

Abstract
Glucose-dependent insulinotropic polypeptide (GIP) is a gastrointestinal hormone that has a potent stimulatory effect on insulin release under conditions of normal glucose tolerance. However, its insulinotropic effect is reduced or even absent entirely in type 2 diabetic patients. In this study, we addressed the role of glucose concentration in the diabetic range of ≥11 mM, i.e., hyperglycemia per se, as a cause of the lack of response to GIP. Culturing rat and human pancreatic islets in ≥11 mM glucose for up to 24 h resulted in prevention of GIP-mediated intracellular cAMP increase compared with culturing in 5 mM glucose. Western blot analysis revealed a selective 67 ± 2% (rat) and 60 ± 8% (human) decrease of GIP-R expression in islets exposed to ≥11 mM glucose compared with 5 mM glucose (P < 0.001). We further immunoprecipitated GIP-R from islets and found that GIP-R was targeted for ubiquitination in a glucose- and time-dependent manner. Downregulation of GIP-R was rescued by treating isolated islets with proteasomal inhibitors lactacystin and MG-132, and the islets were once again capable of increasing intracellular cAMP levels in response to GIP. These results suggest that the GIP-R is ubiquitated, resulting in downregulation of the actions of GIP.
Keywords: pancreatic islet, cyclic adenosine monophosphate, insulin, multivesicular body, glucose-dependent insulinotropic polypeptide
GLUCOSE-DEPENDENT INSULINOTROPIC POLYPEPTIDE (GIP) is an insulinotropic hormone released from duodenum and jejunum into the plasma during a meal. Early studies showed its capacity to increase insulin secretion and subsequent effect on glucose metabolism (11, 21, 38). Interference in the binding of GIP to its receptor (GIP-R) results in impairment of insulin secretion and variable degrees of glucose intolerance. This has been well demonstrated in animal studies (27, 37, 51). GIP-R antagonists caused a marked decrease of insulin secretion with a defective early-phase insulin secretion in response to glucose (27, 51). Although the GIP-R-null mice have pancreatic islets capable of producing an appropriate insulin response to glucose, they exhibited significant glucose intolerance when subjected to an oral glucose tolerance test (37).
In type 2 diabetes there is a functional impairment of the insulinotropic effect of GIP (40, 41). A possible relationship to the etiology of type 2 diabetes and obesity was investigated in studies performed in rodents and humans (3, 30, 35). High-fat diet in ob/ob mice resulted in an increase in both plasma GIP concentrations and the number of GIP-secreting cells in the upper jejunum (3). Increased levels of peptides are seen mostly as a potential explanation for desensitization of receptors. Conversely, studies have reported plasma GIP levels to be increased (9), decreased (45), or just right (8) in diabetic patients. One reason for the diversity of responses may be that plasma GIP levels released in response to nutrients are also a function of the number of years that diabetes has been present. Obesity-linked glucose intolerance results in decreased expression of the GIP-R in Vancouver diabetic fatty Zucker rats, but the upstream mechanism is still unknown (30). A study performed in first-degree relatives of patients with type 2 diabetes described a reduced insulinotropic activity in response to infused GIP, which led those authors to suggest a possible inherited condition for the blunted response to GIP (35). Nevertheless, the impaired response that was seen in the relatives applied to only one-half of this group of subjects, and the effect of hyperglycemia could not be discarded, since the groups were not stratified according to their glucose levels in a glucose tolerance test. A follow-up study by the same investigators showed that the reduced effect of GIP on insulin release in euglycemic relatives of type 2 diabetic patients could not actually be demonstrated (36).
More recent studies concluded that hyperglycemia or a related metabolic condition altered the physiological response to GIP. Decreased expression of the GIP-R mRNA and insulinotropic response were observed in lean Zucker rats following hyperglycemic clamp studies (31), and a diminished response to GIP was demonstrated in diabetic patients whose diabetes was caused by diverse etiologies (52). These newest findings point to a metabolic cause, interfering with GIP-R signaling, rather than a primary GIP-R cause.
The GIP-R is a glycoprotein present in the pancreatic β-cell membrane (2, 32) that, upon binding to GIP, activates adenylyl cyclase and increases intracellular cAMP (19, 32). The rise in cAMP in the presence of glucose is followed by an augmented extracellular calcium influx that ultimately results in potentiation of glucose-induced insulin secretion (29). Inhibition of the GIP-induced cAMP increase blocks the potentiation of glucose-stimulated insulin secretion by GIP (27). therefore, increased cAMP formation is necessary for GIP’s insulinotropic effects.
Regulation of transmembrane proteins, such as tyrosine kinase receptors, G protein-coupled receptors, sodium channels, and others, is widely executed by the multivesicular body (MVB) sorting pathway (12, 15, 48). This pathway uses ubiquitination as its major signal and is responsible for the control of important cellular processes. It works as a regulatory apparatus that ensures proper cell signaling and ultimately proper cell function (22). In this study, we addressed the impact of high glucose levels in cultured islets on GIP-mediated cAMP production and total GIP-R protein levels. We show that there is a decreased response to GIP stimulation and reduced expression of GIP-R in islets exposed to high glucose that can be prevented with proteasomal inhibitors.
MATERIALS AND METHODS
Materials
Bovine serum albumin (BSA), 2-deoxy-D-glucose, and collagenase (XI) were from Sigma (St. Louis, MO). 3-Isobutyl-1-methylxanthine (IBMX), MG-132, and lactacystin were from Calbiochem (San Diego, CA). Exendin-4 (EX-4) as well as porcine and human GIP were from Bachem (King of Prussia, PA). Forskolin was from Calbiochem, anti-ubiquitin antibody was from Santa Cruz Bio-technology, (Santa Cruz, CA), and anti-β-actin antibody was from Sigma. Anti-GIP-R antibody (27) was from Dr. Joel F. Habener (Boston, MA), and anti-GLP-1-R antibody (53) was from Dr. Bernard Thorens (Lausanne, Switzerland). RPMI 1640 medium (RPMI) was from Cellgro (Herndon, VA). Regular and enhanced chemiluminescence (ECL) detection systems were from Amersham Biosciences (Piscataway, NJ). SDS-polyacrylamide precast gels, DNase I, and TRIzol were from Invitrogen (Carlsbad CA). Real-time RT-PCR Kit was from Applied Biosystems (Foster City, CA). Cyclic AMP Correlate-EIA kit was from Assay Designs (Ann Arbor, MI). Rodent and human insulin ELISA assay kits were from Crystal Chem (Downers Grove, IL) and Alpco Diagnostics (Windham, NH), respectively. Protein assay reagent was from Bio-Rad (Hercules, CA). Glucose levels were measured by Analyzer 2 (Beckman Instruments, Fullerton, CA).
Animals
Male Sprague-Dawley rats (4 wk old) were from Harlan (Indianapolis, IN). All animal experiments were carried out on approved protocols and in accordance with the Animal Care and Use Committee of the National Institute on Aging, National Institutes of Health.
