

Abstract
Type 1 von Willebrand disease (VWD), characterized by reduced levels of plasma von Willebrand factor (VWF), is the most common inherited bleeding disorder in humans. Penetrance of VWD is incomplete, and expression of the bleeding phenotype is highly variable. In addition, plasma VWF levels vary widely among normal individuals. To identify genes that influence VWF level, we analyzed a genetic cross between RIIIS/J and CASA/Rk, two strains of mice that exhibit a 20-fold difference in plasma VWF level. DNA samples from F2 progeny demonstrating either extremely high or extremely low plasma VWF levels were pooled and genotyped for 41 markers spanning the autosomal genome. A novel locus accounting for 63% of the total variance in VWF level was mapped to distal mouse chromosome 11, which is distinct from the murine Vwf locus on chromosome 6. We designated this locus Mvwf for “modifier of VWF.” Additional genotyping of as many as 2407 meioses established a high resolution genetic map with gene order Cola1-Itg3a-Ngfr-Mvwf/Gip-Hoxb9-Hoxb1-Cbx·rs2-Cox5a-Gfap. The Mvwf candidate interval between Ngfr and Hoxb9 is ≈0.5 centimorgan (cM). These results demonstrate that a single dominant gene accounts for the low VWF phenotype of RIIIS/J mice in crosses with several other strains. The pattern of inheritance suggests a gain-of-function mutation in a unique component of VWF biosynthesis or processing. Characterization of the human homologue for Mvwf may have relevance for a subset of type 1 VWD cases and may define an important genetic factor modifying penetrance and expression of mutations at the VWF locus.
Von Willebrand factor (VWF) is a multimeric plasma glycoprotein that plays a central role in hemostasis. VWF mediates the adhesion of an initial platelet plug to the subendothelium of injured blood vessels and serves as the carrier for factor VIII in plasma (1). VWF is synthesized in megakaryocytes and endothelial cells as a 309-kDa precursor that initially dimerizes and then multimerizes coincident with cleavage to a mature form. Posttranslational modifications to VWF also include the addition of 12 N-linked and 10 O-linked carbohydrate side chains, some of which are sulfated. Finally, VWF is either secreted or stored in the Weibel–Palade bodies of endothelial cells or in the α-granules of platelets.
Abnormalities of VWF result in von Willebrand disease (VWD), a common inherited bleeding disorder with an estimated prevalence of ≈1% in some populations (2, 3). The most common type of VWD, type 1, is characterized by mild to moderate clinical bleeding, a prolonged bleeding time, and a quantitative reduction in plasma VWF to ≈20–50% of normal. Type 1 VWD is inherited in an autosomal dominant pattern with reduced penetrance and variable expression. VWD diagnosis is difficult because of the low accuracy of laboratory tests (4) and the wide variability in VWF levels contributed by factors including blood group antigens (5, 6), estrogen (7–9), thyroid hormone levels (10, 11), age (9, 12), and stress (13–16). Although type 1 VWD mutations have been identified in a subset of patients, the molecular defects responsible for most cases remain unknown.
In 1990, Sweeney et al. (17) reported a mouse model for type 1 VWD, the inbred mouse strain RIIIS/J. RIIIS/J mice have reduced plasma VWF antigen and factor VIII levels, a prolonged bleeding time, and normal VWF multimers, similar to human type 1 VWD. Genetic crosses of RIIIS/J with C57BL/6J demonstrated autosomal dominant inheritance of the low VWF phenotype. Identification of the RIIIS/J molecular defect may provide insights into the regulation of VWF biosynthesis and processing and might define the genetic basis for a subset of human type 1 VWD cases.
We previously examined the inheritance of VWF antigen levels in a cross between RIIIS/J mice and PWK, a strain of Mus musculus musculus (18). The distribution of VWF levels among (RIIIS/J × PWK)F1 × PWK backcross progeny resembled a bimodal distribution, suggesting that a single dominant gene accounts for most of the RIIIS/J low VWF phenotype. No correlation between low VWF antigen levels and inheritance of the RIIIS/J Vwf gene was observed, demonstrating that the RIIIS/J low VWF phenotype is caused by a defect in a gene(s) distinct from the Vwf locus. We now report the localization of the major gene modifying VWF levels in the RIIIS/J mouse, designated Mvwf for “modifier of von Willebrand factor,” to a 0.5-centimorgan (cM) portion of distal mouse chromosome 11.
