

Abstract
Activation of the p53 tumor suppressor protein has been demonstrated to block cell growth by inducing either a transient cell cycle arrest or programmed cell death (apoptosis). Although evidence exists linking p53’s function as an activator of transcription to its ability to effect cell cycle arrest, the role of this activity in the induction of apoptosis remains unclear. To gain insight into the molecular mechanisms underlying p53-mediated antiproliferative pathways, a study was initiated to explore the functions of a putative p53 signaling domain. This region of the human p53 protein is localized between amino acids 61 and 94 (out of 393) and is noteworthy in that it contains five repeats of the sequence PXXP (where P represents proline and X any amino acid). This motif has been shown to play a role in signal transduction via its SH3 domain binding activity. A p53 cDNA deletion mutant (ΔproAE), which lacks this entire proline-rich domain (deleted for amino acids 62–91), was created and characterized for a variety of p53 functions. The entire domain has been shown to be completely dispensable for transcriptional activation. On the other hand, this deletion of the p53 proline-rich domain impairs p53’s ability to suppress tumor cell growth in culture. Amino acid substitution mutations at residues 22 and 23 of p53 (eliminates transcriptional activity) also impair p53-mediated inhibition of cell growth in culture. Unlike wild-type p53, the ΔproAE mutant cDNA can be stably expressed in tumor derived cell lines with few immediate detrimental effects. These cells express physiologic levels of p53 protein that are induced normally in response to DNA damage, indicating that removal of the proline-rich domain does not disrupt p53’s upstream regulation by DNA damage. These data indicate that, in addition to the transcriptional activation domain, the p53 proline-rich domain plays a critical role in the transmission of antiproliferative signals downstream of the p53 protein and may link p53 to a direct signal transduction pathway.
The p53 tumor suppressor protein plays a pivotal role in the prevention of cellular transformation by curtailing the proliferation of cells harboring potentially oncogenic lesions. The unusually high frequency of p53 mutations observed in human cancers (1) indicates the complexity of the antiproliferative pathways under p53 regulation. Indeed, evidence now suggests that p53 can respond to multiple signals of cellular alarm, including DNA damage (2–4) and perturbations of cell cycle regulation (5–7), by inducing either a transient growth arrest (8–10) or programmed cell death (apoptosis; refs. 11–13). At the molecular level, p53 has been demonstrated to function as a transcriptional activator (14, 15). Transcriptional activation is dependent upon three independent structural domains that mediate: (i) sequence-specific DNA binding (amino acid residues 100–290 out of 393) (16, 17); (ii) interactions with the basal transcription factor TFIID (residues 1–40; refs. 18 and 19); and (iii) homooligomerization (residues 319–360; ref. 20). That the induction of novel gene expression underlies p53’s ability to alter cell cycle progression is strongly supported by evidence that several p53 target genes, such as p21 (21), MDM2 (22), GADD45 (23), and cyclin G (24, 25), encode for potential cell cycle regulatory proteins. Moreover, in cell culture systems, p53 mutants engineered for a loss of transcriptional activation are also defective in activating the G1 cell cycle checkpoint (26). In contrast, the molecular mechanism underlying p53-dependent apoptosis remains less well-defined. Recently, it has been demonstrated that p53-dependent apoptosis can proceed despite the inhibition of ongoing of protein synthesis (27) as well as in response to overexpression of p53 mutant proteins that are transcriptionally inactive (28, 29). These observations lead one to question whether transcriptional activation alone is sufficient for p53-dependent tumor suppression in vivo.
MATERIALS AND METHODS
Cell Lines and Plasmids.
The cell line H1299 (human non-small-cell lung carcinoma; ref. 30) was maintained as described previously (31). Expression plasmids for p53 mutants, pRC/CMV-SN22/23 and pRC/CMV-R175H (31), and human mdm-2, pCHDMΔ222-437 (32), have been previously described. The p53-responsive reporter constructs, WAF1-CAT (21) and BP100CAT (22) have been previously described. The p53-responsive reporter BAX-luciferase was provided by Moshe Oren (Weizmann Institute, Rehovot, Israel) and contains the p53-response element characterized by Miyashita and Reed (33).
Mutagenesis.
The p53 deletion mutant ΔproAE was constructed by using PCR to independently amplify cDNAs encoding amino acids 1–62 and amino acids 92–393. The full-length p53 cDNA cloned into the vector Bluescript KS (Stratagene) was used as a template (31). Primer pairs included the T3 forward primer (Bluescript sequence) plus the reverse primer, 5′-CGGATCCGGACCTGGGTCTTC-3′ (p53 sequence with a 5′ BamHI site) for amino acids 1–62 and the forward primer 5′-CCGGATCCCCTGTCATCTTCTG-3′ (p53 sequences and a 5′ BamHI site) plus the T7 reverse primer (Bluescript sequence) for amino acids 92–393. Individual PCR products were directly cloned into the TA vector (Invitrogen) and subsequently cloned into the expression vector pRC/CMV (Invitrogen) and ligated together at the internal BamHI site.