Isolation and culture of rat pancreatic islets
Rats were anesthetized with isoflurane, and islets of Langerhans were isolated as described (44). Hand-picked islets (100 islets per tube) were cultured for 1 h and 20–24 h in RPMI containing 5, 11, or 22 mM glucose in 1.5-ml Eppendorf tubes. Glucose concentration was measured and adjusted if necessary.
Human pancreatic islets
Human pancreatic islets were provided by the Islet Cell Resource Center (2–3 days postisolation). They were hand-picked upon arrival, and some were immediately used while other islets, 100 per aliquot, were hand-picked into 1.5-ml Eppendorf tubes containing RPMI at various glucose concentrations and then cultured for 24 h. We received four separate isolations (2 female, 2 male).
Intracellular cAMP and insulin release assay
Islets (100 per 1.5-ml Eppendorf tube) were cultured in 5, 11, or 22 mM glucose in 0.3 ml of RPMI for 1 or 20–24 h. Then GIP (10 nM) or vehicle together with the IBMX (100 μM) were added for another 30 min. GIP at 10 nM was used because preliminary experiments had revealed that this was the EC75 for inducing insulin secretion from islets cultured in 11 mM glucose. Intracellular cAMP was extracted with 0.3 ml of 0.1 N HCl. Where indicated, MG-132 (10 or 50 μM), lactacystin (10 or 30 μM), or vehicle was added 2 h before the exposure to glucose concentrations. In all experiments, the supernatant was collected and saved at -20°C for insulin levels, and the cell extracts were saved for total cAMP assay. One set of human islets was first cultured in RPMI supplemented with 5, 11, or 22 mM glucose for 24 h. Then, from each glucose condition, five islets were manually pipetted into new tubes containing fresh RPMI with 5, 11, or 22 mM glucose for 1 h. One-half of the islets were then treated with GIP for another 30 min. The medium was assayed for insulin levels. Insulin release was normalized to one islet. Intracellular cAMP was extracted and assayed.
Rat islet GIP-R and GLP-1R mRNA expression
Rat islets (100–400) were cultured in RPMI containing 5, 11, or 22 mM glucose for 1 h and 20 h. Total RNA was extracted from treated rat islets using TRIzol reagent according to the manufacturer’s instruction. The purity of the RNA was estimated by measuring the 260- to 280-nm absorption ratio with a spectrophotometer. The RNA preparation was treated with DNase I at 37°C for 30 min to remove possible contaminating genomic DNA. DNase was then denatured by heating according to the instructions. Real-time quantitative PCR (qPCR) was performed using a real-time RT-PCR Kit in 96-well plates. Gene expression of 18S ribosomal RNA was used as an internal control. Each plate included reactions for standard curves and quantitation for a control. PCR reactions were performed for 40 cycles on an ABI 7700 Sequence Analyzer (Applied Biosystems) using a SYBR Green Master Mix according to the manufacturer’s instructions. Standard curves were generated on each plate with each type of PCR reaction using a four-point standard dilution of islet cDNA as a template. Quantitative analysis was performed using the SDS 2.0 software (Applied Biosystems). Fluorescence was measured during the annealing/extension steps and used to calculate a cycle threshold (CT), i.e., the point at which the reaction is in the exponential phase and is detectable by the hardware. The minimum CT required for detection was converted into an absolute cDNA amount by extrapolating the CT values onto the four-point standard curve. The level of the relative expression of each gene was calculated by dividing the absolute amount of cDNA for that gene by the amount of ribosomal RNA expression. Primer pairs used were as follows (sense and antisense, respectively): GIP-R: 5′-AGATCCGCCGTCTGCGTCTCA-3′, 3′-GCAGTAACTTTCCAAGACCTCATC-5; GLP-1R: 5′-AGTAGTGTGCTCCAAGGGCAT-3′, 3′-AAGAAAGTGCGTACCCCACCG-5′;18S ribosomal RNA: 5′-ATGCTCTTAGCTGAGTGTCCCG-3′, 3′-ATTCCTAGCTGCG GTATCCAGG-5′. The results were normalized to 18S rRNA levels.
Western blot and immunoprecipitation of GIP-R or GLP-1R
Rat or human islets were cultured in RPMI containing 5, 11, or 22 mM glucose for up to 24 h and lysed for Western blot or immunoprecipitation analysis of GIP-R and GLP-1R (44). Some islets were first cultured in 5 mM glucose with MG-132, lactacystin, or vehicle for 2 h. Then the incubation continued for a total of 20–24 h with various glucose concentrations. For Western blot analysis, islet lysates were separated by 8% Tris-SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Invitrogen). The PVDF membrane was blocked for 1 h using 5% nonfat milk in TBS-T (Tris buffer + 1% Tween) buffer and blotted with the primary antibodies to GIP-R (1:100), GLP-1 R (1:100), or β-actin (1:1,000) overnight at 4°C. Horseradish peroxidase-linked secondary antibodies (1:5,000) were added for 1 h, and then ECL reagents were used for detection. Quantitation of the images was performed with ImageQuant v. 1.2 (Molecular Dynamics). For immunoprecipitation experiments, equal numbers of rat islets (260) and human islets (400) were exposed to to 5, 11, or 22 mM glucose for 4, 8, 16, or 24 h; then the islets were lysed in 200 μl of ice-cold immunoprecipitation assay buffer, containing 50 mM Tris·HCL, pH 8.0, 150 mM NaCl; 1% (vol/vol) NP-40, 0.5% (wt/vol) SDS; 5 μg/ml leupeptin, and 0.1 mM PMSF and supplemented with a protease inhibitor cocktail. These islets were usually lysed for 30 min on ice and then centrifuged at 14,000 rpm for 15 min to remove cell debris. The cell lysates were immunoprecipitated at 4°C overnight with rabbit anti-GIP-R antibody (1:100) or rabbit anti-GLP-1R antibody (1:100) plus 50 μl of washed slurry agarose-linked protein G beads. Parallel control immunoprecipitations were carried out with normal rabbit IgG in all our experiments. The complexes were recovered by centrifugation at 4°C and washed twice with ice-cold lysis buffer and twice with ice-cold wash buffer. Bound antigen was eluted by boiling the beads in 1x SDS-PAGE sample buffer for 10 min. The complexes were resolved by SDS-PAGE, transferred to PVDF membrane, and examined by Western blot using anti-ubiquitin monoclonal antibody (1:100).
Statistical analyses
Results are expressed as means ± SE. Data from multiple groups were analyzed using one-way ANOVA; Data from two groups were compared with an unpaired t-test. P < 0.05 was considered statistically significant.
