

Abstract
It has been demonstrated that CD8+ T cells produce a soluble factor(s) that suppresses human immunodeficiency virus (HIV) replication in CD4+ T cells. The role of soluble factors in the suppression of HIV replication in monocyte/macrophages (M/M) has not been fully delineated. To investigate whether a CD8+ T-cell-derived soluble factor(s) can also suppress HIV infection in the M/M system, primary macrophages were infected with the macrophage tropic HIV-1 strain Ba-L. CD8+ T-cell-depleted peripheral blood mononuclear cells were also infected with HIV-1 IIIB or Ba-L. HIV expression from the chronically infected macrophage cell line U1 was also determined in the presence of CD8+ T-cell supernatants or β-chemokines. We demonstrate that: (i) CD8+ T-cell supernatants did, but β-chemokines did not, suppress HIV replication in the M/M system; (ii) antibodies to regulated on activation normal T-cell expressed and Secreted (RANTES), macrophage inflammatory protein 1α (MIP-1α) and MIP-1β did not, whereas antibodies to interleukin 10, interleukin 13, interferon α, or interferon γ modestly reduced anti-HIV activity of the CD8+ T-cell supernatants; and (iii) the CD8+ T-cell supernatants did, but β-chemokines did not, suppress HIV-1 IIIB replication in peripheral blood mononuclear cells as well as HIV expression in U1 cells. These results suggest that HIV-suppressor activity of CD8+ T cells is a multifactorial phenomenon, and that RANTES, MIP-1α, and MIP-1β do not account for the entire scope of CD8+ T-cell-derived HIV-suppressor factors.
Cells of the monocyte/macrophage (M/M) lineage are major targets of HIV (1, 2). Unlike CD4+ T cells, which are the other major target of HIV and which are depleted during the course of HIV disease, infection of cells of the M/M lineage does not necessarily lead to rapid cytolysis; rather, infection of these cells generally results in latency or persistent low-level chronic infection (3, 4). Different strains of HIV-1 vary markedly in their ability to infect cells belonging to the M/M lineage (5, 6). HIV-1 strains tend to lose macrophage tropism during laboratory adaptation or progression of clinical disease. Thus, the M/M system seems to play a substantial role in the pathogenesis of HIV disease, especially during primary infection and the clinically latent period; however, the mechanisms involved in establishment of viral latency or persistent infection in the M/M system are not well understood.
A number of cytokines, including tumor necrosis factor α, interleukin 1 (IL-1), IL-6, granulocyte/macrophage colony-stimulating factor (GM-CSF), and macrophage CSF, have been shown to upregulate HIV replication in the M/M system (reviewed in ref. 7). In contrast, interferons (IFNs) α and β and IL-13 (8) suppress, and IL-10 (9), IFN-γ (10), and transforming growth factor β (11) have dichotomous effects on HIV replication in the M/M system. Interestingly, these anti-HIV cytokines have little or no effect on HIV replication in primary lymphocytes, suggesting that regulation of HIV replication by cytokines is dependent on the cell type in question.
It was initially demonstrated by Walker et al. (12) and subsequently confirmed by others (13–15) that CD8+ T cells are capable of suppressing in vitro HIV replication in CD4+ T cells or peripheral blood mononuclear cells (PBMCs) in a noncytolytic, major histocompatibility complex nonrestricted manner (reviewed in ref. 16). This suppressive effect is mediated, at least in part, by a soluble factor(s) produced by CD8+ T cells (17). It is unclear whether a CD8+ T-cell-derived soluble factor(s) is also capable of suppressing HIV infection in cells belonging to the M/M lineage. Recently, Cocchi et al. (18) reported that the β-chemokines RANTES (regulated on activation, normal T-cell expressed and secreted), macrophage inflammatory protein 1α (MIP-1α), and MIP-1β, derived from CD8+ T cells, suppressed HIV replication in a CD4+ T-cell clone and in PBMCs. Several laboratories have recently identified CCR5, a receptor for RANTES, MIP-1α, and MIP-1β as a coreceptor for HIV-1 macrophage tropic strains, indicating that the β-chemokines inhibit HIV-1 infection by interfering with viral entry (19–23). However, their activity in cells of the M/M lineage is still in question.
In this study, we have examined the relative effects of crude supernatants from CD8+ T cells compared with purified RANTES, MIP-1α, MIP-1β, and a number of other cytokines on the regulation of HIV-1 Ba-L replication in acutely infected M/M and primary PBMCs as well as on the regulation of HIV expression in chronically infected promonocytic cell lines. Our results indicate that the HIV-suppressor effects of CD8+ T-cell supernatants are complex and multifactorial and that these effects cannot be accounted for exclusively by RANTES, MIP-1α, and MIP-1β.