Transfections and Transactivation Assays.
For transactivation assays, cells were plated at ≈20% confluency for 18–24 hr. Transfections were carried out with 1 μg of reporter plasmid and 200 ng of p53 expression plasmid (unless otherwise indicated) and adjusted to a total of 15 μg of DNA with sheared salmon sperm DNA that was prepared in a calcium phosphate precipitate (34). Cells were exposed to calcium phosphate/DNA precipitates for ≈12 hr, then washed twice with phosphate-buffered saline (PBS), refed with 10 ml of completed medium, and incubated for an additional 48 hr. For chloramphenicol acetyltransferase (CAT) assays, cells were harvested, lysed, and analyzed as previously described (35).
Growth Suppression Assay.
H1299 cells grown in 10-cm dishes were transfected with 10 μg of plasmid using lipofectin reagent (BRL) and serum-free medium (Optimem; BRL) according to manufacturer’s specifications. Lipofectin–DNA precipitates were left in the culture medium for 12 hr. Cells were then washed twice with PBS and refed with 10 ml of completed DMEM. Twenty-four hours following completion of transfection, cells were counted and seeded at three different cell densities: 1 × 105, 5 × 104, and 2.5 × 104 cells per 10-cm plate in 15 ml of completed DMEM. One day later, cells were placed under drug selection by adding 10 ml of completed DMEM containing geneticin (BRL) at 0.8 mg/ml. Cells were refed every 4–5 days with fresh drug-supplemented medium. Colonies were stained and scored ≈2 weeks following transfection.
Immunoprecipitation/Western Blot Analysis.
Whole-cell lysates were prepared as described previously (31), and the p53 antibody, pAb421, and 30 μl of protein A–Sepharose, which were incubated for 2–8 hr at 4°C, were used for p53 immunoprecipitations. Immunocomplexes were washed three times in 0.5% Nonidet P-40 lysis buffer and resolved in SDS/10% polyacrylamide gels. For Western blot analysis, gels were transferred to Immobilon-P membrane (Millipore) and probed with pAb421 (for p53) for 2–6 hr. Bound antibody was detected using 125I-conjugated protein A–Sepharose and autoradiography. Data were quantitated using PhosphorImager analysis (Molecular Dynamics) and imagequant software.
RESULTS
Structural Definition of a Putative p53 Signaling Domain.
Previous studies have divided the p53 protein into four structural and functional domains [see Ko and Prives (36); Fig. 1A]. However, the p53 protein contains a potentially unique fifth structural domain, defined approximately by amino acids 61–94 of human p53, whose function is explored in this communication (see open region in Fig. 1A). This region is rich in the amino acid proline (12/34 residues) and contains five repeats of the amino acid sequence PXXP (P designating proline and X designating any amino acid). Sequences containing multiple PXXP repeats have been detected in a variety of proteins, which comprise diverse signal transduction pathways [see Cohen et al. (38) and Pawson (39) for reviews]. Detailed structural studies have revealed that the PXXP residues form a left-handed polyproline type II helix, which creates a binding site for SH3 (src homology 3) domains (40). The fundamental importance of the PXXP sequence is supported by the fact that these motifs have been found in all proteins known to bind directly to SH3 domains, despite the fact that specificity of such interactions appears to be determined by variable adjacent residues (41). Because of the well-documented role that PXXP-rich domains play in the formation of signaling complexes, a reasonable hypothesis is that the proline-rich domain of p53 may function as a discrete signaling module. Support for this idea comes from the fact that the overall structural features of p53 (residues 61–94) have been maintained throughout evolution. Fig. 1B demonstrates that both the high proline content of this region and, in particular, the PXXP motif are conserved in p53 molecules from divergent species. Therefore, the hypothesis that the proline-rich domain of p53 may participate in the transmission of antiproliferative signals through a mechanism independent of transcriptional activation was explored.
Figure 1.