RESULTS
Intracellular cAMP formation and insulin secretion in rat islets
We first examined whether glucose concentration impacts rat islet intracellular cAMP levels and insulin secretion over short (1 h) and long (20–24 h) periods of culture. GIP (10 nM) induced cAMP formation and insulin secretion in a glucose-dependent manner in freshly isolated islets (Fig. 1 A and B). Even in 5 mM glucose, GIP increased cAMP formation approximately twofold (from 4.17 ± 0.33 to 8.69 ± 1.78 fmol/islet, P < 0.05), whereas in 11 and 22 mM glucose, there was a therefold increase in response to GIP [5.13 ± 0.09 (11 mM glucose alone) vs. 15.38 ± 1.90 (11 mM glucose + GIP), 6.23 ± 0.89 (22 mM glucose) vs. 17.36 ± 1.30 (22 mM glucose + GIP) fmol/islet]. Although there was a tendency for increasing the glucose alone to increase cAMP levels, this was not statistically significant in three separate islet experiments (Fig. 1A). Glucose increased insulin secretion in a concentration-dependent manner, and addition of GIP to each glucose concentration further increased insulin secretion ∼1.6-fold above that achieved with glucose alone (Fig. 1B).
Fig. 1.

cAMP formation and insulin secretion in rat islets. Freshly isolated rat islets were incubated in glucose, as labeled, for 1 h (A and B) or 24 h (C and D). Glucose-dependent insulinotropic polypeptide (GIP, 10 nM) or vehicle were added for a further 30 min. Intracellular cAMP levels (A and C) and insulin secretion (B and D) were then measured. Results are expressed as means ± SE (n = 3–4 independent islet preparations). *P < 0.05 and **P < 0.01, GIP treatment vs. corresponding vehicle group; #P < 0.05 and ##P < 0.01 vs. 5 mM glucose + GIP treatment group; ^P < 0.05 and ^^P < 0.01 vs. 5 mM glucose + vehicle group.
When rat islets were cultured in ≥11 mM glucose for 20–24 h, there was no cAMP increase in response to GIP (Fig. 1C). Additionally, insulin secretion in response to both glucose and GIP was blunted (Fig. 1D). Only in islets maintained in 5 mM glucose was there a significant increase in cAMP formation (P < 0.05) and insulin secretion (P < 0.01) in response to GIP.
Intracellular cAMP formation and insulin secretion in human islets
We next examined the impact of increasing glucose concentrations on human islets to see whether responses were similar to thiose of rat islets. GIP induced cAMP formation and insulin secretion in a glucose-dependent manner in human islets 2–3 days postisolation that had been maintained in 5 mM glucose (Fig. 2, A and B). In 5 mM glucose, GIP increased cAMP formation approximately fourfold (from 2.53 ± 0.77 to 10.71 ± 1.7 fmol/islet, P < 0.01), and in islets in 11 and 22 mM glucose for just 1 h, intracellular cAMP levels were increased three- and fourfold, respectively (5.72 ± 0.55 vs. 18.35 ± 2.43 and 5.01 ± 1.00 vs. 20.59 ± 1.44 fmol/islet) by GIP. When human islets were cultured in ≥11 mM glucose for 24 h, there was no cAMP increase in response to GIP (Fig. 2C). As islets appeared responsive to glucose and GIP only when cultured in 5 mM glucose, in one set of islets we sought to further investigate the veracity of this finding (Fig. 2, D and E). Islets were cultured in 5, 11, or 22 mM glucose for 24 h. Then, five islets from each culture condition were individually hand-picked and placed into new Eppendorf tubes. They were then subjected to increased glucose concentration for 1 h with or without GIP for the last 30 min. Only islets cultured for 24 h in 5 mM glucose showed increased cAMP formation (P < 0.05) and insulin secretion (P < 0.05) in response to GIP and glucose (Fig. 2, D and E).
Fig. 2.

cAMP formation and insulin secretion in human islets. Human islets were incubated in glucose, as labeled, for 1 h on arrival (A and B) or for a further 24h(C, D, and E). GIP (10 nM) or vehicle was then added for a further 30 min. Intracellular cAMP levels (A, C, and D) and insulin secretion (B and E) were then measured. Intracellular cAMP levels (D) and insulin secretion (E) in human islets that were cultured in glucose for 24 h (top-labeled glucose concentrations), and then 5 islets from each group were individually hand-picked and subjected to glucose (bottom-labeled glucose concentrations), GIP, or vehicle for 1 h. Results are expressed as means ± SE (results shown in A, B, and C are from 3 independent islet preparations; results shown in D and E are from a single islet preparation). *P < 0.05 and **P < 0.01 GIP vs. corresponding vehicle group; #P < 0.05 and ##P < 0.01 vs. 5 mM glucose + GIP treatment group; ^^P < 0.01 vs. 5 mM glucose + vehicle group.
To ascertain whether ≥11 mM glucose for 24 h had a global effect on adenylyl cyclase activity, we treated human islets with forskolin (10 μM), an adenylyl cyclase activator. Regardless of the glucose concentration or the time islets were in the presence of glucose (1 h, Fig. 3A or 24 h, Fig. 3B), cAMP levels increased in response to forskolin. GLP-1R activation, similar to GIP-R activation, induces insulin secretion in a cAMP-dependent manner. Islets cultured in ≥11 mM glucose for 24 h still responded to the GLP-1R agonist EX-4, in that it induced a clear increase in cAMP levels (Fig. 3C) and insulin secretion (data not shown).
Fig. 3.

Adenylylyl cyclase function and GLP-1 receptor (R)-mediated response in human islets. Human islets on arrival at National Institute on Aging were cultured with glucose for 1 h (A) or 24 h (B and C), as labeled. At the end of incubation, vehicle, forskolin (10 μM), or exendin (EX)-4 (1 nM) were added to the culture for 30 min, and intracellular cAMP levels were measured. Results are expressed as means ± SE from 3 independent islet preparations. *P < 0.05 and **P < 0.01 vs. corresponding vehicle group; #P < 0.05 vs. 5 mM glucose + EX-4 treatment group.
To eliminate any osmotic effects of glucose, human islets were cultured for 24 h in 5 mM glucose or 5 mM glucose plus 15 mM 2-deoxy-D-glucose and then stimulated with 10 nM GIP. Intracellular cAMP increases were similar with both sugars (data not shown). We also cultured human islets in 5 mM glucose in the presence or absence of 10 nM GIP for 24 h. We then manually pipetted the islets into fresh RPMI with 5 mM glucose and stimulated one-half of them with GIP (10 nM). cAMP levels still significantly increased in response to fresh GIP (data not shown).
mRNA expression of GIP and GLP-1R
The mRNA expression levels of GIP and GLP-1R in isolated rat islets were assessed. Similar expression was found whether the islets were cultured for 20 h in 5 or 11 mM glucose (Fig. 4A), thus indicating that the transcription of the GIP-R gene was not compromised by exposure to high glucose. The modest increased GIP-R mRNA that we observed with 11 mM glucose was not statistically significant (n = 5). The relative mRNA expression of the GIP-R was quantified as 1.45 ± 0.65 for 5 mM glucose and 2.89 ± 1.47 for 11 mM glucose and that of the GLP-1R was 3.43 ± 1.33 and 5.42 ± 1.97 for 5 and 11 mM glucose, respectively (Fig. 4B).