MATERIALS AND METHODS
Isolation and Culture of Peripheral Monocytes and Lymphocytes.
PBMCs were obtained from HIV-negative, healthy donors by Ficoll/Hypaque centrifugation, and seeded on plastic tissue culture plates. After 3–4 hr incubation at 37°C in humidified 5% CO2/95% air atmosphere, nonadherent cells were removed by vigorous pipetting, and adherent cells were maintained in DMEM supplemented with 10% human male AB serum (Sigma) and GM-CSF (2 ng/ml; R & D Systems) for 10–14 days. The media, sera, and cytokines were determined to be endotoxin free. More than 98% of the adherent cells obtained by this procedure were identified as monocyte-derived macrophages (MDM) by their morphology and nonspecific esterase activity.
PBMCs from HIV-infected or uninfected individuals were depleted of monocytic cells and CD8+ T cells with immunomagnetic beads specific for CD14 and CD8 (Dynal, Lake Success, NY), respectively, following the plastic adherence procedure as described above. CD8+ T cells were positively selected with immunomagnetic beads specific for CD8 (Dynal). CD8- and monocyte-depleted PBMCs were stimulated in RPMI 1640 medium (BioWhittaker) containing 10% heat-inactivated fetal bovine serum (FBS; HyClone), phytohemagglutinin (3 μg/ml; Sigma), and IL-2 (10 units/ml; Boehringer Mannheim) for 3 days before infection with HIV.
Cell Lines.
The chronically HIV-infected U1 cells were described (24). Upregulation of HIV expression was induced by phorbol 12-myristate 13-acetate (PMA; 10−8 M; Sigma).
Establishment of Herpesvirus Saimiri-Transformed CD8+ T Cells.
CD8+ T cells were positively selected as described above from PBMCs derived from an HIV-1-infected, asymptomatic individual. Approximately 5 × 106 cells were infected with approximately 106 plaque-forming units of herpesvirus saimiri (HVS) strain 488–779 (kindly provided by R. C. Desrosiers, New England Regional Primate Center, Harvard Medical School, Southborough, MA), as described (25). HVS-transformed CD8+ T cells (HVS/HIV+/CD8+ T cells) were maintained in long-term culture (more than 6 months) in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and recombinant human IL-2 (2.5 units/ml; Boehringer Mannheim). The transformed cells were tested for HVS production by coculture with permissive owl monkey kidney cells, and for HIV infection by polymerase chain reaction using SK38/SK39 primers; neither HVS nor HIV was detected.
Preparation of CD8+T-Cell Culture Supernatants.
HVS/HIV+/CD8+ T cells were stimulated with IL-2 (10 units/ml) alone, whereas primary CD8+ T cells were stimulated with phytohemagglutinin and IL-2. Cell-free supernatants were collected from CD8+ T-cell cultures, passed through a 0.45-μm (pore size) filter, and kept at 4°C for a short period (<2 weeks) or at −70°C for longer periods.
Infection of Primary MDMs and Lymphocytes with HIV-1.
Approximately 1 × 105 primary MDMs were exposed to the macrophage-tropic Ba-L strain of HIV-1 (American Biotechnologies, Columbia, MD) or the primary isolate AU (derived in our laboratory and provided by M. Ostrowski) at a multiplicity of infection of approximately 0.01 at 37°C for 2 hr, washed twice, refed with DMEM supplemented with 10% HS and GM-CSF, and incubated at 37°C in 5% CO2 in humidified air. Approximately 2 × 105 CD8- and monocyte-depleted PBMCs were exposed to HIV-1 Ba-L or IIIB at a multiplicity of infection of approximately 0.01 at 37°C for 2 hr, washed twice, and refed with RPMI 1640 medium supplemented with 10% FBS and rIL-2 (10 units/ml). These cultures were maintained in the presence of cytokines as indicated or supernatants from CD8+ T-cell cultures. Where indicated, goat IgG, mouse IgG1, or antibodies to RANTES, MIP-1α, MIP-1β, IL-10, IL-13, IFN-α, or IFN-γ were added to the CD8+ T-cell supernatants.
Approximately one-half of the culture supernatants were replaced with the same medium containing the indicated cytokine or antibody every 3–4 days for several weeks after infection. Aliquots of cell-free supernatants were kept at −20°C for measurement of reverse transcriptase (RT) activity and/or p24 antigen.
RT Assays and p24 Antigen Assays.
RT assays were performed as described (26), and p24 antigen levels were determined by an enzyme-linked immunosorbent assay (Coulter).
Northern Blot Analysis.