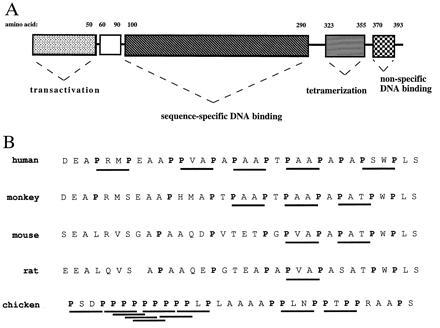
Structural features of the p53 protein. (A) Schematic representation of the domain structure of the human p53 protein indicating regions mediating transcriptional activation (amino acids 1–50), sequence-specific DNA binding (amino acids 100–290), tetramerization (amino acids 311–393), and non-specific DNA binding (amino acids 370–393). (B) Comparison of the predicted p53 amino acid sequence of the proline-rich domains from human (residues 61–94), monkey (residues 61–94), mouse (residues 55–88), rat (residues 60–92), and chicken (residues 56–89) as determined by Soussi et al. (37). Proline residues are indicated by boldface type and PXXP motifs are underlined.
The Proline-Rich Domain of p53 Is Dispensable for Transactivation.
To gain insight into the possible functions of p53’s proline-rich domain, p53 cDNAs that were deleted in this region of the protein were produced. A PCR-based method was used to create an internal deletion of amino acids 62–91, removing all five PXXP motifs from the human p53 protein. The deletion mutant, designated ΔproAE, was placed under the control of the cytomegalovirus promoter in the expression vector pRC-CMV so as to express the mutant protein in cells. To determine the relative contributions to p53 function made by different regions of the molecule, the transcriptionally impaired double point mutant, 22-23 (containing two amino acid substitutions in the transcriptional activation domain; ref. 31) and the tumor-derived, DNA binding-defective mutant, R175H (with a single amino acid substitution in the DNA binding domain; ref. 26), were analyzed in parallel with ΔproAE. As a first test of p53 ΔproAE function, these cDNAs were transfected into cells without any endogenous p53 (H1299) along with a p53-responsive reporter construct, WAF1-CAT, containing the regulatory elements derived from the p53-responsive p21 gene. A cDNA for the human wild-type p53 protein served as a positive control. As shown in Fig. 2A, both the 22-23 and R175H mutants demonstrate severe defects in transactivation, confirming the requirement for both intact DNA binding and transactivation domains. In contrast, the ΔproAE mutant activates the expression of WAF1-CAT to a level greater than the wild-type p53 (Fig. 2B). These results have been recapitulated with the p53-responsive reporters derived from the regulatory regions of the MDM2 and BAX genes (data not shown), demonstrating the generality of the reporter sequence employed to test the ΔproAE mutant. Moreover, when assayed in the p53-null cell lines SAOS-2 (human osteosarcoma) and (10)1 (immortalized murine), the activity of the ΔproAE mutant was also greater than or indistinguishable from that of the wild-type protein (data not shown), showing cell-type independence. Together, these observations suggest that p53’s proline-rich domain is dispensable for transactivation and establish ΔproAE as an unusual p53 mutant in having sustained a dramatic structural change without a consequent loss of transactivation potential.
Figure 2.
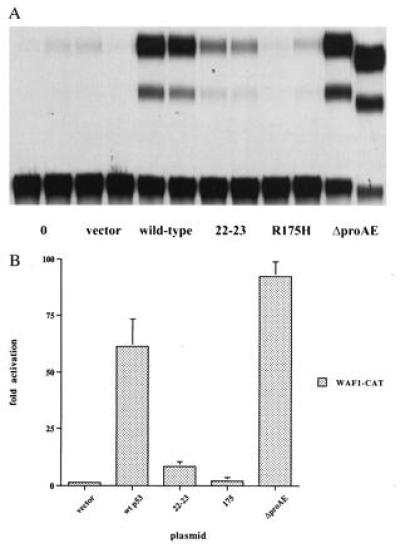
Transcriptional transactivation activity of p53 mutants in H1299 cells. (A) Transactivation of the p53-responsive reporter, WAF1-CAT (21). cDNAs encoding for wild-type p53, the double point mutant 22-23, the tumor-derived mutant R175H, and the deletion mutant ΔproAE (deleted of residues 62–91) cloned into the mammalian expression vector, pRC-CMV (Invitrogen) were used for transactivation assays. Cells were cotransfected with 100 ng of p53 expression plasmid or the empty vector and 1 μg of reporter plasmid. Each transfection is presented in duplicate. (B) Quantitation of transactivation data displayed graphically as the fold activation of the WAF1-CAT reporter plasmid over its basal level of expression.
The Proline-Rich Domain of p53 Is Necessary for Efficient Growth Suppression.