Fig. 4.
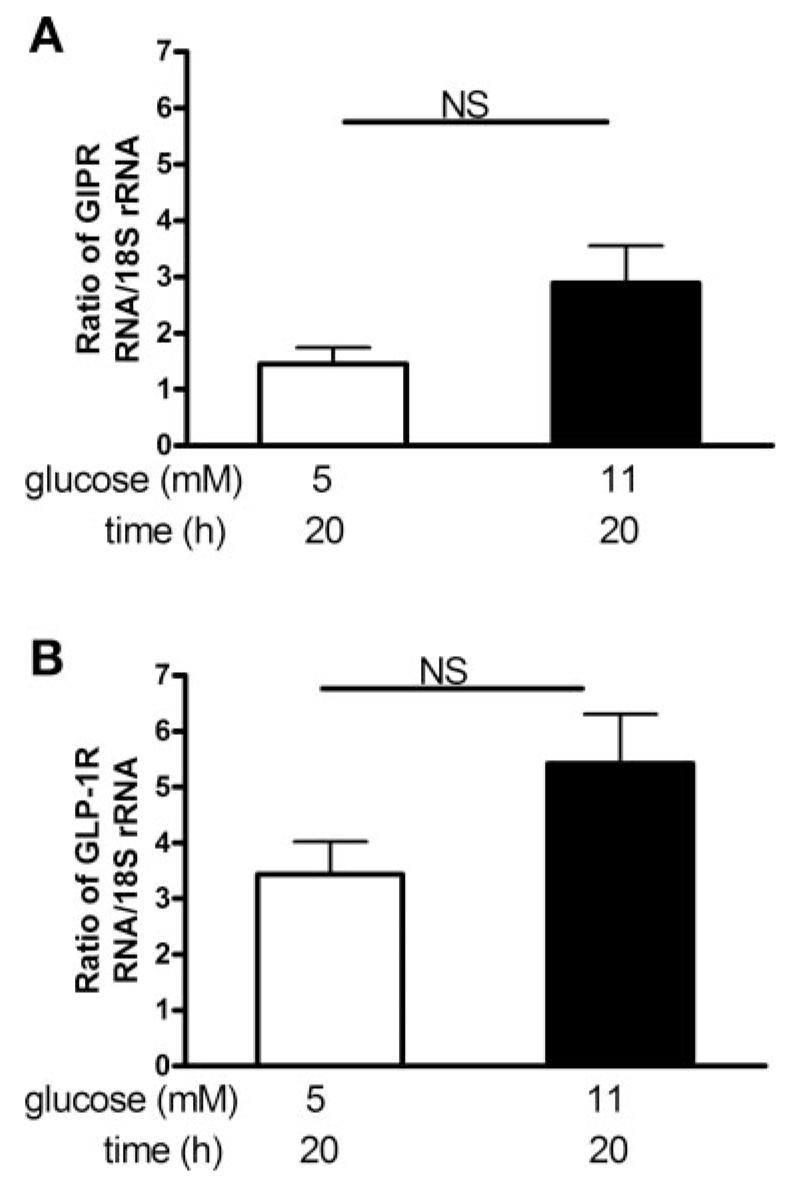
mRNA expression of GIP-R and GLP-1R in rat islets. mRNA levels were measured by real-time RT-PCR and normalized with 18S rRNA. Bars depict GIP-R (A) and GLP-1 R (B) mRNA expression in islets cultured in 5 or 11 mM glucose for 20 h. Results are expressed as means ± SE (n = 5 separate islet preparations).
GIP-R protein expression in rat and human pancreatic islets
By Western blotting, GIP-R protein expression was decreased 67 ± 2% in rat islets that were cultured in 11 mM glucose compared with culturing in 5 mM glucose for 20–24 h (Fig. 5, A, B, and E, P < 0.01), but the intensity of the GLP-1R protein expression was the same with both glucose concentrations (Fig. 5, C, D, and F).
Fig. 5.

Western blot analysis of GIP-R and GLP-1R protein expression in rat islets. GIP-R (A and B) and GLP-1 R (C and D) protein expression in whole cell lysates from islets cultured in 5 or 11 mM glucose for 20–24 h. Relative protein expressions of GIP-R (E) and GLP-1 R (F) were normalized to β-actin (B and D). Results are expressed as means ± SE (n = 7 separate islet preparations, ***P < 0.001).
A similar phenomenon was found in human islets. GIP-R protein expression was significantly reduced (60 ± 8%) in human islets that were exposed to ≥11 mM glucose for 24 h compared with those exposed to 5 mM glucose for 24 h (Fig. 6, A and C, P < 0.01), but GLP-1R expression was not altered under the same conditions (Fig. 6, B and D).
Fig. 6.

Western blot analysis of GIP-R and GLP-1R protein expression in human islets. GIP-R (A) and GLP-1 R (B) protein expression in whole cell ly-sates of human islets that were cultured in glucose, as labeled, for 24 h. Protein expression levels of GIP-R (C) and GLP-1 R (D) were normalized to β-actin. Results are expressed as means ± SE (n = 3 separate islet preparations, **P < 0.01).
Mechanisms of GIP-R downregulation
To test the hypothesis that GIP-R was being continuously degraded when islets were exposed to high glucose for a prolonged period, we investigated whether GIP-R in rat and in human islets were being ubiquitinated. We found that glucose at ≥11 mM caused GIP-R ubiquitination as early as4hinrat islets (2.5-fold increase compared with 5 mM glucose; Fig. 7A: 173,570 ± 3,226 vs. 429,796 ± 5,768, 5 vs.11 mM glucose). After rat islets were exposed to ≥11 mM glucose for 8 h, the ubiquitin-targeted GIP-R level was increased about sevenfold compared with that in 5 mM glucose (Fig. 7A, P < 0.001: 72,531 ± 3,195 vs. 520,427 ± 5,773 arbitrary units, 5 vs. 11 mM glucose). 11 mM glucose increased Ub-GIP-R 2.7-fold at 16 h (191,185 ± 3,701 vs.518,133 ± 17,547 arbitrary units, 5mM glucose vs.11 mM glucose). Even cultured in 5 mM glucose, some GIP-R ubiquitinization in freshly isolated rat islets was observed by 16 h (Fig. 7B). In Fig. 7C, 11 mM glucose increased rat islet Ub-GIP-R threefold at 24 h (226,832 ± 30,804 vs. 714,081 ± 107,461 arbitrary units, 5 vs. 11 mM glucose). In parallel experiments of rat islets isolated at the same time, we compared GIP-R and GLP-1R ubiquitination (Fig. 7, C and D): GLP-1 R in rat islets was not ubiquitinated after 24 h in high glucose (n = 3–5 separate islet preparations).
Fig. 7.