MATERIALS AND METHODS
Animals.
Inbred strains of RIIIS/J and CASA/Rk (a strain of Mus musculus castaneus) mice were obtained from The Jackson Laboratory. Mice were housed in microisolator cages under specific pathogen-free conditions and bred at the University of Michigan. Reciprocal matings generated F1 progeny. To confirm Mvwf phenotype classification, selected F2 progeny were progeny-tested by mating to CASA/Rk mice or to mice homozygous for the CASA/Rk allele across the Mvwf interval. The (RIIIS/J × PWK)F1 × PWK backcross was described previously (18).
Genotype Analysis.
Genomic DNA was prepared from liver or tail samples as described (19). To create the DNA pools, 2 μg of five DNA samples was combined at a final concentration of 50 ng/μl, and 200 ng was used in PCRs. PCR amplification of simple sequence repeats and gel electrophoresis were performed either as described (20) or with unlabeled primers followed by electrophoresis on 3% Metaphor agarose gels (FMC). The initial 41 markers for the genome scan were selected based on data reported by Taylor et al. (21). Additional markers were selected from the Massachusetts Institute of Technology (22, 23) (MIT) and European Collaborative Interspecific Mouse Backcross maps (http://www.hgmp.mrc.ac.uk/MBx/MBxHomepage.html), supplemented by additional markers in: Whitehead Institute/MIT Center for Genome Research, Genetic Map of the Mouse, database release 10, April 28, 1995. All MIT primer pairs were purchased from Research Genetics (Huntsville, AL).
Calculation of Genetic Distances.
Map distances were calculated assuming that genetic distance is equivalent to recombination fraction. Maximum likelihood estimates for detection of two independent (maternal and paternal) crossovers were not used because the greatest distance between two genotyped markers was <4 cM and because the corresponding frequency of such events would be small. Because the RIIIS/J allele at Mvwf is dominant and RIIIS/J homozygotes at Mvwf have a phenotype similar to heterozygotes, only half of the potential recombinants are phenotypically informative. Mice that are homozygous for the CASA/Rk allele at one flanking marker and heterozygous at the other flanking marker are informative whereas mice that are homozygous for the RIIIS/J allele at one flanking marker and heterozygous at the other flanking marker are not informative. Recombination fractions were calculated only for the informative class of mice. The number of informative meioses is slightly greater than the number of F2 progeny typed because 2 of 1047 F2 progeny carried two recombination events in a 7-cM interval. For these two mice, both meioses are informative. SDs of genetic distances also were calculated based on the number of informative recombinants and meioses. Standard analysis of variance calculations were performed using spss for Windows, 6.1.
Phenotype Analysis.
The RIIIS/J low VWF phenotype was defined by the presence of reduced plasma VWF antigen. For the initial 106 (RIIIS/J × CASA/Rk)F2 progeny, collection of blood by cardiac puncture and measurement of VWF levels were performed as described for the (RIIIS/J × PWK)F1 × PWK backcross progeny (18). For additional intercross progeny, blood samples were collected by orbital plexus puncture at 4–8 weeks of age. One to three samples of ≈100 μl of blood were collected into heparinized Natelson blood-collecting tubes (Fisher) for each mouse. When multiple samples were collected from the same mouse, collections were 1-week apart with blood drawn from the opposite eye each week. Plasma VWF antigen was measured by ELISA using antibodies against human VWF as described (18) except that eight dilutions of plasma were measured for each mouse and detection of VWF was performed with a 1:2000 dilution of peroxidase-tagged antibody. A standard curve for each plate was constructed by log–log transformation of the OD readings for pooled plasma from C57BL/6J mice. Individual VWF levels were the average of six values (duplicate samples at each of three dilutions) corresponding to the linear portion of the standard curve.
PCR and Restriction Analysis for Polymorphism Detection.
Most primers were designed based on mouse, rat, or human sequences available through the references listed in Table 1. One of the primers for Ngfr was based on RIIIS/J mouse sequence as determined by sequencing of PCR product (∗ in Table 1); 40 ng of genomic DNA from RIIIS/J, CASA/Rk, and F1 mice was amplified by PCR using the primers listed in Table 1. PCR products were digested with a series of restriction enzymes and were separated by electrophoresis on 3% Metaphor agarose gels (FMC) to detect polymorphisms. Selected reactions were performed using the Taq-Start antibody (CLONTECH).
