAF was cotransfected with either myc-tagged K375MCRAF or D486ACRAF into craf−/− cells. Protein lysates were generated and either K375MCRAF/D486ACRAF was immunoprecipitated with an antibody for the myc tag or WTC-RAF was immunoprecipitated with an antibody for HA. Immunoprecipitated material was analyzed with the antibodies indicated.
(E) Endogenous WTCRAF cannot transphosphorylate transfected KDCRAF. Vectors expressing either myc-tagged WTCRAF, K375MCRAF, or D486ACRAF were transfected into craf+/+ cells. CRaf was immunoprecipitated and analyzed with the antibodies indicated. In (D) and (E), protein loading of the immunoprecipitates was adjusted to obtain equivalent levels of immunoprecipitated CRaf so that levels of S621 phosphorylation between wild-type and mutant CRaf proteins could be directly compared.
We also investigated whether S621 autophosphorylation occurs in cis or in trans by cotransfecting myc-tagged K375MCRAF or D486ACRAF with HA-tagged WTCRAF into craf−/− cells. However, even in the presence of WTCRAF, S621 phosphorylation of these kinase-inactive versions of CRAF was severely abrogated (Figure 5D). S621 phosphorylation of both of these kinase inactive mutants was also disrupted when expressed in craf+/+ cells (Figure 5E). These results strongly suggest that phosphorylation of CRaf on S621 by itself occurs in cis.
DISCUSSION
In a well-established model of CRAF regulation, inactive CRAF is held in the cytoplasm and is phosphorylated on S259 and S621 that allow binding to 14-3-3 scaffold/adaptor molecules (Kolch, 2000; Mercer and Pritchard, 2003). Induction of the MEK kinase activity of CRAF by extracellular signals is achieved by RAS.GTP-mediated displacement of 14-3-3, resulting in recruitment of CRAF to the plasma membrane, where it is converted to its active state by a succession of critical regulatory steps, including phosphorylation of key residues. It is also known that CRAF is an HSP90 client protein and that the chaperone activity of HSP90, in conjunction with other chaperones such as HSP70 and p50cdc37, is responsible for either establishing the correct tertiary structure of the protein or for targeting CRAF molecules that remain misfolded for degradation by the proteasome (Kolch, 2000; Powers and Workman, 2006). The data presented here neatly combine these two modes of CRAF regulation to provide a comprehensive model (Figure 6). CRAF is a misfolded protein unless it undergoes autophosphorylation in cis of S621, thus preventing it from being degraded by the proteasome. Without this stabilization, CRAF levels are prohibitively low and it is unable to participate in normal signaling.
Figure 6. A Comprehensive Model of CRAF Autoregulation.
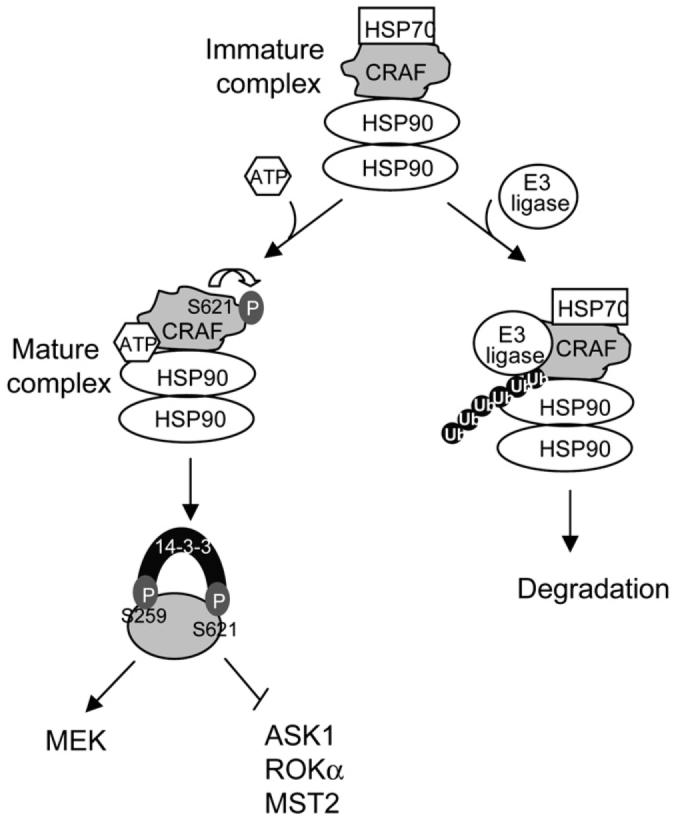
CRAF is part of an immature multiprotein complex that includes the chaperones HSP90 and HSP70. CRAF then has one of two fates: activation of the ATPase activity of HSP90 and HSP90 dimerization leads to the dissociation of HSP70, and, in conjunction with S621 autophosphorylation, CRAF acquires its correct tertiary structure and becomes competent to either activate MEK or inactivate ASK1/MST2/ROKα. Alternatively, if CRAF remains misfolded, an E3 ubiquitin ligase is recruited to the complex, leading to ubiquitylation of CRAF and its degradation via the proteasome. A key modulator of this binary switch is autophosphorylation of S621. For simplicity, a range of other chaperones thought to be involved in this process such as p50cdc37 and HSP40 are not included on this figure.