Immunoprecipitation and Western blot analysis of ubiquitin-targeted GIP-R and GLP-1R. Rat (A, B, C, and D) and human (E and F) islets were cultured in glucose concentrations for the times as labeled. Whole cell lysates of islets were immunoprecipitated with anti-GIP-R antibody and anti-GLP-1R antibody, subjected to SDS-PAGE, blotted with ubiquitin antibody, and exposed overnight (A), exposed for 10 min (B), or exposed for 5 min (C and D). E and F indicate exposure for 1 h.
GIP-R ubiquitination level in human islets was also increased after culturing in ≥11 mM glucose for 24 h. There was threefold (11 mM glucose, 24 h) and fivefold (22 mM glucose, 24 h) more ubiquitinated GIP-R compared with 5 mM glucose, respectively (Fig. 7E, P < 0.01: 65,011 ± 41.28 vs. 187,794 ± 4,744 vs. 309,762 ± 5,842 arbitrary units, 5 vs. 11 vs. 22 mM glucose). We did not find any ubiquitination of GLP-1 R in human islets (Fig. 7F; n = 3 separate islet preparations).
We next treated rat islets with the reversible proteasomal inhibitor MG-132 or with the irreversible proteasome inhibitor lactacystin. Optimal concentrations for both compounds were first determined. Both inhibitors were added to islets that were maintained for2 h in 5 mM glucose before the addition of more glucose to reach a final concentration ≥11 mM glucose in some of these islets. MG-132 (10 and 50 μM) and lactacystin (10 and 30 μM) rescued the glucose-mediated reduction in GIP-R expression (Fig. 8A). GIP-R immunoprecipitates from rat islets that were cultured in 5 or 11 mM glucose for 20 h, and with the proteasome inhibitor MG-132, showed similar amounts of GIP-R ubiquitination (Fig. 8B). No ubiquitin signal was observed when control immunoprecipitations were carried out with normal rabbit IgG (data not shown). Most importantly, isolated rat islets cultured in 11 mM glucose with the proteasome inhibitor MG-132 (10 μM) for 24 h still possessed the ability to significantly increase cAMP levels in response to GIP (Fig. 8C, P < 0.05 vs. vehicle control).
Fig. 8.
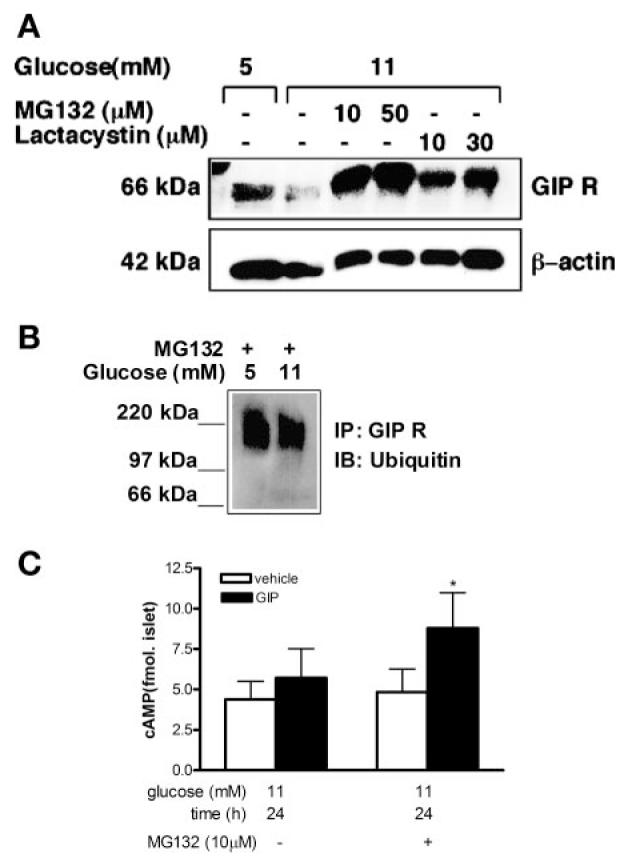
Effect of proteasomal inhibitors in rat islets. A: rat islets were cultured in glucose, as labeled, without or with MG-132 or lactacystin for 2 h followed by 20 h incubation in 11 mM glucose. Whole cell lysates were probed with anti-GIP-R antibody, and β-actin was used as loading control. Representative blot is shown (n = 3 separate islet preparations). B: MG-132 (10 μM) was added to islets cultured for 6 h followed by 20-h incubation in glucose concentrations as labeled. GIP-R immunoprecipitates were reblotted with ubiquitin antibody. Representative blot is shown (n = 3 separate islet preparations). C: cAMP formation in rat islets that were cultured in 11 mM glucose with or without MG-132 (10 μM) for 2 h followed by 24-h incubation in 11 mM glucose. GIP (10 nM) or vehicle was then added to the cultures for 30 min. Results are expressed as means ± SE (n = 3 separate islet preparations, *P < 0.05).
DISCUSSION
Here, we show that GIP-induced cAMP production is significantly decreased in islets cultured in glucose levels in the diabetic range of ’11 mM glucose for 20–24 h. The reduction in GIP-R protein expression in islets cultured in this manner seems specific for the GIP-R. In islets cultured for 24 h in ≥11 mM glucose, cAMP could still be generated in response to forskolin and EX-4. Therefore, the defect in GIP signaling was not due to a global effect of high glucose on adenylyl cyclase activity. These results are also in agreement with previous in vivo observations, which show that type 2 diabetes is accompanied by defective insulinotropic response to GIP but not to GLP-1 (41). It has often been postulated that GIP itself induces homologous desensitization of the GIP receptor, but depending on the study, plasma levels of GIP in type 2 diabetes have been reported to be increased (9), not changed (8) and decreased (45). We checked plasma levels of GIP in response to oral glucose in newly diagnosed diabetic patients in the Baltimore Longitudinal Study of Aging and found that plasma levels of GIP were similar to those in nondiabetic subjects (49). So we concluded that it is hyperglycemia and/or its metabolic consequences, and not elevated GIP plasma levels, that lead to the well-described unresponsivity of β-cells to exogenous GIP. This is also in line with what has been shown in diabetic fatty Zucker rats, in which plasma levels of GIP were reported to be similar to leels in lean animals but the GIP-R expression and GIP-induced cAMP responsivity of islets were, respectively, greatly diminished and absent (30). The same authors also subsequently reported that perfused pancreata from lean rats rendered hyperglycemic at 25 mM plasma glucose levels for 6 h prior to exposure to GIP (25 pM) also did not have an insulinotropic response and had reduced GIP-R expression (31).
Xu et al. (54) recently showed also that hyperglycemia per se impacted incretin receptor expression by using two models of hyperglycemia: 90% pancreatectomy as well as glucose challenge in rats (96 h) and rat islets (48 h). They found that isolated islets exposed to ≥10 mM glucose for 48 h had reduced mRNA levels of GLP-1R but increased GIP-R mRNA levels. They did not, unfortunately, study protein levels of the receptors under the same conditions. We found similar results to theirs as regards GIP-R mRNA levels: a tendency toward increased GIP-R expression with ≥11 mM glucose that did not quite reach statistical significance. However, our treatment schedule was for 24 h, and therefore most likely there would have been significant differences with a longer incubation time. Their contrary results to ours as regards GLP-1R expression may also be a time-of-exposure issue.