Based on the prevalent idea that transcriptional activation correlates with p53-mediated growth suppression, it became of interest to compare the activity of the ΔproAE mutant to that of the transactivation mutant, 22-23, and the DNA binding mutant, R175H, in a growth suppression assay. The ability of these mutants to suppress the growth of tumor cells in culture was determined by colony formation assay. This assay enables growth suppression in its most general sense to be measured as it scores for either p53-dependent growth arrest and/or apoptosis in tumor cells that receive the p53 protein. Plasmids containing wild-type p53 or the various mutants in cis with a G418 resistance marker were transfected into the H1299 line (null for p53) and geneticin-resistant colonies were scored ≈2 weeks later. Results from four independent colony formation assays are presented in Table 1. As a potent suppressor of tumor cell growth in culture, the wild-type p53 plasmid reduces the number of drug-resistant colonies 10-fold, yielding an average ratio of 0.1 colonies relative to the empty vector control. The tumor-derived mutant, R175H, in contrast, is completely defective in this assay, demonstrating no suppression of colony formation. Interestingly, the ΔproAE mutant demonstrates a significant defect in growth suppression, achieving a <2-fold decrease in colony number compared with the R175H mutant. Therefore, the removal of the proline-rich domain from the p53 protein reduces the efficiency of growth suppression in culture ≈5- to 6-fold as compared with the wild-type p53 cDNA. These same results have also been repeated in the p53-null tumor cell line SAOS-2, arguing that the defect is not cell type-specific (data not shown). Because the ΔproAE mutant maintains its activity as a transactivator, these results indicate that p53-mediated growth suppression can, in fact, be partially uncoupled from transcriptional activation. That these two activities may be separable has precedence in the observations that p53 mutants defective for transcriptional activation still maintain activity in assays measuring suppression of oncogene-mediated transformation (42, 43) or apoptosis (28, 29). However, this is the first report that a structural change outside the DNA binding domain can reduce p53’s growth suppression activity without apparently altering its transactivation properties. These data strongly suggest that p53’s proline-rich domain mediates an activity critical for the effective transmission of antiproliferative signals.
Table 1.
Growth suppression of H1299 cells following transfection with mutant p53 expression plasmids
| Plasmid | No. of drug-resistant
colonies*
|
Average† | |||
|---|---|---|---|---|---|
| Exp 1 | Exp 2 | Exp 3 | Exp 4 | ||
| Vector | 410 (1.00) | 510 (1.00) | 325 (1.00) | 540 (1.00) | |
| Wild-type p53 | 48 (0.12) | 53 (0.10) | 28 (0.09) | 55 (0.10) | 0.10 |
| 22-23 | 250 (0.61) | 334 (0.65) | 195 (0.60) | 273 (0.51) | 0.59 |
| R175H | 425 (1.04) | 525 (1.03) | 390 (1.20) | 510 (0.94) | 1.03 |
| ΔproAE | 310 (0.76) | 260 (0.51) | 180 (0.55) | 253 (0.47) | 0.57 |
The data shown represent the number of drug-resistant colonies counted 2 weeks following transfection with 10 μg of the indicated plasmid.
Numbers in parentheses represent the calculated ratio of colony number relative to the number scored on the empty vector control plate.
The average of the four ratio values determined for each plasmid is presented in the last column.
Although it has sustained a significant loss of growth suppression activity, it must be noted that the ΔproAE mutant does not demonstrate a complete loss of function, in contrast to the tumor-derived mutant, R175H. Interestingly, the transactivation mutant, 22-23, also displays this same intermediate phenotype. In fact, the activities of the 22-23 and ΔproAE mutants are virtually indistinguishable in this growth suppression assay, despite the fact that their transactivation properties are diametrically opposed. Together, these observations suggest that the proline-rich domain and the transactivation domain are both necessary for p53-dependent growth suppression, but that each alone is not sufficient. Therefore, it is likely that p53’s transcriptional regulatory activity must be complemented by an additional function directly or indirectly mediated by its proline-rich domain to effectively transmit antiproliferative signals.