Autophosphorylation Is a Key First Step in CRAF Regulation
We show here that the first essential role of CRAF kinase activity is to autophosphorylate S621. In an initial examination of CRAF phosphorylation sites using phosphopeptide mapping, Morrison et al. (1993) provided evidence that S621 phosphorylation was not compromised following the expression of the K375MCRAF mutant in insect cells. However, using phosphoantibodies, there has been growing evidence in favor of S621 being a site of autophosphorylation (Hekman et al., 2004; Mischak et al., 1996; Thorson et al., 1998), and this view is corroborated by our findings here. Indeed, it has been shown that S621 autophosphorylation occurs within seconds after growth factor stimulation in the cytoplasm of the cell (Hekman et al., 2004), which confirms that it occurs at an earlier stage prior to plasma membrane translocation of the protein. In this regard, CRAF shows similarities to DYRKs and GSK3β, which have been shown to autophosphorylate tyrosine residues within their activation loops via intramolecular reactions during translation and folding (Lochhead et al., 2005, 2006). In the case of GSK3β, HSP90 chaperone activity was shown to be required for this event.
The autokinase activity of CRAF does not involve homo- or heterodimerization and transphosphorylation but occurs in cis (Figure 5). Teleologically, this must invoke a model in which a C-terminal fragment of CRAF spanning S621 folds into the CRAF catalytic cleft where it can be phosphorylated. The current crystal structure of the RAF kinase domain was established for an inactive version of BRAF bound to the RAF inhibitor sorafenib and does not accommodate this possibility, since a C-terminal fragment spanning the equivalent serine in BRAF, S729, was deleted in order to allow the protein to be crystallized (Wan et al., 2004). It is known that S729 of BRAF is not autophosphorylated like S621 of CRAF (C.N. and C.A.P., unpublished data; Hekman et al., 2004), and so the BRAF and CRAF kinase domains must fold quite differently to allow autophosphorylation in one isoform but not the other. This critical difference in the modulation of the two RAFs can only be further clarified by solving the crystal structure of CRAF without deleting the C-terminal fragment containing S621 and comparing it to that established for BRAF.
CRAF Suppresses Apoptosis Independently of Its MEK Kinase Activity
A wealth of biochemical data have established CRAF as a MEK kinase that is induced by RAS.GTP (Kolch, 2000), but its relative contribution to ERK activation is low, and it is known that BRAF has considerably stronger activity as a MEK kinase in many physiological settings. A recent study showed that CRAF:BRAF heterodimers have considerably higher MEK kinase activity than either BRAF or CRAF homodimers, even though they are present at very low concentrations in the cell (Rushworth et al., 2006). Therefore, a key role of CRAF in MEK/ERK activation may be as a cofactor for BRAF. The importance of such crossregulation is highlighted by the fact that many cancer samples with oncogenic BRAF mutations possess deregulated CRAF activity that can contribute to tumor induction via ERK activation in some situations (Wan et al., 2004). Our results presented here show that the overriding role of CRAF during embryonic development is to suppress apoptosis, and this is mediated through the deregulation of other CRAF effectors, particularly ASK1, a proapoptotic kinase whose activity is negatively regulated by CRAF (Figure 6) (Chen et al., 2001; Yamaguchi et al., 2004). The MEK kinase activity of CRAF does not seem to be involved here, since ERK activation is not disrupted under these circumstances (Figure 2) (Huser et al., 2001; Mikula et al., 2001).