Our data indicate for the first time that GIP-R is ubiquitinated and that this likely leads to degradation of the targeted GIP-R. Even in 5 mM glucose the GIP-R seems susceptible to ubiquitination over time. However, with glucose levels in the diabetic range the degree of ubiquitination is increased and the speed of ubiquitination is accelerated. Therefore, we propose that GIP-R is regulated, at least partially, by the MVB-sorting pathway in islets, that it is degraded and not recycled following prolonged exposure of islets to glucose in the diabetic range, and that this explains the profound decrease in GIP-R levels seen after several hours of exposure to elevated glucose levels. The facts that the cAMP response was restored and GIP-R protein expression was preserved by the presence of proteasome inhibitors MG-132 and lactacystin (4, 26, 28) in islets exposed to high glucose support this assertion. Ubiquitination targets membrane proteins for degradation, and it is the foremost signal in the MVB sorting pathway, a conserved system that is present from yeast (15) to mammals (12) and plays a major role in many basic cellular functions (22). Polyubiquitinated proteins that have specific sequences or trans-acting components for targeting are usually degraded (6). The down-regulation of transmembrane proteins such as tyrosine kinase and G protein-coupled receptors has been proposed to occur by endosomal or lysosome-mediated degradation (22, 28, 47). Proteasomal inhibitors (e.g., MG-132 and lactacystin) act at the level of these structures, thus preventing degradation of ubiquitinated proteins that are ferried from the plasma membrane. Consequently, these compounds also prevent deubiquitination of the formerly targeted proteins, resulting in their accumulation inside the cell and depletion of free ubiquitin molecules or chains (14, 24). The G protein-coupled receptors regulating mechanisms (5, 20) were described in muscarinic M1, M3, and M5 receptors (25), as well as in the receptors for GnRH (42). The MVB-sorting pathway is a complex system with a large number of different signals and components. Even though several aspects of this sorting pathway have been identified, there are still important questions related to upstream and downstream ubiquitination pathways that remain unsolved.
The synthesis of GIP receptors is not likely to be altered by high glucose, as mRNA levels were not decreased when assessed by quantitative PCR. This contrasts with the results of a previous study showing that hyperglycemia and its metabolic consequences caused a reduction in GIP-R mRNA and protein levels in lean Zucker rats (31). The disparity with our findings might be explained by some genetic susceptibility of the Zucker strain or the use of a different control gene. Herein, 18S rRNA was selected as a control because its measurement seems to be very accurate according to recent publications that evaluated real-time RT-PCR techniques (1).
Our data introduce a new concept to downregulation of receptors on β-cells; the metabolic state results in GIP-R ubiquitination in a ligand-independent manner. In general, ubiquitin is covalently bound to target proteins by linkage between the carboxy glycine of ubiquitin and usually the ε-amino group of lysine in the target protein (39). In some proteins, polyubiquitination may occur at the amino-terminal residue (7). There are three lysines in the intracellular portion of the rat GIP receptor, of which Lys250 is the most likely to be ubiquitinated. Preceeding this lysine is a potential serine phosphorylation motif, 246Arg-Arg-Ser-Glu249, that may be either a cAMP-dependent protein kinase A (PKA) or PKC motif. This motif shows a highly significant serine phosphorylation prediction, using the neural network phosphorylation predictor NetPhos 2.0 (Technical University of Denmark). Despite this sequence’s difference to the canonical PKA phosphorylation motif, i.e., Arg/Lys-Arg/Lys-X-Ser/Thr, similar Arg/Lys-Arg/ Lys-Ser/Thr motifs in the proteins RFX-5 and Wee-1 have been shown, using mass spectrometry, to be direct PKA phosphorylation sites (17). We propose that PKA activity (and also PKC activity, as it has been shown to be elevated in many tissues, i.e., kidney, in the diabetic state) is slightly increased in the presence of elevated glucose, as glucose itself causes a slight increase in intracellular cAMP levels. cAMP-mediated activation of PKA may then lead to a subsequent phosphorylation of the receptor. Although GIP treatment alone (in 5 mM glucose for 24 h) did not result in increased ubiquitination of the GIP-R, it is possible that glucose-mediated ubiquination would have been accelerated by GIP itself in high glucose; this remains to be tested when we receive more human islets. Ubiquitination of several receptors has been shown to be highly dependent on their stoichiometric Ser/Thr phosphorylation (16, 33). Mutagenic removal of these Ser/Thr phosphorylation sites resulted in an inability of the agonist stimulation to induce ubiquitination and eventual internalization (33). These early studies of receptor ubiquitination indicated that, despite the coincidence of agonist-induced internalization and ubiquitination, the ubiquitination of the receptor was not involved in the sequestration process itself but was subsequently required for targeting of the internalized receptor to proteolytic lysosomes (50). More recent data have implicated a role of β-arrestins in the receptor ubiquitination process. Hence, Shenoy et al. (46) demonstrated that ubiquitination of the β2-adrenergic receptor could be mediated by its interaction with a multiprotein complex including β-arrestin and the E3-ubiquitin ligase mdm2. In this scenario, agonist stimulation results in receptor phosphorylation by PKA and G protein-coupled receptor kinases, causing the subsequent uncoupling from Gsα and the binding of a β-arrestin-mdm2 complex. The receptor and also the arrestin subsequently become ubiquitinated, thus facilitating a rapid downregulation of the receptor.
It is of interest for our experimental paradigm that elevated intracellular cAMP levels have also been shown to actually potentiate the expression of β-arrestin in multiple cell types (18). β-Arrestin molecules have previously been shown by Gurevich et al. (13) to possess the capacity to interact stably with phosphorylated receptors. In addition, recent evidence has specifically demonstrated that PKA-mediated phosphorylation may be sufficient to induce β-arrestin association with G protein-coupled receptors (23). This phosphorylation-dependent association of β-arrestin with the GIP-R may thereby facilitate a mechanism by which mdm2 could induce ubiquiti-nation of the GIP-R in response to high glucose levels. Also, as stated previously, it is considered that ubiquitination may not be of specific importance for receptor sequestration but controls subcellular trafficking. This aspect of receptor posttranslational modification then allows the possibility that there may still be GIP receptors present at the plasma membrane surface in type 2 diabetic patients; these receptors consequently may be tonically associated with β-arrestin and therefore unable to induce Gsα activation in response to GIP. GLP-1R localizes in lipid rafts and interacts with caveolin-1. This interaction is necessary for GLP-1R’s normal functions. However, considering recent growing evidence of non-G protein-mediated receptor signaling (34), it is possible that the extant GIP-R are still functional and activate transduction pathways other than Gsα. Reinforcing this, data have been generated that indicate that PKA phosphorylation of the intracellular loops of G protein-coupled receptors can deleteriously affect their affinity for Gsα and even elevate their affinity for Giα (10, 43). Under these proposed scenarios, GIP-R agonists, unlike GLP-1R agonists, are unlikely to ever be useful in increasing insulin secretion in type 2 diabetes.