To confirm that the defect in growth suppression manifest by the ΔproAE mutant does not merely reflect an instability of the protein in tumor cells, clonal cell lines generated from the stable transfection of each p53 plasmid were tested for p53 protein expression by Western blot analysis of p53-specific immunocomplexes. An analysis of five randomly selected drug-resistant cell lines generated from each individual p53 plasmid is presented in Fig. 3. As a control, the cell line (10)1val5 (an immortalized, p53-null, murine embryo fibroblast line stably transfected with the temperature-sensitive murine mutant p53 A135V) was subjected to the same immunoprecipitation/Western blot analysis. As shown in Fig. 3, full-length p53 is immunoprecipitated from (10)1val5 lysates with the p53-specific monoclonal antibody, pAb421 (lane 21), but not the negative control antibody, pAb419 (lane 22), which does not recognize p53. Of the five drug-resistant cell lines generated from transfection with the plasmid encoding wild-type p53, none were found to express p53 protein (Fig. 3, lanes 1–5), confirming previous observations that the wild-type p53 protein cannot be stably reintroduced into fully transformed cells (26). In contrast, expression of the R175H mutant is readily detectable in all cell lines stably transfected with this plasmid (see lanes 11–15), indicating that the amino acid substitution at residue 175 in the DNA binding domain abrogates the toxicity of wild-type p53 in the H1299 cell line. Analysis of cell lines stably transfected with the the 22-23 (Fig. 3, lanes 6–10) and the ΔproAE (lanes 16–20) mutant p53 plasmids reveals that both proteins can, in fact, be stably expressed in H1299 cells (see lanes 9, 17, and 19). As predicted, the ΔproAE protein migrates faster than the full-length protein, consistent with the decrease in molecular weight resulting from the deletion of the proline-rich domain. Successful expression of both the 22-23 and the ΔproAE mutant proteins indicates that the biological consequences of these mutations are not due to mere protein destabilization. Interestingly, both these mutant plasmids fail to generate the high percentage of p53-positive cell lines obtained following transfection with the DNA binding mutant, R175H. Instead, each yields an intermediate number of stable cell lines, with 20% (1/5) of the 22-23 lines and 40% (2/5) of the ΔproAE lines expressing detectable p53 protein. These numbers corroborate the intermediate phenotypes displayed by the ΔproAE and 22-23 mutants in the growth suppression assay. Importantly, the stable introduction of the ΔproAE mutant into fully transformed cells strongly suggests that the removal of the proline-rich domain of p53 critically impairs some aspect of growth suppression or apoptosis.
Figure 3.
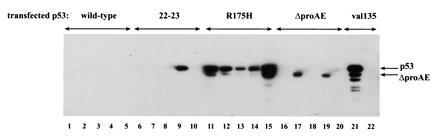
Analysis of H1299 cell lines stably transfected with mutant p53 plasmids. Clonal cell lines were generated by stable transfection with the plasmid pRC-CMV containing p53 cDNAs encoding the wild-type p53 protein (lanes 1–5), the transactivation mutant, 22-23 (lanes 6–10), the DNA binding mutant, R175H (lanes 11–15), the proline deletion mutant, ΔproAE (lanes 16–20), or the temperature-sensitive murine DNA binding mutant, A135V (lanes 21 and 22). Cell lines were analyzed for p53 protein expression by Western blot analysis of p53-specific immunocomplexes. Cell lysates were immunoprecipitated with the panspecific antibody, pAb421, which recognizes both mutant and wild-type conformations of p53 (lanes 1–21) or the negative control antibody, pAb419 (lane 22). Immunocomplexes were resolved by 10% SDS/PAGE, transferred to an Immobilon-P membrane (Millipore), and immunoblotted with pAb421. The positions of full-length p53 and the ΔproAE proteins are indicated.
Stably Expressed ΔproAE Maintains Its Transactivation Activity.
One possibility to account for the unusual fact that the ΔproAE protein is tolerated by tumor cells is that it has become functionally inactivated during the establishment of stable cell lines. This possibility was tested by demonstrating that the Δpro lines express an endogenous activity capable of activating transcription in a p53-dependent manner. Initially, this was approached by determining whether the Δpro lines were capable of transactivating a transiently transfected p53-responsive reporter construct. The parental cell line, H1299, and two of the ΔproAE derivatives were transfected with the WAF1-CAT reporter containing a p53-response elements or as a negative control, Gal4-CAT to determine the level of reporter activation 48 hr post-transfection. As shown in Fig. 4, each of the Δpro lines is able to transactivate the WAF1-CAT reporter (see lanes 7 and 11), but not the Gal4-CAT reporter (lanes 9 and 13). The parental line, in contrast, expresses this activity only when a plasmid containing the ΔproAE cDNA is included in the transfection (lanes 1 and 2). These results indicate that stable expression of the ΔproAE mutant confers to cells the capacity to activate expression of a reporter under the regulation of a p53-response element. That this activity is directly p53-dependent is supported by the fact that the p53-dependent CAT activity is inhibited by cotransfection of the human MDM2 cDNA clone, a known negative regulator of p53’s transactivation function (lanes 8 and 12; ref. 44). This same result was obtained when an MDM2 responsive element, BP100CAT, was employed or the endogenous steady-state levels of p21 protein were examined in these cells. Western blot analysis demonstrated that the Δpro.1 cell line produced 4-fold more p21 protein while the Δpro.3 cell line synthesized 7-fold more p21 protein than the levels found in the parental cell line (results not shown).
Figure 4.