The Role of S621 Phosphorylation and 14-3-3 Binding Is to Stabilize CRAF
In the past, the role of S621 phosphorylation and 14-3-3 binding to the C terminus of CRAF has been controversial, with some studies suggesting that 14-3-3 binding is necessary for activity, whereas others show that dissociation of 14-3-3 from CRAF does not alter its kinase activity (Kolch, 2000; Wellbrock et al., 2004). The work presented here provides important clarification on this issue by showing that S621 phosphorylation is absolutely critical to ensure the correct tertiary structure of CRaf so that it is not targeted for degradation. As such, therefore, our results support the view that that 14-3-3 binding is essential for CRAF activity. The fact that WTCRaf moves to the insoluble fraction if it is not phosphorylated on S621 (Figure 4A) provides evidence that, if this regulatory step does not occur, CRAF is sequestered from its normal cytoplasmic and plasma membrane-associated substrates.
CRAF Is Degraded by the Proteasome
Several previous reports have demonstrated CRAF ubiquitylation and degradation by the proteasome (Du et al., 2006; Manenti et al., 2002; Schulte et al., 1997), and, consistent with the data presented here, disruption of the CRAF-HSP90 interaction with geldanamycin leads to destabilization of CRAF by the proteasome (Schulte et al., 1997). PEST domains within proteins frequently serve to promote degradation (Rechsteiner and Rogers, 1996), and, indeed, CRAF contains a PEST sequence in the variable hinge region between CR2 and CR3 at residues 284–309, and a lysine at residue 309 may serve as a ubiquitin attachment site. This PEST domain in CRAF is not conserved in either ARAF or BRAF, although BRAF contains a PEST sequence at a different position within residues 298–338 in the variable region between CR1 and CR2, suggesting that it may also be regulated by protein degradation.
It is well known that CRAF is an HSP90 client protein and can also bind various other chaperones including HSP70 and p50cdc37 (Kolch, 2000; Wartmann and Davis, 1994). Based on analysis of other HSP90 client proteins, a model for how these various chaperones ensure the correct folding of their substrates has been proposed (Figure 6) (Powers and Workman, 2006). Initially, client proteins interact with HSP70 and HSP90 in a complex with other chaperones to form an immature complex. When the ATPase activity of HSP90 is activated, it undergoes a conformational change that includes transient dimerization. This leads to the dissociation of HSP70 and its associated chaperones and allows the ATP-dependent association of other chaperones such as p50cdc37 (Powers and Workman, 2006). It is while in this mature state that the client protein is folded such that it can become an active protein, and, in the case of CRAF, we have shown that autophosphorylation of S621 is also a key step in this process. If the client protein does not fold, then HSP70 mediates targeting to the proteasome for degradation (Connell et al., 2001; Demand et al., 2001).
The CHIP protein has been shown to interact with HSP70 and stimulates the degradation of chaperone substrates. It is thought to do this by recruiting E2 ubiquitin-conjugating enzymes of the Ubc4/5 family to the chaperone complex and acting as a E3 ubiquitin ligase to add ubiquitin residues to the chaperone substrate, thus inducing its targeting to the proteasome. The targeting process is also facilitated by the ubiquitin domain-binding protein BAG1 (Demand et al., 2001; Song et al., 2001). Since CRAF is a HSP90 client protein and is known to bind BAG1, CHIP was considered to be the best-candidate E3 ubiquitin ligase that mediates the increased degradation of kinase-inactive CRAF described here. However, our data showing that CHIP and BAG1 knockdown do not restore the expression level of D486ACRAF suggest that CRAF can be targeted for degradation by alternative mechanisms. The nature of these other pathways, and particularly the E3 ubiquitin ligases involved, is currently unknown.