Table 1.
Primers and restriction enzymes for polymorphisms
| Gene | Primer sequences (5′-3′) | Product size, kb | Restriction enzyme | GenBank name (ref.) |
|---|---|---|---|---|
| Cola1 | AGCCCGGTAGTAGCGGCCAC | 2.0 | AluI | HUMCG1PA1 |
| GCAAGTGGTGAACGTGGTCC | ||||
| Itg3a | GGGGCTCATCATCCTCCTCTTGTG | 0.4 | none | HUMINTA3A (24) |
| CTTCATCTCCGCCTTCTGCCTCTT | S66292 (25) | |||
| Ngfr | GTTCTCCTGCCAGGACAAACAGAACAC | 0.2 | RsaI | RNNGFRR (26) |
| GCATTCAGCATCAGCCCAGGGCGTGCA | ||||
| Ngfr | AGCCGCCCTGCGACGCATCCA | 1.4 | HinfI | RNNGFRR (26) |
| GCCATGAAACTGAGGAGAAAGTTG | ∗ | |||
| Gip | AGAGAGAGGCCCGGGCTTTGGA | 0.9 | HaeIII | MMU34295 |
| CCAGGCCAGTAGCTCTTGAATCAG | HUMGIPX5 (27) | |||
| Hoxb9 | GCATGAAGTGGCCAGACTCCTCAAT | 1.2 | AluI | MUSHOX25A (28) |
| CAATGGGAGACTTGTCTTTGCTGGT | RsaI | |||
| Hoxb1 | CTGGATGAAGGTCAAGAGAAACCCA | 1.3 | DdeI | MMHOX29 (29) |
| TATGTCACAAGGCAGTCGGTGCTAT | HphI | |||
| Cox5a | CAGGCAACTATTTCAATCAAGTAGC | 0.5 | DdeI | MMCOXVA (30) |
| GTTCCATTCGGTGCTATTCTCATG | ||||
| Itgb3 | TCCAGCTCATTGTTGATGCTTATGG | 1.1 | RsaI | HUMGP3A (31) |
| GGTCACGCACTTCCAGCTCGACTTT |
Restriction enzymes listed detect polymorphic sites between RIIIS/J and CASA/Rk mice. When two enzymes are listed, both were polymorphic between strains. Product sizes are approximate. Itg3a is a length polymorphism; the RIIIS/J product is ≈50 bp larger than the CASA/Rk product. When two GenBank references are listed, both were used to design primers. ∗, Based on direct sequence analysis of murine PCR product (see text). ref., reference number.
RESULTS
A Major Gene Influences Murine VWF Levels.
To study the molecular basis for reduced plasma VWF levels in the RIIIS/J mouse, we established an intercross between RIIIS/J and CASA/Rk mice. Mean plasma VWF levels in the parental strains and F1 progeny are shown in Table 2. The RIIIS/J and F1 mice both have markedly reduced VWF antigen levels of 5% and 13%, respectively. Although low VWF level is clearly dominant, as observed previously in crosses of RIIIS/J with C57BL/6J or PWK (17, 18), the average level observed here in F1 mice is significantly higher than that observed in the RIIIS/J mice, as determined by Student’s t test (P < 0.0005). This difference suggests the presence of a dosage effect at a single major locus or contributions from additional modifying loci.
Table 2.
Comparison of plasma VWF levels of parental strains and 205 F2 progeny grouped by genotype
| n | VWF level, % ± SD | |
|---|---|---|
| Parental strains | ||
| RIIIS/J | 14 | 5 ± 1 |
| (RIIIS/J × CASA/Rk)F1 | 30 | 13 ± 5 |
| CASA/Rk | 24 | 100 ± 33 |
| F2 progeny | ||
| Homozygous RIIIS/J at D11Mit54 | 52 | 11 ± 7 |
| Heterozygous at D11Mit54 | 98 | 14 ± 7 |
| Homozygous CASA/Rk at D11Mit54 | 55 | 49 ± 21 |
VWF levels of mice were determined by ELISA. The average VWF level of parental CASA/Rk mice is arbitrarily defined as 100%. n, number of mice. F2 progeny are grouped by genotype at D11Mit54 near the Mvwf locus.