Northern blot analysis was performed as described (27). The same membrane was hybridized with probes specific for the CCR5 (28), CXCR4 (29), or β-actin gene.
RESULTS
A Soluble Factor(s) Produced from CD8+T Cells, but Not Exogenous β-Chemokines RANTES, MIP-1α, and MIP-1β, Suppress Replication of HIV-1 Ba-L in M/M.
Anti-HIV activity of cell-free supernatants from CD8+ T cells was investigated in either M/M or PBMCs. As a source of CD8+ T-cell supernatants, we isolated and transformed CD8+ T cells from an HIV-infected, asymptomatic individual with HVS. HVS-transformed T cells are known to retain essential properties of conventionally cultured T cells, including cytokine production, and express only two viral proteins of unknown function (30). A 50% dilution of a supernatant derived from cultures of HVS/HIV+/CD8+ T cells was added every 4 days to MDMs that had been acutely infected with HIV-1 Ba-L, and RT activity was monitored. Viral replication was inhibited by more than 90% (Fig. 1A), and the suppression was dose-dependent (Fig. 1B). Cell viability as determined by trypan blue staining was comparable among cultures, making it highly unlikely that the CD8+ T-cell supernatants had suppressed HIV replication by a toxic effect on the MDMs (data not shown). Infection of MDMs with the primary isolate AU was also inhibited by the HVS/HIV+/CD8+ T-cell supernatants (data not shown).
Figure 1.
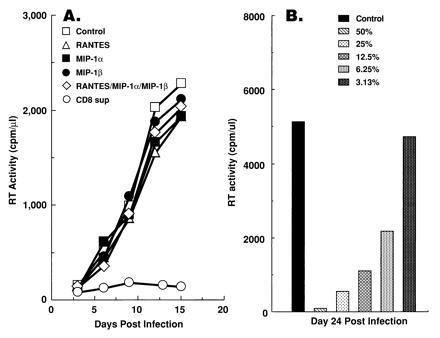
CD8+ T-cell supernatants, but not β-chemokines RANTES, MIP-1α, and MIP-1β, are capable of suppressing HIV-1 Ba-L replication in MDMs. (A) HVS/HIV+/CD8+ T-cell supernatants, but not RANTES, MIP-1α, and MIP-1β, are capable of suppressing HIV-1 Ba-L replication in MDMs. PBMCs were differentiated into macrophages for 14 days under the influence of GM-CSF, infected with HIV-1 Ba-L, and maintained in culture medium containing either 50% HVS/HIV+/CD8+ T-cell supernatants or the indicated chemokine (200 ng/ml; R & D Systems). Cell-free supernatants were collected every 4 days, replaced with the same medium, and RT activity was determined. (B) Anti-HIV activity of the supernatants is dose-dependent. Different concentrations (50%, 25%, 12.5%, 6.25%, and 3.13%) of HVS/HIV+/CD8+ T-cell supernatants were prepared and tested for their anti-HIV activity in MDMs. One-half of the culture medium was replaced with the same concentrates every 4 days. RT activity in the cell-free supernatants was determined on day 28 after infection
To determine whether the ability to inhibit HIV-1 replication in MDMs is restricted to CD8+ T cells or HVS-transformed CD8+ T cells derived from HIV-1 infected individuals, other sources of CD8+ T cells were also tested for their anti-HIV activity in the M/M system. Cell-free supernatants from primary CD8+ T cells, derived from HIV-negative as well as HIV-infected individuals, variably suppressed HIV replication in MDMs (data not shown). Thus, although the numbers of individuals tested was small, it appears that the ability of CD8+ T cells to produce an HIV-suppressor soluble factor(s) is not restricted to HVS-transformed cells or to cells derived from HIV-infected individuals. In contrast, β-chemokines RANTES, MIP-1α, and MIP-1β did not inhibit infection of MDMs with Ba-L (Fig. 1A). HIV-suppressor activity was not seen in this system even when as much as 1 μg/ml of the chemokines were used (data not shown).
To confirm that β-chemokines are not the major suppressors of HIV replication in the M/M system, neutralizing antibodies to RANTES, MIP-1α, and MIP- 1β were added to the CD8+ T-cell supernatants. The concentrations of RANTES, MIP-1α, and MIP-1β in the CD8+ T-cell supernatants were typically 1–5 ng/ml, 20–30 ng/ml, and 25–30 ng/ml, respectively, as determined by ELISA (R & D Systems) (data not shown). The selected antibody concentrations were at least 5 times the neutralization dose50, and were sufficient to completely block the β-chemokines present in the supernatants. As shown in Fig. 2A, the ability of the supernatants to inhibit Ba-L replication in the M/M system was not neutralized by adding these antibodies individually or in combination. In contrast, treatment of Ba-L-infected, CD8-depleted PBMC cultures to which CD8+ T-cell supernatants had been added with the combination of antibodies against RANTES, MIP-1α, and MIP-1β had a pronounced, but not complete, blocking effect on the HIV-suppressor activity of the supernatants (Fig. 2B). These results indicate that the β-chemokines, which are capable of suppressing HIV-1 Ba-L replication in CD4+ T cells, do not account for the anti-HIV activity of CD8+ T-cell supernatants in the M/M system.