In summary, we have shown that there is a glucose-dependent downregulation of the GIP-R in vitro that could explain the defective response to GIP that is seen during GIP infusions in diabetic patients. It is probable that hyper-glycemia triggers the association of GIP-R and ubiquitin ligase complexes and stimulates several cellular responses whose aims are to maintain cell homeostasis and to adapt to this new metabolic demand. Therefore, similar changes might occur in other cells that are sensitive to hyperglycemia, and they might be relevant for the pathogenesis of certain complications in diabetes.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Aerts JL, Gonzales MI, Topalian SL.Selection of appropriate control genes to assess expression of tumor antigens using real-time RT-PCR BioTechniques 20043684–86., 88, 90–81, [DOI] [PubMed] [Google Scholar]
- 2.Amiranoff B, Couvineau A, Vauclin-Jacques N, Laburthe M. Gastric inhibitory polypeptide receptor in hamster pancreatic beta cells. Direct cross-linking, solubilization and characterization as a glycoprotein. Eur J Biochem. 1986;159:353–358. doi: 10.1111/j.1432-1033.1986.tb09875.x. [DOI] [PubMed] [Google Scholar]
- 3.Bailey CJ, Flatt PR, Kwasowski P, Powell CJ, Marks V. Immunore-active gastric inhibitory polypeptide and K cell hyperplasia in obese hyperglycaemic (ob/ob) mice fed high fat and high carbohydrate cafeteria diets. Acta Endocrinol. 1986;112:224–229. doi: 10.1530/acta.0.1120224. [DOI] [PubMed] [Google Scholar]
- 4.Balan KV, Wang Y, Chen SW, Chen JC, Zheng LF, Yang L, Liu ZL, Pantazis P, Wyche JH, Han Z. Proteasome-independent down-regulation of estrogen receptor-alpha (ERalpha) in breast cancer cells treated with 4,4′-dihydroxy-trans-stilbene. Biochem Pharmacol. 2006;72:573–581. doi: 10.1016/j.bcp.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 5.Bohm SK, Grady EF, Bunnett NW. Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem J. 1997;322:1–18. doi: 10.1042/bj3220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 7.Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 2004;14:103–106. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Creutzfeldt W, Talaulicar M, Ebert R, Willms B. Inhibition of gastric inhibitory polypeptide (GIP) release by insulin and glucose in juvenile diabetes. Diabetes. 1980;29:140–145. doi: 10.2337/diab.29.2.140. [DOI] [PubMed] [Google Scholar]
- 9.Crockett SE, Mazzaferri EL, Cataland S. Gastric inhibitory polypeptide (GIP) in maturity-onset diabetes mellitus. Diabetes. 1976;25:931–935. doi: 10.2337/diab.25.10.931. [DOI] [PubMed] [Google Scholar]
- 10.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the [beta]2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 11.Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37:826–828. doi: 10.1210/jcem-37-5-826. [DOI] [PubMed] [Google Scholar]
- 12.Futter CE, Pearse A, Hewlett LJ, Hopkins CR. Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J Cell Biol. 1996;132:1011–1023. doi: 10.1083/jcb.132.6.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- 14.Hanna J, Leggett DS, Finley DE. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol Cell Biol. 2003;23:9251–9261. doi: 10.1128/MCB.23.24.9251-9261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- 16.Hicke L, Zanolari B, Riezman H. Cytoplasmic tail phosphorylation of the alpha-factor receptor is required for its ubiquitination and internalization. J Cell Biol. 1998;141:349–358. doi: 10.1083/jcb.141.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hjerrild M, Stensballe A, Rasmussen TE, Kofoed CB, Blom N, Sicheritz-Ponten T, Larsen MR, Brunak S, Jensen ON, Gammeltoft S. Identification of phosphorylation sites in protein kinase A substrates using artificial neural networks and mass spectrometry. J Proteome Res. 2004;3:426–433. doi: 10.1021/pr0341033. [DOI] [PubMed] [Google Scholar]
- 18.Iacovelli L, Franchetti R, Grisolia D, De Blasi A. Selective regulation of G protein-coupled receptor-mediated signaling by G protein-coupled receptor kinase 2 in FRTL-5 cells: analysis of thyrotropin, alpha(1B)-adrenergic, and A(1) adenosine receptor-mediated responses. Mol Pharmacol. 1999;56:316–324. doi: 10.1124/mol.56.2.316. [DOI] [PubMed] [Google Scholar]
- 19.Irwin N, Gault VA, Green BD, Greer B, Harriott P, Bailey CJ, Flatt PR, O’Harte FP. Antidiabetic potential of two novel fatty acid derivatised, N-terminally modified analogues of glucose-dependent insulino-tropic polypeptide (GIP): N-AcGIP(LysPAL16) and N-AcGIP(Lys-PAL37) Biol Chem. 2005;386:679–687. doi: 10.1515/BC.2005.079. [DOI] [PubMed] [Google Scholar]
- 20.Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, Coughlin SR. Inhibition of thrombin receptor signaling by a G-protein coupled receptor kinase. Functional specificity among G-protein coupled receptor kinases. J Biol Chem. 1994;269:1125–1130. [PubMed] [Google Scholar]
- 21.Jackson RA, Blix PM, Matthews JA, Morgan LM, Rubenstein AH, Nabarro JD. Comparison of peripheral glucose uptake after oral glucose loading and a mixed meal. Metabolism. 1983;32:706–710. doi: 10.1016/0026-0495(83)90128-2. [DOI] [PubMed] [Google Scholar]
- 22.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nature. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 23.Kilianova Z, Basora N, Kilian P, Payet MD, Gallo-Payet N. Human melanocortin receptor 2 expression and functionality: effects of protein kinase A and protein kinase C on desensitization and internalization. Endocrinology. 2006;147:2325–2337. doi: 10.1210/en.2005-0991. [DOI] [PubMed] [Google Scholar]
- 24.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 25.Koenig JA, Edwardson JM. Kinetic analysis of the trafficking of muscarinic acetylcholine receptors between the plasma membrane and intra-cellular compartments. J Biol Chem. 1994;269:17174–17182. [PubMed] [Google Scholar]
- 26.Lee AV, Gooch JL, Oesterreich S, Guler RL, Yee D. Insulin-like growth factor I-induced degradation of insulin receptor substrate 1 is mediated by the 26S proteasome and blocked by phosphatidylinositol 3′-kinase inhibition. Mol Cell Biol. 2000;20:1489–1496. doi: 10.1128/mcb.20.5.1489-1496.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis JT, Dayanandan B, Habener JF, Kieffer TJ. Glucose-dependent insulinotropic polypeptide confers early phase insulin release to oral glucose in rats: demonstration by a receptor antagonist. Endocrinology. 2000;141:3710–3716. doi: 10.1210/endo.141.10.7750. [DOI] [PubMed] [Google Scholar]