The distribution of VWF levels among 106 (RIIIS/J × CASA/Rk)F2 progeny supports the hypothesis that a major dominant gene causes the RIIIS/J low VWF phenotype (Fig. 1A). Approximately three-quarters of the progeny have VWF levels in the lower tail of the distribution, and about one-quarter are present in the upper tail. Vwf genotype analysis of selected mice demonstrated no correlation between low VWF level and inheritance of an RIIIS/J allele at Vwf (data not shown), which is consistent with our previous analysis of an independent cross between RIIIS/J and PWK (18).
Figure 1.
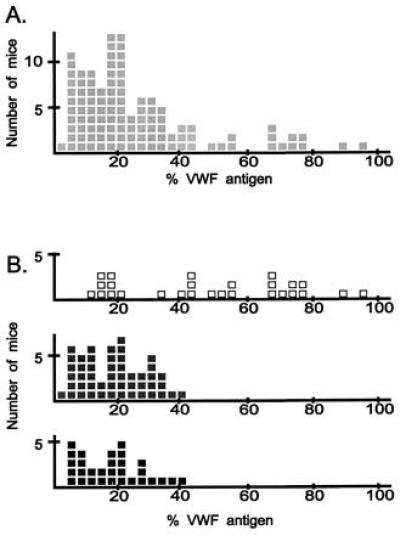
VWF levels in 106 (RIIIS/J × CASA/Rk)F2 progeny. VWF levels were determined by ELISA; 100% VWF is defined as the mean VWF level of CASA/Rk parental mice. Each square represents a single mouse. (A) All progeny. (B) Progeny genotyped at D11Nds7. Open squares, homozygous for the CASA/Rk allele; gray squares, heterozygotes; black squares, homozygous for the RIIIS/J allele.
Mapping of Mvwf.
We next sought to localize Mvwf through a genotype analysis of F2 progeny at markers across the genome. We initially used the approach described by Taylor et al. (21) in which DNA samples from individual mice are pooled for analysis, reducing the number of genotypes required for the scan. DNA samples were combined from the 5% of progeny with the highest and lowest VWF levels from a distribution of 106 mice to create the high and low VWF pools, respectively. The average percentage of VWF for mice in the high VWF pool was 84 ± 10% (mean ± SD) and in the low VWF pool was 4 ± 1%. In this assay, a linked marker would be observed as an excess of CASA/Rk alleles in the high VWF pool genotype and an excess of RIIIS/J alleles in the low VWF pool genotype.
For the initial genome scan, the DNA pools were typed for 41 simple sequence repeat markers. Excess CASA/Rk alleles were observed in the high VWF pool at four loci: D11Nds7, D11Mit5, D4Mit7, and D13Mit36. For marker D11Nds7 (Fig. 2 Right), the high VWF pool (lane 4) contains only the CASA/Rk allele whereas the low pool (lane 5) contains an excess of RIIIS/J alleles. Thus, D11Nds7 appears to map near the Mvwf locus. Less significantly skewed pool genotypes were observed for marker D11Mit5 (Fig. 2 Left), which is located ≈30 cM proximal to D11Nds7 (32), as well as for markers D4Mit7 and D13Mit36 (data not shown). For the latter two markers, individual genotyping of animals in the upper and lower quartiles of the phenotypic distribution did not confirm the apparent association of marker genotype with low VWF levels. In addition, the chromosome 6 marker closest to the Vwf gene (33) did not show skewing, confirming our published observation that the RIIIS/J low VWF level is not linked to the Vwf gene (18).
Figure 2.