Figure 2.
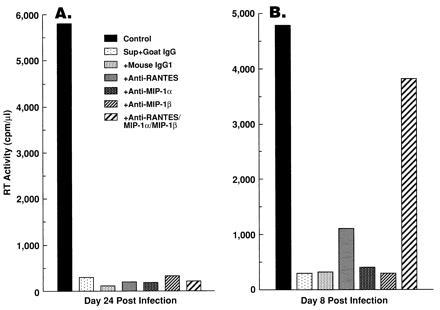
Antibodies to β-chemokines markedly neutralized the ability of CD8+ T-cell supernatants to suppress HIV-1 Ba-L replication in PBMCs, but not in MDMs. MDMs differentiated for 14 days with GM-CSF (A) or CD8- and monocyte-depleted PBMCs (B) were infected with HIV-1 Ba-L, and 50% HVS/HIV+/CD8+ T-cell supernatants were added to the culture. Where indicated, antibodies to β-chemokines [monoclonal anti-RANTES antibody (20 μg/ml; R & D Systems), polyclonal anti-MIP-1α antibody (50 μg/ml; R & D Systems), and monoclonal anti-MIP-1β antibody (20 μg/ml; R & D Systems)] were added individually or in combination to the supernatants. Either goat IgG (50 μg/ml; R & D Systems) or mouse IgG1 (20 μg/ml; R & D Systems) were added as controls.
CD8+ T-Cell-Derived Factor(s), but Not β-Chemokines, Suppress Replication of HIV-1 IIIB in PBMCs as Well as HIV Expression in U1 Cells.
The HVS/HIV+/CD8+ T-cell supernatants also inhibited replication of HIV-1 IIIB or Ba-L in CD8- and monocyte-depleted PBMCs (Fig. 3). In contrast, the chemokines inhibited replication of HIV-1 Ba-L but not IIIB in this system.
Figure 3.
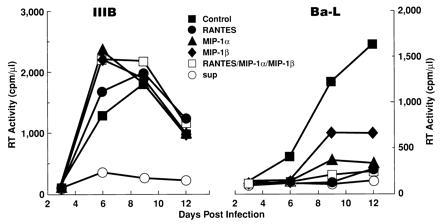
CD8+ T-cell supernatants are capable of suppressing HIV-1 IIIB or Ba-L in PBMCs, whereas β-chemokines are capable of suppressing HIV-1 Ba-L but not IIIB. PBMCs from HIV-negative individuals were depleted of CD8+ T cells and monocytes as described. Approximately one-half of the culture medium was replaced with medium containing either 50% HVS/HIV+/CD8+ T-cell supernatants or the indicated β-chemokine (200 ng/ml; R & D Systems) every 4 days after infection. RT activity was measured for the cell-free medium collected.
Anti-HIV activity of the supernatants was also tested in chronically HIV-infected U1 promonocytic cells. U1 cells harbor the integrated HIV provirus and exhibit persistent, low level viral expression that is inducible by various stimuli including PMA, tumor necrosis factor α, IL-1, or IL-6 (7). PMA-induced HIV expression was significantly reduced by the HVS/HIV+/CD8+ T-cell supernatants (Fig. 4), indicating that the CD8+ T-cell-derived factor(s) is capable of suppressing viral replication at a point in the virus life cycle after the integration event. However, the β-chemokines did not significantly suppress HIV expression in this system.
Figure 4.
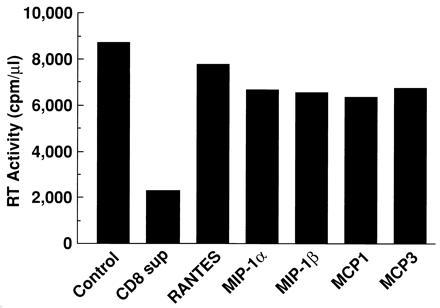
CD8+ T-cell supernatants are capable of suppressing HIV expression from chronically HIV-infected promonocytic U1 cells. U1 cells were stimulated with PMA (10−8 M), and maintained in culture medium containing the indicated β-chemokine (200 ng/ml; R & D Systems) or 50% HVS/HIV+/CD8+ T-cell supernatants. RT activity in cell-free supernatants was measured on day 7 after stimulation when HIV-1 expression had peaked.