- 28.Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002;156:843–854. doi: 10.1083/jcb.200106056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu M, Wheeler MB, Leng XH, Boyd AE., 3rd The role of the free cytosolic calcium level in beta-cell signal transduction by gastric inhibitory polypeptide and glucagon-like peptide I(7-37) Endocrinology. 1993;132:94–100. doi: 10.1210/endo.132.1.8380389. [DOI] [PubMed] [Google Scholar]
- 30.Lynn FC, Pamir N, Ng EH, McIntosh CH, Kieffer TJ, Pederson RA. Defective glucose-dependent insulinotropic polypeptide receptor expression in diabetic fatty Zucker rats. Diabetes. 2001;50:1004–1011. doi: 10.2337/diabetes.50.5.1004. [DOI] [PubMed] [Google Scholar]
- 31.Lynn FC, Thompson SA, Pospisilik JA, Ehses JA, Hinke SA, Pamir N, McIntosh CH, Pederson RA. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J. 2003;17:91–93. doi: 10.1096/fj.02-0243fje. [DOI] [PubMed] [Google Scholar]
- 32.Maletti M, Altman JJ, Hoa DH, Carlquist M, Rosselin G. Evidence of functional gastric inhibitory polypeptide (GIP) receptors in human insulinoma. Binding of synthetic human GIP 1-31 and activation of adenylate cyclase. Diabetes. 1987;36:1336–1340. doi: 10.2337/diab.36.11.1336. [DOI] [PubMed] [Google Scholar]
- 33.Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- 34.Maudsley S, Martin B, Luttrell LM. The origins of diversity and specificity in g protein-coupled receptor signaling. J Pharmacol Exp Ther. 2005;314:485–494. doi: 10.1124/jpet.105.083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meier JJ, Hucking K, Holst JJ, Deacon CF, Schmiegel WH, Nauck MA. Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes. 2001;50:2497–2504. doi: 10.2337/diabetes.50.11.2497. [DOI] [PubMed] [Google Scholar]
- 36.Meier JJ, Nauck MA, Siepmann N, Greulich M, Holst JJ, Deacon CF, Schmidt WE, Gallwitz B. Similar insulin secretory response to a gastric inhibitory polypeptide bolus injection at euglycemia in first-degree relatives of patients with type 2 diabetes and control subjects. Metabolism. 2003;52:1579–1585. doi: 10.1016/s0026-0495(03)00327-5. [DOI] [PubMed] [Google Scholar]
- 37.Miyawaki K, Yamada Y, Yano H, Niwa H, Ban N, Ihara Y, Kubota A, Fujimoto S, Kajikawa M, Kuroe A, Tsuda K, Hashimoto H, Yamashita T, Jomori T, Tashiro F, Miyazaki J, Seino Y. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:14843–14847. doi: 10.1073/pnas.96.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan LM, Tredger JA, Madden A, Kwasowski P, Marks V. The effect of guar gum on carbohydrate-, fat- and protein-stimulated gut hormone secretion: modification of postprandial gastric inhibitory polypeptide and gastrin responses. Br J Nutr. 1985;53:467–475. doi: 10.1079/bjn19850056. [DOI] [PubMed] [Google Scholar]
- 39.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 40.Nauck M, Stockmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52. doi: 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- 41.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–307. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neill JD, Duck LW, Musgrove LC, Sellers JC. Potential regulatory roles for G protein-coupled receptor kinases and beta-arrestins in gonadotropin-releasing hormone receptor signaling. Endocrinology. 1998;139:1781–1788. doi: 10.1210/endo.139.4.5868. [DOI] [PubMed] [Google Scholar]
- 43.Okamoto T, Murayama Y, Hayashi Y, Inagaki M, Ogata E, Nishimoto I. Identification of a Gs activator region of the beta 2-adrenergic receptor that is autoregulated via protein kinase A-dependent phosphorylation. Cell. 1991;67:723–730. doi: 10.1016/0092-8674(91)90067-9. [DOI] [PubMed] [Google Scholar]
- 44.Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology. 2000;141:4600–4605. doi: 10.1210/endo.141.12.7806. [DOI] [PubMed] [Google Scholar]
- 45.Service FJ, Rizza RA, Westland RE, Hall LD, Gerich JE, Go VL. Gastric inhibitory polypeptide in obesity and diabetes mellitus. J Clin Endocrinol Metab. 1984;58:1133–1140. doi: 10.1210/jcem-58-6-1133. [DOI] [PubMed] [Google Scholar]
- 46.Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 47.Stang E, Johannessen LE, Knardal SL, Madshus IH. Polyubiquitination of the epidermal growth factor receptor occurs at the plasma membrane upon ligand-induced activation. J Biol Chem. 2000;275:13940–13947. doi: 10.1074/jbc.275.18.13940. [DOI] [PubMed] [Google Scholar]
- 48.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Theodorakis MJ, Carlson O, Muller DC, Egan JM. Elevated plasma glucose-dependent insulinotropic polypeptide associates with hyperinsulinemia in impaired glucose tolerance. Diabetes Care. 2004;27:1692–1698. doi: 10.2337/diacare.27.7.1692. [DOI] [PubMed] [Google Scholar]
- 50.Torrecilla I, Tobin AB. Co-ordinated covalent modification of G-protein coupled receptors. Curr Pharm Des. 2006;12:1797–1808. doi: 10.2174/138161206776873716. [DOI] [PubMed] [Google Scholar]
- 51.Tseng CC, Zhang XY, Wolfe MM. Effect of GIP and GLP-1 antagonists on insulin release in the rat. Am J Physiol Endocrinol Metab. 1999;276:E1049–E1054. doi: 10.1152/ajpendo.1999.276.6.E1049. [DOI] [PubMed] [Google Scholar]
- 52.Vilsboll T, Knop FK, Krarup T, Johansen A, Madsbad S, Larsen S, Hansen T, Pedersen O, Holst JJ. The pathophysiology of diabetes involves a defective amplification of the late-phase insulin response to glucose by glucose-dependent insulinotropic polypeptide-regardless of etiology and phenotype. J Clin Endocrinol Metab. 2003;88:4897–4903. doi: 10.1210/jc.2003-030738. [DOI] [PubMed] [Google Scholar]
- 53.Widmann C, Dolci W, Thorens B. Desensitization and phosphorylation of the glucagon-like peptide-1 (GLP-1) receptor by GLP-1 and 4-phorbol 12-myristate 13-acetate. Mol Endocrinol. 1996;10:62–75. doi: 10.1210/mend.10.1.8838146. [DOI] [PubMed] [Google Scholar]
- 54.Xu GKH, Laybutt DR, Duvivier-Kali VF, Trivedi N, Suzuma K, King GL, Weir GC, Bonner-Weir S.Downregulation of GLP-1 and GIP-receptor expression by hyperglycemia: possible contribution to impaired incretin effects in diabetes Diabetes in press, 2007 [DOI] [PubMed] [Google Scholar]