There is a growing link between phosphorylation, ubiquitylation, and proteasomal degradation, and many kinases are now known to be involved in regulatory steps that generally promote proteasome-mediated degradation of their target proteins (Hoeller et al., 2006). There are also numerous examples of phosphorylation preventing proteasome-mediated degradation such as p53 phosphorylation by ATR and ATM damage response kinases. However, CRAF is the first case of autophosphorylation preventing degradation of a kinase. Although BRAF is also a HSP90 client protein, current evidence suggests that it is regulated differently to CRAF, as it is far less sensitive to HSP90 inhibition (da Rocha Dias et al., 2005). In addition, autoregulation is not involved in stabilizing BRAF, as by creating mice expressing the analogous kinase inactive mutant D594ABRaf we have found that its kinase activity does not affect either S729 phosphorylation or the expression level of the protein (C.N. and C.A.P., unpublished data).
In summary, our data have identified an important mode of regulation of CRaf and have elucidated that autophosphorylation is a key step in stabilizing the protein. These results have important implications in normal growth factor signaling as well as in cancer and suggest that S621 autophosphorylation is critical for this protein to perform its function within these contexts. Our data should also be taken into consideration in experiments using kinase-inactive CRAF as a dominant interfering protein.
EXPERIMENTAL PROCEDURES
Generation of crafDA/DA Mice and Embryo Analysis
craf+/DA mice with the D486ACRaf knockin mutation were generated by standard procedures. Inheritance of the targeted allele was assessed by PCR genotyping using primers A (5′-CTCCTGGAATTAGCATCTTAGAACC-3′) and B (5′-GGTTTACCACCCAACTGGTC-3′). To delete the neoR cassette, craf+/DA+neo mice were crossed to CMV-Cre mice (Schwenk et al., 1995), and a breeding colony of craf+/DA−neo mice was established on the C57BL6 background. Embryos were harvested at E10.5–E14.5 and used for MEFs, protein lysates, or histology as described (Huser et al., 2001).
Cell Treatments
MEFs were transfected with expression vectors using a nucleofector under the conditions recommended by the manufacturer (Amaxa Biosystems, Germany). siRNA was transfected using oligofectamine according to the manufacturer's instructions (Invitrogen). CHIP-specific siRNA (5′-GGGAUGAUAUUCCUAGUGCUU-3′; Dharmacon) or a Bag-1 siRNA pool (Dharmacon) was used. A siCONTROL nontargeting siRNA pool (Dharmacon) was used as a control. For protease inhibition, cells were treated either with 0.5 μM epoxomicin, 0.5 μM lactacystin, or 30 μM MG132 for 5 hr prior to preparation of protein lysates. Apoptosis was induced by treating MEFs with either 50 ng/ml α-CD95 antibody with 0.5 μM cycloheximide for 20 hr, 75 μM etoposide for 20 hr, or serum-free medium (SFM) for 48 hr and assessed by annexin V staining. Cells were treated with 0–20 μm sorafenib with 0–300 μm A-769662 in DMSO or with 25 μm forskolin/500 μm IBMX in DMSO.
Protein Analysis
Triton X-100 soluble proteins were prepared by taking the supernatants following 13,000 rpm centrifugation of total protein lysates as described (Luckett et al., 2000), and insoluble proteins were obtained by treating the pellets with 2% (w/v) SDS. The primary antibodies used were the following: ARaf (Santa Cruz Biotechnology, Inc., SC-408), CRaf (BD Biosciences, 610152), BRaf (Santa Cruz Biotechnology, Inc., SC-5284), actin (Sigma, A2103), GAPDH (Millipore, MAB374), MEK1/2 (Cell Signaling Technology, 9122), ERK2 (Santa Cruz Biotechnology, Inc., SC-154), Thr202/Tyr204 phosphop44/42 ERK1/2 (Cell Signaling Technology, 9101), phosphoser621 (Santa Cruz Biotechnology, Inc., SC-16807-R), phosphoser259 (Cell Signaling Technology, 9421), phosphoser338 (Serotec, MCA 1852), GAPDH (Chemicon International, MAB374), HA (Santa Cruz Biotechnology, Inc., SC-805), myc tag (Cell Signaling Technology, 2276), HSP90 (Stressgen, SPA-830), HSP70 (Stressgen, SPA-820), CHIP (Abcam, AB2917), BAG1 (Abcam, AB7976), phosphoACC (Cell Signaling Technology, 3661), ACC (Cell Signaling Technology, 3662), and phosphoserine 43 (Dumaz et al., 2002). The ERK2 antibody was a kind gift from Prof. Chris Marshall (ICR, London). The Santa Cruz Biotechnology, Inc., SC-227 CRaf antibody was used for immunoprecipitation, and kinase assays were performed using the kinase cascade assay (Marais et al., 1997). ERK2 kinase assays were performed and quantitated as previously described (Wan et al., 2004).