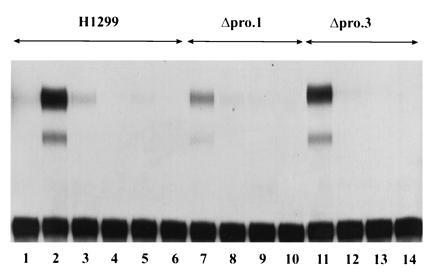
Analysis of the ability of stably expressed ΔproAE to activate transcription. Transactivation of transiently transfected CAT reporter constructs. The H1299 cell line (lanes 1–6) and two independent derivatives stably expressing ΔproAE protein, Δpro.1 (lanes 7–10), and Δpro.3 (lanes 11–14), were transfected with 1 μg of the p53-responsive reporter WAF1-CAT (lanes 1–3, 7 and 8, and 11 and 12) or the negative control reporter Gal4-CAT (lanes 4–6, 9 and 10, and 13 and 14) in the presence (lanes 3, 6, 8, 10, 12, and 14) or absence (lanes 1, 2, 4, 5, 7, 9, 11, and 13) of 10 μg of the human MDM2 expression vector, HDM2. As a positive control for transactivation, 200 ng of the ΔproAE expression plasmid was transfected into the H1299 parental line (lanes 2 and 3).
p53’s Proline-Rich Domain Does Not Participate in p53’s Upstream Regulation by DNA Damage.
There are several possible ways in which the proline-rich domain of p53 could mediate the flow of growth-inhibitory information. For instance, it could participate in the reception of upstream alarm signals, which lead to posttranslational stabilization and activation of the p53 protein; conversely, it could function in the transmission of downstream signals that alter cell growth. To address the first possibility, the effects of DNA damage on the levels of stably expressed ΔproAE protein were tested. The cell line Δpro.3 was irradiated with a high dose of UV irradiation (20 J/m2) and the steady-state levels of p53 protein were determined at 0, 5, 9, and 12 hr postirradiation by immunoprecipitation followed by Western blot analysis using the p53-specific antibody, pAb421. As a control, the induction of wild-type p53 was monitored in the immortalized murine cell line (12)1, which expresses endogenous wild type (45). As shown in Fig. 5, wild-type p53 demonstrates characteristic increases in concentration after UV treatment. Quantitation of these data indicates that the levels of wild-type p53 protein increase ≈6-fold by 5 hr and another 2-fold by 9 hr posttreatment. Under identical experimental conditions, levels of the ΔproAE protein increase 20-fold by 5 hr and another 2-fold by 9 hr post-treatment. Likewise, the ΔproAE protein is similarly induced by gamma irradiation and the cross-linking agent, etoposide (data not shown). These results provide clear evidence that p53’s upstream regulation—i.e., communication with damaged DNA intermediates—is intact in the Δpro.3 line and suggest that the functional defect associated with the loss of the proline-rich domain reflects an inability to transmit downstream antiproliferative signals.
Figure 5.
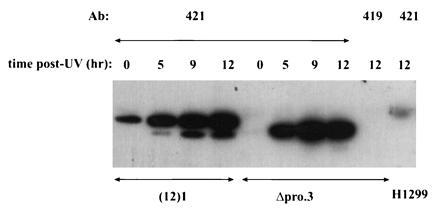
UV induction of ΔproAE protein. The cell lines (12)1 (immortalized murine fibroblast expressing endogenous wild-type p53) and Δpro.3 (derivative of the tumor line H1299 stably expressing ΔproAE) were irradiated with a high dose of UV light (20 J/m2). Cells were collected at 0, 5, 9, and 12 hr posttreatment. Steady-state levels of p53 protein were determined by immunoprecipitation/Western analysis. Cell lysates were immunoprecipitated with the p53-specific monoclonal antibody, pAb421. Negative controls included precipitating the Δpro.3 12-hr lysate with the antibody pAb419, which does not recognize p53 and the p53-null H1299 lysate with pAb421. Immunocomplexes were resolved by 10% SDS/PAGE and transferred to an Immobilon membrane (Millipore). The membrane was probed with the p53-specific antibody pAb421 and bound antibody was detected with 125I-conjugated protein A. Data were quantitated using PhosphorImager analysis (Molecular Dynamics) and imagequant software. The positions of wild-type and ΔproAE p53 are indicated.
DISCUSSION
The p53 protein has been divided into four domains; (i) a transcriptional activation domain that contacts TAF components of TFIID (residues 1–40; refs. 18 and 19); (ii) a sequence-specific DNA binding domain (residues 120–290; refs. 16 and 17); (iii) a tetramerization domain (residues 310–360; ref. 20); and (iv) a domain that recognizes and binds to damaged DNA nonspecifically (residues 364–390; refs. 46 and 47). This manuscript describes a new fifth functional domain, localized between residues 61–94, containing a putative proline-rich signaling domain. Deletion of this domain from the p53 protein leaves a normal p53 protein with respect to transcriptional transactivation. Therefore, despite its physical juxtaposition between the transcriptional activation and DNA binding domains, the proline-rich region of p53 is not essential to the function of these domains. Because the proline-rich domain of p53 is dispensable for transactivation, the ΔproAE mutant serves as a useful tool to address the question of whether transcriptional activation alone is sufficient for the transmission of p53-dependent antiproliferative signals.