Expression of a Chemokine Receptor(s) in MDMs.
Recently, a number of groups have reported that the β-chemokine receptor CCR5 is critical for viral entry during infection of T cells with macrophage-tropic strains of HIV-1 (19–23). The β-chemokines RANTES, MIP-1α, and MIP-1β competitively bind to this receptor, resulting in marked inhibition of viral entry into its target cell. Lack of susceptibility of MDMs to the anti-HIV activity of the β-chemokines could conceivably result from an absence of the appropriate β-chemokine receptor. To address this possibility, we investigated the expression of mRNA specific for the CCR5 gene in MDMs. CCR5 mRNA was detected in macrophages derived from two of three individuals by Northern blot analysis (Fig. 5); in the third individual in whom CCR5 mRNA was not detectable by Northern blot analysis (Fig. 5), expression was detectable when analyzed by RT-PCR (data not shown). Expression of mRNA specific for the CXCR4 or β-actin gene was comparable in the MDM RNA preparations among all three individuals tested (Fig. 5). Of note is the fact that the difference in level of expression of the CCR5 gene did not seem to markedly influence HIV infectivity or susceptibility to CD8+ T-cell supernatants or β-chemokines (data not shown). These results suggest that HIV-1 macrophage-tropic strains either use a coreceptor(s) other than CCR5 or do not require a coreceptor to enter the macrophage, thereby lacking susceptibility to the anti-HIV effects of the β-chemokines. The lack of susceptibility of MDMs to T-cell-tropic strains despite the fact that CXCR4, a coreceptor for T-cell-tropic strains in various cell types, is expressed in MDMs also suggests that the process of virus entry into macrophages may be quite different from that for other cell types.
Figure 5.
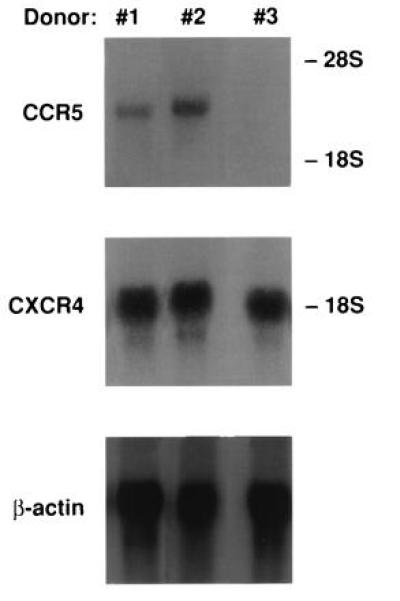
Expression of β-chemokine receptors in MDMs. Total RNA was extracted from 14-day culture of MDMs derived from three HIV-negative donors (nos. 1–3), and 15 μg of total RNA was subjected to Northern blot analysis. The same blot was hybridized with a probe specific to CCR5, CXCR4, or β-actin gene.
Neutralization of IL-10, IL-13, IFN-α, and IFN-γ Reduces, but Does Not Totally Abolish, Anti-HIV Activity of CD8+T-Cell Supernatants in M/M.
Several cytokines (IL-4, IL-10, IL-13, IFN-α, IFN-β, IFN-γ, and TGF-β) are known to inhibit HIV infection in the M/M system (reviewed in ref. 31), and CD8+ T cells are capable of producing some of these cytokines (IL-10, IL-13, IFN-α, and IFN-γ) (reviewed in ref. 31). When exogenously added to cultures of M/M, IL-10, IL-13, IFN-α, and IFN-γ variably suppressed HIV replication (Fig. 6A).
Figure 6.