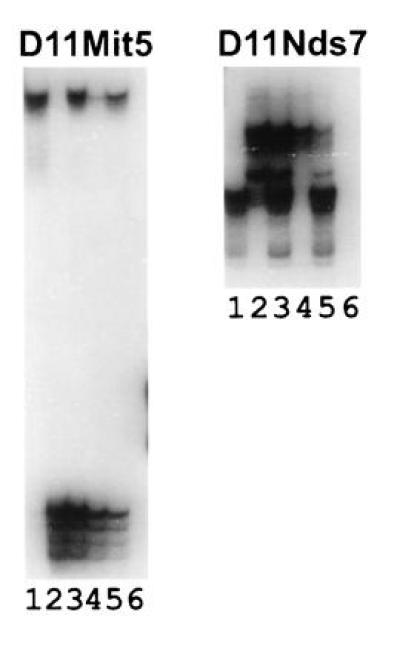
Pooled DNA samples from the extremes of a phenotypic distribution of (RIIIS/J × CASA/Rk)F2 progeny genotyped at D11Mit5 and D11Nds7. Lanes: 1, RIIIS/J; 2, CASA/Rk; 3, 1:1 mix of parental DNAs; 4, high VWF pool; 5, low VWF pool; 6, zero DNA control.
For the marker showing the strongest initial evidence for linkage, D11Nds7, individual genotypes of 106 F2 progeny are shown in Fig. 1B. Most of the mice with the highest VWF levels inherited two CASA/Rk alleles (open squares) whereas the animals with the lowest VWF levels inherited at least one RIIIS/J allele (gray and black squares), confirming linkage of Mvwf to this marker on chromosome 11. This linkage was also confirmed by genotyping 110 (RIIIS/J × PWK)F1 × PWK backcross progeny, as shown in Fig. 3. The single outlier mouse with the highest VWF level among the backcross progeny was heterozygous at markers spanning 30 cM of chromosome 11, suggesting that a factor other than the Mvwf genotype accounts for the high VWF level in this mouse.
Figure 3.
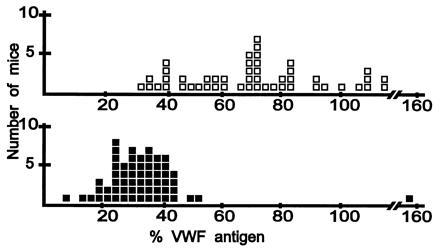
VWF levels in (RIIIS/J × PWK)F1 × PWK progeny genotyped at D11Nds7. VWF levels were determined by ELISA; 100% VWF is defined as the mean VWF level of PWK parental mice. Each square represents a single mouse. Open squares, homozygous for the PWK allele; black squares, heterozygous for the RIIIS/J and PWK alleles.
At this point in the analysis, it was particularly important to precisely define the low VWF phenotype. A large overlap of VWF levels was observed between the D11Nds7 genotypic classes (Fig. 1B). Among mice with low VWF levels, no clear boundary separates mice inheriting RIIIS/J allele(s) from mice inheriting only CASA/Rk alleles. Although some of the latter mice could represent recombinants between D11Nds7 and Mvwf, a VWF level cutoff excluding animals with intermediate phenotypes would include only 10% of the progeny. High resolution mapping using only mice at the extreme ends of the distribution would require significantly larger numbers of mice. Because VWF levels in humans are known to be influenced by several environmental factors, we altered the plasma sample collection from subsequent intercross progeny to limit the potential contribution of additional variables. Samples were collected by orbital plexus puncture to allow multiple samples to be collected from individual mice. Blood was drawn weekly, only when the mice were between 4 and 8 weeks of age and at approximately the same time each day.
To determine the range of VWF levels that could be considered high, genotypes and VWF levels were determined for 205 additional F2 mice. For each mouse, VWF level was determined at least once. For 55 mice homozygous CASA/Rk at marker D11Mit54, which is closer to Mvwf than D11Nds7, three plasma samples were collected, and VWF levels were averaged. The distribution of VWF levels among the 205 animals, determined with this revised approach, showed a clearer separation between the genotypic classes, as shown in Fig. 4. Based on this distribution, VWF levels above 39% were defined as high, and VWF levels below 19% were defined as low. Initially, animals with VWF levels between 19 and 39% were not used to determine Mvwf location. As the candidate interval narrowed, recombinant mice with intermediate VWF levels were progeny-tested to assign phenotype. Among test progeny, phenotype was assigned to the tested mouse by the presence of high or low VWF levels in two or more progeny inheriting the recombinant chromosome. One to four litters of test progeny were required to assign phenotype.
Figure 4.