Quantitative RT-PCR
Primers for craf were 5′-AATACTATCCGGGTTTTCTTGCC-3′ (forward) and 5′-GCGTGCTTTCTTACCTTTGTGT-3′ (reverse). Primers for gapdh were 5′-AGGTCGGTGTGAACGGATTTG-3′ (forward) and 5′-TGTAGACCATGTAGTTGAGGTCA (reverse). cDNA was PCR amplified using 300 nM of each primer and SYBR Green (Bio-Rad) using a Bio-Rad MiniOpticon Real Time PCR system. Each sample was amplified in triplicate with each primer set, and mean CT values were obtained. ΔCT for each sample was calculated by normalizing the CT value for craf to the CT value for gapdh. ΔΔCT was calculated by normalizing the ΔCT value for the crafDA/DA sample to the ΔCT value for the craf+/+ sample, and the expression ratio was calculated as 2−ΔΔCT.
35S-Methionine Labeling of Proteins
MEFs were cultured in DMEM media lacking cysteine and methionine (Invitrogen). Cells were pulsed with 1175 Ci/mmol Tran35S-Label (MP Biomedicals Inc.) for 1–24 hr and the 35S-containing media replaced with fresh media. Protein lysates were collected over a time course of between 0 and 200 min. For analysis of total protein synthesis, protein lysates collected after the pulse were counted using a scintillation counter. For pulse-chase experiments, CRaf was immunoprecipitated and electrophoresed through an SDS-PAGE gel, and gels were dried and exposed to X-ray film. The optical density of bands on X-ray film was quantitated using NIH ImageJ software. The percent reduction in optical density compared to optical density at t = 0 was determined and plotted on a graph of log optical density versus time. The half-life of CRaf (t1/2) was calculated as the time required for the optical density to decrease by 50%.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Biotechnology and Biochemical Sciences Research Council studentships to C.N. and J.H. and by a Cancer Research UK project grant (C1362) to C.A.P. and R.M. We thank Terry Herbert for advice on pulse-chase experiments, Caroline Springer for providing sorafenib, and Grahame Hardie for AMPK reagents. We also thank the Division of Biomedical Services at Leicester for their invaluable support. This paper is dedicated to the memory of my father, Tony Pritchard (C.A.P.).
REFERENCES
- Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, Zhang XF. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res. 2001;56:127–155. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl. Acad. Sci. USA. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, Lee J, Guan KL. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001;20:3716–3727. doi: 10.1093/emboj/20.14.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686–10691. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Demand J, Alberti S, Patterson C, Hohfeld J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001;11:1569–1577. doi: 10.1016/s0960-9822(01)00487-0. [DOI] [PubMed] [Google Scholar]
- Du J, Zeng J, Ou X, Ren X, Cai S. Methylglyoxal downregulates Raf-1 protein through a ubiquitination-mediated mechanism. Int. J. Biochem. Cell Biol. 2006;38:1084–1091. doi: 10.1016/j.biocel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Light Y, Marais R. Cyclic AMP blocks cell growth through Raf-1-dependent and Raf-1-independent mechanisms. Mol. Cell. Biol. 2002;22:3717–3728. doi: 10.1128/MCB.22.11.3717-3728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Hekman M, Wiese S, Metz R, Albert S, Troppmair J, Nickel J, Sendtner M, Rapp UR. Dynamic changes in C-Raf phosphorylation and 14-3-3 protein binding in response to growth factor stimulation: differential roles of 14-3-3 protein binding sites. J. Biol. Chem. 2004;279:14074–14086. doi: 10.1074/jbc.M309620200. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer. 2006;6:776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Huser M, Luckett J, Chiloeches A, Mercer K, Iwobi M, Giblett S, Sun XM, Brown J, Marais R, Pritchard C. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001;20:1940–1951. doi: 10.1093/emboj/20.8.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000;351:289–305. [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Sibbet G, Morrice N, Cleghorn V. Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell. 2005;121:925–936. doi: 10.1016/j.cell.2005.03.034. [DOI] [PubMed] [Google Scholar]
- Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghorn V. A chaperone-dependent GSK3β transitional intermediate mediates activation-loop autophosphorylation. Mol. Cell. 2006;24:627–633. doi: 10.1016/j.molcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Luckett JC, Huser MB, Giagtzoglou N, Brown JE, Pritchard CA. Expression of the A-raf proto-oncogene in the normal adult and embryonic mouse. Cell Growth Differ. 2000;11:163–171. [PubMed] [Google Scholar]
- Manenti S, Delmas C, Darbon JM. Cell adhesion protects c-Raf-1 against ubiquitin-dependent degradation by the proteasome. Biochem. Biophys. Res. Commun. 2002;294:976–980. doi: 10.1016/S0006-291X(02)00594-6. [DOI] [PubMed] [Google Scholar]
- Marais R, Marshall CJ. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv. 1996;27:101–125. [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Mason CS, Marshall CJ. Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J. Biol. Chem. 1997;272:4378–4383. doi: 10.1074/jbc.272.7.4378. [DOI] [PubMed] [Google Scholar]
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18:2137–2148. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Mikula M, Schreiber M, Husak Z, Kucerova L, Ruth J, Wieser R, Zatloukal K, Beug H, Wagner EF, Baccarini M. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J. 2001;20:1952–1962. doi: 10.1093/emboj/20.8.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischak H, Seitz T, Janosch P, Eulitz M, Steen H, Schellerer M, Philipp A, Kolch W. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol. 1996;16:5409–5418. doi: 10.1128/mcb.16.10.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK, Heidecker G, Rapp UR, Copeland TD. Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem. 1993;268:17309–17316. [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–2270. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
- Piazzolla D, Meissl K, Kucerova L, Rubiolo C, Baccarini M. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J. Cell Biol. 2005;171:1013–1022. doi: 10.1083/jcb.200504137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers MV, Workman P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer. 2006;13(Suppl 1):S125–S135. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- Pritchard CA, Samuels ML, Bosch E, McMahon M. Conditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells. Mol. Cell. Biol. 1995;15:6430–6442. doi: 10.1128/mcb.15.11.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK. CK2 is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr. Biol. 2007;17:179–184. doi: 10.1016/j.cub.2006.11.061. [DOI] [PubMed] [Google Scholar]
- Rushworth LK, Hindley AD, O'Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol. Cell. Biol. 2006;26:2262–2272. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- Schulte TW, An WG, Neckers LM. Geldanamycin-induced destabilization of Raf-1 involves the proteasome. Biochem. Biophys. Res. Commun. 1997;239:655–659. doi: 10.1006/bbrc.1997.7527. [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Takeda M, Morimoto RI. Bag1-Hsp70 mediates a physiological stress signalling pathway that regulates Raf-1/ERK and cell growth. Nat. Cell Biol. 2001;3:276–282. doi: 10.1038/35060068. [DOI] [PubMed] [Google Scholar]
- Sprenkle AB, Davies SP, Carling D, Hardie G, Sturgill TW. Identification of Raf-1 Ser621 kinase activity from NIH 3T3 cells as AMPK-activated protein kinase. Mol. Cell. 1997;403:254–258. doi: 10.1016/s0014-5793(97)00062-8. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- Thorson JA, Yu LW, Hsu AL, Shih NY, Graves PR, Tanner JW, Allen PM, Piwnica-Worms H, Shaw AS. 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol. Cell. Biol. 1998;18:5229–5238. doi: 10.1128/mcb.18.9.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Project CG, Jones CM, Marshall CJ, Springer CJ, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Wartmann M, Davis RJ. The native structure of the activated Raf protein kinase is a membrane-bound multi-subunit complex. J. Biol. Chem. 1994;269:6695–6701. [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Yamaguchi O, Watanabe T, Nishida K, Kashiwase K, Higuchi Y, Takeda T, Hikoso S, Hirotani S, Asahi M, Taniike M, et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Invest. 2004;114:937–943. doi: 10.1172/JCI20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.