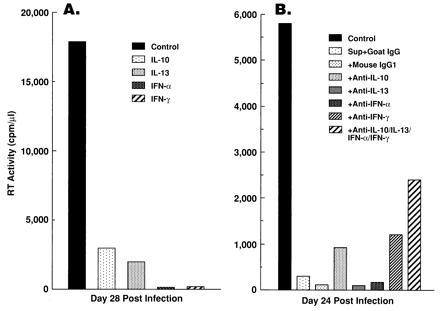
CD8+ T-cell-derived cytokines IL-10, IL-13, IFN-α, and IFN-γ may contribute to, but do not seem to fully account for, the anti-HIV activity of CD8+ T-cell supernatants. (A) Exogenously added cytokines IL-10, IL-13, IFN-α, and IFN-γ suppressed HIV replication in MDMs. Peripheral blood monocytes were differentiated into macrophages for 14 days in the presence of GM-CSF, infected with HIV-1 Ba-L, and maintained in the presence of the indicated cytokines [IL-10 (10 ng/ml; R & D Systems), IL-13 (10 ng/ml; R & D Systems), IFN-α (1000 units/ml; Endogen, Cambridge, MA), or IFN-γ (1000 units/ml; R & D Systems)]. RT activity in cell-free supernatants was determined on day 28 after infection. (B) Antibodies to IL-10, IL-13, IFN-α, or IFN-γ modestly, but never completely, neutralized the ability of the supernatants to suppress growth of HIV-1 Ba-L. MDMs were differentiated for 14 days with GM-CSF, infected with HIV-1 Ba-L, maintained in culture medium containing 50% HVS/HIV+/CD8+ T-cell supernatants, and RT activity in cell-free supernatants was monitored. Where indicated, antibodies to IL-10 (mouse monoclonal, 10 μg/ml; R & D Systems), IL-13 (mouse monoclonal, 10 μg/ml; R & D Systems), IFN-α (100 neutralizing units/ml; Endogen), or IFN-γ (mouse monoclonal, 10 μg/ml; R & D Systems) were added to the CD8+ T-cell supernatants. Either goat IgG (10 μg/ml; R & D Systems) or mouse IgG1 (10 μg/ml; R & D Systems) were added as controls.
To investigate whether the ability of CD8+ T-cell supernatants to suppress HIV replication in the M/M system is related to the HIV-suppressor capability of these cytokines, approximately 100 times the neutralization dose50 of antibodies to IL-10, IL-13, IFN-α, or IFN-γ were added individually or in combination to CD8+ T-cell supernatants. MDMs were infected with HIV-1 Ba-L and cultured in the presence of either untreated or antibody-treated supernatants. Neutralization of each of these cytokines individually had minimal, if any, effect on replication of Ba-L in MDMs. When all of these cytokines were neutralized together, HIV infectivity was modestly, but never completely, restored in some donors (Fig. 6B), whereas in others there was no effect (data not shown).
DISCUSSION
This study was undertaken to determine whether a crude CD8+ T-cell-derived soluble factor(s) is able to suppress HIV infection in cells of the M/M lineage and to delineate the relationship between the effects of this soluble factor(s) and the recently described HIV-suppressor effects of the β-chemokines RANTES, MIP-1α, and MIP-1β (18). Our results clearly indicate that HIV infection in the M/M system is markedly suppressed by a CD8+ T-cell-derived soluble factor(s) other than RANTES, MIP-1α, and MIP-1β. Several lines of evidence support our conclusion. CD8+ T-cell culture supernatants suppressed HIV-1 Ba-L replication in MDMs in a dose-dependent manner. Reduced virus replication was not due to cytotoxic effects of the supernatants, since cell viability was comparable in the presence or absence of the supernatants. This anti-HIV activity was found in the supernatants of CD8+ T cells derived from both HIV-infected and uninfected individuals. These results indicate that a CD8+ T-cell-derived soluble factor(s) is capable of suppressing HIV replication in the M/M system.
RANTES, MIP-1α, and MIP-1β, which suppressed HIV-1 Ba-L replication in PBMCs, did not inhibit infectivity of HIV-1 Ba-L in the M/M system; as much as 1 μg/ml of these chemokines had no inhibitory effect on HIV-1 Ba-L replication in MDMs. Furthermore, treatment of the CD8+ T-cell supernatants with neutralizing antibodies to RANTES, MIP-1α, and MIP-1β did not remove the HIV-suppressor effects of the supernatants. Thus, CD8+ T cells produce a soluble factor(s) other than RANTES, MIP-1α, and MIP-1β, which suppress HIV infection in the M/M system. Recently, Schmidtmayerova et al. also observed the inability of β-chemokines to inhibit replication of primary HIV-1 isolates in MDMs (32).
Finally, CD8+ T-cell supernatants suppressed HIV-1 IIIB infection in PBMCs, as well as HIV expression from chronically infected U1 cells, whereas HIV-1 infection was not suppressed in either of those systems by RANTES, MIP-1α, and MIP-1β. Taken together, these results indicate that HIV-suppressor activity of CD8+ T-cell supernatants is a multifactorial phenomenon, and that a CD8+ T-cell-derived factor(s) other than RANTES, MIP-1α, and MIP-1β has potent suppressor effects on replication of various HIV strains in different target cells. We are currently investigating the mechanisms whereby the CD8+ T-cell supernatants block HIV-1 infection in cells of the M/M lineage.