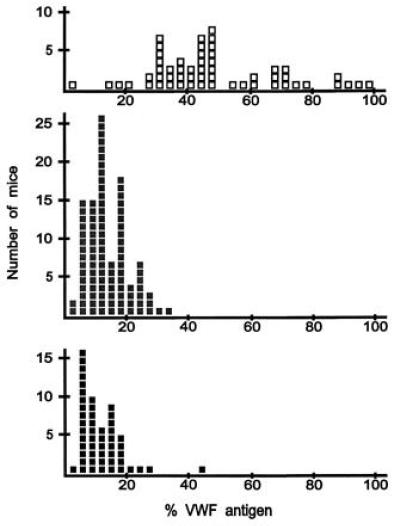
VWF levels in 205 (RIIIS/J × CASA/Rk)F2 progeny genotyped at D11Mit54. VWF levels were determined by ELISA; 100% VWF is defined as the mean VWF level of CASA/Rk parental mice. Each square represents a single mouse. Open squares, homozygous for the CASA/Rk allele; gray squares, heterozygotes; black squares, homozygous for the RIIIS/J allele.
Using this definition of low VWF phenotype, we continued to narrow the Mvwf candidate interval by analyzing 21 additional chromosome 11 markers on an expanded cross of F2 progeny. Phenotypes were only determined for mice inheriting an informative recombination event. Recombination events were only realistically informative when the nonrecombinant allele was CASA/Rk because only mice inheriting this type of recombinant would be expected to have significantly different VWF levels across the interval. At this stage, 1047 F2 mice were genotyped at three markers spanning an ≈7-cM portion of chromosome 11. By genotyping simple sequence repeat markers, the possible Mvwf location was limited to the region between markers D11Mit263 and D11Mit54 (Fig. 5).
Figure 5.
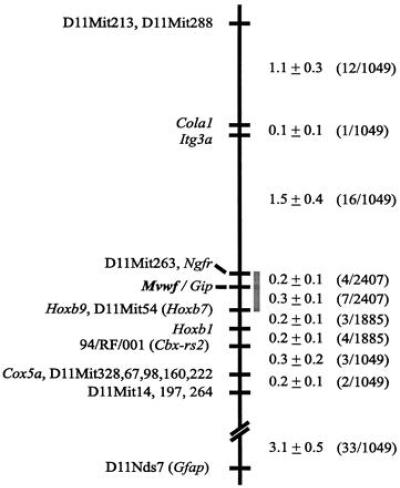
Genetic map of a portion of distal mouse chromosome 11. Markers are indicated to the left and recombination fraction (% ± SD) to the right. At the far right is the fraction of recombinants among informative meioses (see Materials and Methods) in each interval. The gray bar indicates the Mvwf candidate interval.
Exclusion of Candidate Genes.
Several known genes in this region were potential candidates for Mvwf. Of particular interest, Itgb3 is the β chain of the glycoprotein IIb/IIIa platelet surface integrin, for which VWF is an important ligand (34). In addition, Cola1, the α-1 chain of type 1 collagen, is a potential ligand for VWF in the vessel wall (35). Both of these genes were placed on our map by analysis of informative mice. For this purpose, primers were designed based on available sequence to amplify across introns or untranslated regions for each candidate gene. PCR products were amplified from genomic DNA of the parental strains and digested with a series of restriction enzymes to identify informative polymorphisms (Table 1). Selected recombinant mice were then genotyped for these polymorphisms to determine the map location of each gene. Based on map position, Itgb3, Cola1, and Itg3a (integrin α-3) were excluded as potential candidate genes. The positions of Cola1 and Itg3a are shown in Fig. 5. Itgb3 was confirmed to map distal to D11Nds7 (32).
Polymorphisms were then identified in other genes known to map in the region of mouse chromosome 11 or in the syntenic region in humans 17q21-q22. Map positions were determined for nerve growth factor receptor (Ngfr, two polymorphisms), cytochrome oxidase 5a (Cox5a), homeobox b1 (Hoxb1), homeobox b9 (Hoxb9), and gastric inhibitory peptide (Gip). The ≈160-bp PCR product amplified by the first set of Ngfr primers (Table 1) was sequenced (data not shown) and found to be 95% identical to rat mRNA for fast nerve growth factor receptor (26) and 82% identical to human nerve growth factor receptor mRNA (36). The intragenic markers identified here defined new limits to the Mvwf candidate interval—between Ngfr/D11Mit263 and Hoxb9. Of note, Mvwf is nonrecombinant with Gip, indicating that Gip is a candidate for Mvwf. The map in Fig. 5 shows the position of Mvwf compared with other genes and markers based on an analysis of as many as 2407 informative meioses (see Materials and Methods).