Analysis of the ΔproAE mutant in assays designed to measure biological activity has revealed that it is severely compromised for growth suppression. This defect manifests in both the plating efficiency assay (in which the ΔproAE mutant displays a <2-fold reduction in colony number) and in the ability to generate tumor cell lines which stably express this transcriptionally active p53 protein. These observations strongly suggest that transcriptional activation can be partially uncoupled from growth suppression and, moreover, that the proline-rich domain likely mediates an activity critical for p53-dependent tumor suppression in vivo. The results presented here have also demonstrated that cell lines stably expressing the ΔproAE mutant show a strong induction in the levels of this p53 mutant protein in response to DNA damage. Thus, p53’s upstream regulation is intact in these cell lines, suggesting that p53’s proline-rich domain is likely to participate in the transmission of downstream antiproliferative signals.
It has previously been demonstrated that cell lines in which p53 activation is achieved through the conditional expression of high levels of wild-type p53 protein can be blocked in a G1 cell cycle arrest (9). However, these same cells simultaneously expressing high levels of E2F-1 or c-Myc will, instead, undergo apoptosis (48). Thus, p53-mediated events in a cell have some method to detect and appropriately respond to signals generated from a variety of nuclear signal transduction pathways. Transmission of antiproliferative signals in 1299 cells then are likely to require the functions of both p53’s transactivation and proline-rich domains. Indeed, abrogation of just one of these domains does not completely eliminate p53’s ability to suppress the growth of tumor cells in culture, supporting the conclusion that both the transcriptional activation and proline-rich domains are necessary but neither is sufficient for wild-type function. In fact, continued passage of tumor cells lines that stably express either the 22-23 or ΔproAE mutants has revealed that the expression of both these proteins is selected against over time (unpublished observations). Thus, neither of these mutants is functionally inert in the H1299 cell line, in contrast to the tumor-derived mutant, R175H.
The p53 tumor suppressor protein has been implicated in the regulation of multiple cellular antiproliferative pathways, including those resulting in transient cell cycle arrest as well as that which leads to permanent cell death. The specificity of the p53-dependent response appears to be determined by multiple factors, including the nature of the signal as well as the physiologic status of the cell. This manuscript identifies a new domain of the p53 protein that possesses a new activity mediated by its proline-rich domain, which is involved in signaling for growth suppression. A reasonable hypothesis of how p53’s proline-rich domain may signal is via contacting an SH3 domain of another protein. Indeed, both full-length p53 and the domain corresponding to residues 61–94 display SH3 domain binding activity in vitro (K.K.W., A. Muller, and A.J.L., unpublished results). Thus, the p53 protein may use multiple mechanisms to inhibit cell growth, and the degree to which transcriptional regulation and direct signaling contribute to signal transmission will vary depending on the particular conditions.
Footnotes
Abbreviation: CAT, chloramphenicol acetyltransferase.
References
- 1.Hollstein M, Sidransky D, Vogelstein B, Harris C C. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 2.Maltzman W, Czyzyk L. Mol Cell Biol. 1984;4:1689–1694. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R W. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 4.Lu X, Lane D P. Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 5.Lowe S W, Ruley H E. Genes Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 6.Debbas M, White E. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- 7.Symonds H, Krall L, Remington L, Saenz-Robies M, Lowe S, Jacks T, Van Dyke T. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 8.Diller L, Kassel J, Nelson C E, Gryka M A, Litwak G, Gebhardt M, Bressac B, Ozturk M, Baker S J, Vogelstein B, Friend S H. Mol Cell Biol. 1990;10:5772–5781. doi: 10.1128/mcb.10.11.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez J, Georgoff I, Martinez J, Levine A J. Genes Dev. 1991;5:151–159. doi: 10.1101/gad.5.2.151. [DOI] [PubMed] [Google Scholar]