Recently, CCR5, a receptor for the β-chemokines RANTES, MIP-1α, and MIP-1β, was identified as a coreceptor for HIV-1 macrophage-tropic strains (19–23), indicating that the β-chemokines inhibit HIV-1 infection by interfering with virus entry. We demonstrated that MDMs express CCR5 mRNA, making it unlikely that lack of susceptibility of MDMs to the HIV-suppressor activity of the β-chemokines is due to an absence of CCR5. We also showed that MDMs express CXCR4, a coreceptor for HIV-1 T-cell-tropic strains. Thus, lack of susceptibility of MDMs to T-cell-tropic strains cannot be explained by an absence of the CXCR4 coreceptor. These data suggest that the process of virus entry into macrophages may be quite different from that for T cells.
It has been shown that a CD8+ T-cell-derived soluble factor(s) is capable of suppressing HIV replication in PBMCs or CD4+ T-cell lines (reviewed in ref. 16). This study indicates that CD8+ T-cell supernatants are also effective suppressors of HIV replication in the M/M system. There are certain critical differences in the anti-HIV activity of CD8+ T-cell supernatants when compared with the β-chemokines RANTES, MIP-1α, and MIP-1β. The β-chemokines cannot suppress certain HIV strains (e.g., IIIB) and are apparently specific for macrophage-tropic strains of HIV-1, whereas CD8+ T-cell supernatants can suppress replication of all HIV strains tested (i.e., IIIB, Ba-L, primary isolates). Furthermore, while the anti-HIV activity of the β-chemokines is effective in PBMCs but not in the M/M system, CD8+ T-cell supernatants can suppress HIV replication in both PBMC and M/M systems. Finally, CD8+ T-cell supernatants can, but the β-chemokines cannot, inhibit HIV expression from chronically infected U1 cells. Thus, the anti-HIV activity of the β-chemokines is viral strain-specific as well as cell type-specific, and the major site of action of these factors is prior to the integration event. In contrast, the anti-HIV activity of the CD8+ T-cell supernatants is broader, and is able to inhibit HIV replication following the integration event, as indicated by the fact that the supernatants suppress HIV expression in the U1 cell system, where each cell is already infected and HIV provirus is integrated into host cell genome.
We have further demonstrated that cytokines such as IL-10, IL-13, and the IFNs, which are well-established inhibitors of HIV infection in the M/M system (reviewed in ref. 31), contribute to, but did not seem to fully account for, the anti-HIV activity of CD8+ T-cell supernatants. Here again, the data strongly indicate that CD8+ T-cell supernatants contain as yet unidentified factors that are capable of suppressing HIV replication.
Thus, it appears that the HIV-suppressor activity of CD8+ T-cell supernatants is multifactorial and that various factors within these supernatants including, but not limited to the β-chemokines, may affect HIV replication at various stages of the life cycle of the virus. Further delineation of the spectrum of endogenous factors that are involved in the regulation of HIV replication will be critical for a more comprehensive understanding of the pathogenic mechanisms of HIV disease as well as for the design of strategies for the potential therapeutic manipulation of these factors.
Acknowledgments
We thank R. C. Desrosiers, M. V. Pavlis, D. Goletti, and M. Ostrowski for providing materials; W. Turner for helpful discussion; P. Walsh for editorial assistance; and J. Weddle for graphic work. H.M. and M.M. contributed equally to this project, which M.M. performed for the partial fulfillment of the requirements of the Ph.D. program of the Department of Microbiology at Howard University (Washington, DC).
Footnotes
Abbreviations: M/M, monocyte/macrophages; MDM, monocyte-derived macrophages; PBMC, peripheral blood mononuclear cells; RANTES, regulated on activation normal T-cell expressed and secreted; MIP, macrophage inflammatory protein; IL, interleukin; IFN, interferon; GM-CSF, granulocyte/macrophage colony-stimulating factor; HVS, herpesvirus saimiri; RT, reverse transcriptase.
References
- 1.Gartner S, Markovits P, Markovits D M, Kaplan M H, Gallo R C, Popovic M. Science. 1986;233:215–219. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 2.Koenig S, Gendelman H E, Orenstein J M, Dal Canto M C, Pezeshpour G H, Yungbluth M, Janotta F, Aksamit A, Martin M A, Fauci A S. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 3.Gendelman H E, Orenstein J M, Martin M A, Ferrua C, Mitra R, Phipps T, Wahl L M, Lane H C, Fauci A S, Burke D S, Skillman D, Meltzer M S. J Exp Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orenstein J M, Meltzer M S, Phipps T, Gendelman H E. J Virol. 1988;62:2578–2586. doi: 10.1128/jvi.62.8.2578-2586.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Brien W A, Koyanagi Y, Namazie A, Zhao J-Q, Diagne A, Idler K, Zack J A, Chen I S Y. Nature (London) 1990;348:69–73. doi: 10.1038/348069a0. [DOI] [PubMed] [Google Scholar]
- 6.Shioda T, Levy J A, Cheng-Mayer C. Nature (London) 1991;349:167–169. doi: 10.1038/349167a0. [DOI] [PubMed] [Google Scholar]
- 7.Poli G, Fauci A S. Pathobiology. 1992;60:246–251. doi: 10.1159/000163729. [DOI] [PubMed] [Google Scholar]
- 8.Montaner L J, Doyle A G, Collin M, Herbein G, Illei P, James W, Minty A, Caput D, Ferrara P, Gordon S. J Exp Med. 1993;178:743–747. doi: 10.1084/jem.178.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weissman D, Poli G, Fauci A S. AIDS Res Hum Retroviruses. 1994;10:1199–1206. doi: 10.1089/aid.1994.10.1199. [DOI] [PubMed] [Google Scholar]
- 10.Biswas P, Poli G, Kinter A L, Justement J S, Stanley S K, Maury W J, Bressler P, Orenstein J M, Fauci A S. J Exp Med. 1992;176:739–750. doi: 10.1084/jem.176.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poli G, Kinter A L, Justement J S, Bressler P, Kehrl J H, Fauci A S. J Exp Med. 1991;173:589–597. doi: 10.1084/jem.173.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker C M, Moody D J, Stites D P, Levy J A. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 13.Brinchmann J E, Gaudernack G, Vartdal F. J Immunol. 1990;144:2961–2966. [PubMed] [Google Scholar]
- 14.Kannagi M, Masuda T, Hattori T, Kanoh T, Nasu K, Yamamoto N, Harada S. J Virol. 1990;64:3399–3406. doi: 10.1128/jvi.64.7.3399-3406.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsubota H, Lord C I, Watkins D I, Morimoto C, Letvin N L. J Exp Med. 1989;169:1421–1434. doi: 10.1084/jem.169.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy J A, Mackewics C E, Baker E. Immunol Today. 1996;17:217–222. doi: 10.1016/0167-5699(96)10011-6. [DOI] [PubMed] [Google Scholar]
- 17.Walker C M, Levy J A. Immunology. 1989;66:628–630. [PMC free article] [PubMed] [Google Scholar]
- 18.Cocchi F, DeVico A L, Garzino-Demo A, Arya S K, Gallo R C, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 19.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, DiMarzio P, Marmon S, Sutton R E, Hill C M, Davis C B, Peiper S C, Schall T J, Littman D R, Landau N R. Nature (London) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 20.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 21.Alkhatib G, Combadiere C, Broder C C, Keng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 22.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath P D, Wu L, Mackay C R, LaRose G, Newman W, Gerard N, Gerard C, Sodroski J. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 23.Doranz B J, Rucker J, Yi Y, Smyth R J, Samson M, Peiper S C, Parmentier M, Collman R G, Doms R W. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 24.Folks T M, Justement J, Kinter A L, Dinarello C A, Fauci A S. Science. 1987;238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- 25.Fleckenstein H, Fleckenstein B. Methods Mol Genet. 1994;4:345–362. [Google Scholar]
- 26.Poli G, Kinter A L, Fauci A S. Proc Natl Acad Sci USA. 1994;91:108–112. doi: 10.1073/pnas.91.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moriuchi H, Moriuchi M, Smith H A, Straus S E, Cohen J I. J Virol. 1992;66:7303–7308. doi: 10.1128/jvi.66.12.7303-7308.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Combadiere C, Ahuja S K, Tiffany H L, Murphy P M. J Leukocyte Biol. 1996;60:147–152. doi: 10.1002/jlb.60.1.147. [DOI] [PubMed] [Google Scholar]
- 29.Feng Y, Broder C C, Kennedy P E, Berger E A. Science. 1996;272:872–877. [Google Scholar]
- 30.Meinl E, Hohlfeld R, Wekerle H, Fleckenstein B. Immunol Today. 1995;16:55–58. doi: 10.1016/0167-5699(95)80087-5. [DOI] [PubMed] [Google Scholar]
- 31.Poli G, Fauci A S. In: Human Cytokines: Their Role in Disease and Therapy. Aggarwal B B, Puri R, editors. Oxford: Blackwell Scientific; 1995. pp. 421–449. [Google Scholar]
- 32.Schmidtmayerova H, Sherry B, Bukrinsky M. Nature (London) 1996;382:767. doi: 10.1038/382767a0. [DOI] [PubMed] [Google Scholar]