- 10.Kuerbitz S J, Plunkett B S, Walsh W V, Kastan M B. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Nature (London) 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 12.Lowe S W, Schmitt E M, Smith S W, Osborne B A, Jacks T. Nature (London) 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 13.Clarke A R, Purdie C A, Harrison D J, Morris R G, Bird C C, Hooper M L, Wyllie A H. Nature (London) 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 14.Fields S, Jang S K. Science. 1990;249:1046–1049. doi: 10.1126/science.2144363. [DOI] [PubMed] [Google Scholar]
- 15.Raycroft L, Wu H, Lozano G. Science. 1990;249:1049–1051. doi: 10.1126/science.2144364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bargonetti J, Manfredi J J, Chen X, Marshak D R, Prives C. Genes Dev. 1994;7:2565–2574. doi: 10.1101/gad.7.12b.2565. [DOI] [PubMed] [Google Scholar]
- 17.Cho Y, Gorina S, Jeffrey P D, Pavletich N P. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 18.Thut C J, Chen J L, Klemin R, Tjian R. Science. 1995;267:100–104. doi: 10.1126/science.7809597. [DOI] [PubMed] [Google Scholar]
- 19.Lu H, Levine A J. Proc Natl Acad Sci USA. 1995;92:5154–5158. doi: 10.1073/pnas.92.11.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clore G M, Ernst J, Clubb R, Omichinski J G, Kennedy W M P, Sakaguchi K, Appella E, Gronenborn A M. Struct Biol. 1994;2:321–332. doi: 10.1038/nsb0495-321. [DOI] [PubMed] [Google Scholar]
- 21.El-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, Bayle J H, Olson D, Levine A J. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 23.Kastan M B, Zhan Q, El-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J., Jr Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 24.Okamoto K, Beach D. EMBO J. 1994;13:4816–4822. doi: 10.1002/j.1460-2075.1994.tb06807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zauberman A, Lupo A, Oren M. Oncogene. 1995;10:2361–2366. [PubMed] [Google Scholar]
- 26.Hinds P W, Finlay C A, Quartin R S, Baker S J, Fearon E R, Vogelstein B, Levine A J. Cell Growth Differ. 1990;1:571–580. [PubMed] [Google Scholar]
- 27.Caelles C, Heimberg A, Karin M. Nature (London) 1994;370:220–223. doi: 10.1038/370220a0. [DOI] [PubMed] [Google Scholar]
- 28.Wagner A J, Kokontis J M, Hay N. Genes Dev. 1994;8:2817–2830. doi: 10.1101/gad.8.23.2817. [DOI] [PubMed] [Google Scholar]
- 29.Haupt Y, Rowan S, Shaulian E, Vousden K, Oren M. Genes Dev. 1995;9:2170–2183. doi: 10.1101/gad.9.17.2170. [DOI] [PubMed] [Google Scholar]
- 30.Mitsudomi T, Steinberg S M, Nau M M, Carbone D, D’Amico D, Bodner S, Oie H K, Linnoila R I, Mulshine J L, Minna J D, Gazdar A F. Oncogene. 1992;7:171–180. [PubMed] [Google Scholar]
- 31.Lin J, Chen J, Elenbaas B, Levine A J. Genes Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Lin J, Levine A J. Mol Med. 1995;1:142–152. [PMC free article] [PubMed] [Google Scholar]
- 33.Miyashita T, Reed J C. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 34.Graham F L, van der Eb A J. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 35.Zambetti G P, Olson D, Labow M, Levine A J. Proc Natl Acad Sci USA. 1992;89:3952–3956. doi: 10.1073/pnas.89.9.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko L J, Prives C. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 37.Soussi T, Caron De Fromentel C, May P. Oncogene. 1990;5:945–952. [PubMed] [Google Scholar]
- 38.Cohen G B, Ren R, Baltimore D. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- 39.Pawson T. Nature (London) 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 40.Yu H, Chen J C, Feng S, Dalgarno D C, Brauer A W, Schreiber S L. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 41.Rickles R J, Botfield M C, Weng Z, Taylor J A, Green O M, Brugge J S, Zoller M A. EMBO J. 1994;13:5598–5604. doi: 10.1002/j.1460-2075.1994.tb06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unger T, Mietz J A, Scheffner M, Yee C, Howley P M. Mol Cell Biol. 1993;13:5186–5194. doi: 10.1128/mcb.13.9.5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crook T, Marston N J, Sara E A, Vousden K H. Cell. 1994;79:817–827. doi: 10.1016/0092-8674(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 44.Momand J, Zambetti G P, Olson D C, George D, Levine A J. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 45.Harvey D, Levine A J. Genes Dev. 1991;5:2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- 46.Wang B Q, Kostrub C F, Finkelstein A, Burton Z F. Protein Expr Purif. 1993;4:207–214. doi: 10.1006/prep.1993.1027. [DOI] [PubMed] [Google Scholar]
- 47.Lee S, Elenbaas B, Levine A J, Griffith J. Cell. 1995;81:1–20. [Google Scholar]
- 48.Wu X, Levine A J. Proc Natl Acad Sci USA. 1994;91:3602–3606. doi: 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]