Other Potential Minor Modifying Genes.
By standard one-way analysis of variance, genotype at D11Mit54 near the Mvwf locus accounted for 63% of the total variance in VWF levels. To examine the potential effects on murine VWF levels of loci distinct from Mvwf, the phenotypic distributions of the parental strains were compared using Student’s t test to the distributions among 205 F2 progeny grouped by genotype at D11Mit54 (Table 2). The average VWF level observed in RIIIS/J parental mice was 5%, which is significantly lower than the 11% observed in F2 progeny homozygous for the RIIIS/J allele (P < 0.0005). In addition, the 100% level in CASA/Rk parental mice is significantly higher than the 50% observed in F2 progeny homozygous for the CASA allele (P < 0.0005). These values indicate that, in addition to the major chromosome 11 locus, at least one or more minor modifying loci also contribute to the determination of plasma VWF levels in mice.
DISCUSSION
We have determined the location of a gene distinct from Vwf that causes an up to 20-fold reduction in VWF levels in the RIIIS/J mouse relative to other strains. The successful mapping of this locus confirms our previous hypothesis (18) that a single dominant gene unlinked to Vwf is the major modifier of murine VWF levels. These results suggest that the human homologue of Mvwf, or other functionally related genes, may contribute to the low penetrance and highly variable expression of human VWD.
Analysis of multiple simple sequence repeat markers, as well as a number of newly identified intragenic polymorphisms in progeny of a large cross, provides a more detailed genetic map for a portion of mouse chromosome 11. The order and distances we observed between markers are consistent with most previously reported maps of chromosome 11 (32). In addition, several genes can now be ordered that were not previously separable. Recombinants were identified among Cola1, Itg3a, Ngfr, and Hoxb. In addition, the murine Hoxb cluster has been oriented on chromosome 11 based on three recombinants between Hoxb1 and Hoxb9.
A discrepancy exists between our genetic map and a previously reported physical map of the murine Hoxb/Ngfr region (37). Bentley et al. (37) localized the Hoxb cluster and Ngfr to a 50-kb interval. Taken together with our genetic map, this 50-kb physical segment would have to contain at least the 0.6-cM interval from Hoxb to Ngfr, including Mvwf. In addition, the orientation we observed of the Hoxb cluster in comparison to Ngfr (Hoxb1/b9/Ngfr) also differs from the data obtained by Bentley et al (37) (Hoxb9/b1/Ngfr). Although we confirm the presence of Hoxb on the reported yeast artificial chromosomes (37), we were unable to detect a signal for Ngfr in the relevant clones by either PCR (both Ngfr primer pairs) or Southern blotting using as probe the 1.4-kb, amplified portion of Ngfr described earlier (data not shown). Sequence analysis of the 160-bp Ngfr product used for genotyping confirms that the polymorphism we mapped corresponds to murine Ngfr. However, we cannot exclude the presence of another related gene in the region of Hoxb.
The order of genes we observed on mouse chromosome 11 differs from the order of homologous genes previously reported in the syntenic region on human chromosome 17q. By linkage analysis, Anderson et al. (38) determined a gene order of cen-HOX2B-GP3A-GIP-qter. This order differs from the mouse, in which the GP3A homologue Itgb3 is located more than 10 cM away from both Hoxb and Gip and is interrupted by at least 30 known genes. Lewis et al. (39) identified a human gene order of cen-HOXB6-NGFR-GIP-tel by genetic and radiation hybrid mapping. In comparison, our map places Gip between Hoxb and Ngfr in the mouse. Taken together, these results suggest that significant rearrangement may have occurred in this region since the divergence of mice and humans.
Significant variation in plasma VWF was still observed both within and between individual indeterminate mice even though we could obtain triplicate samples and readily control for age and time of sample collection. The VWF levels observed in a small number of mice (≈3/1000) were inconsistent with Mvwf typing. In these mice, factors other than Mvwf could have influenced VWF level. Perhaps the extremely high VWF level in the outlier mouse in Fig. 3 was caused by nongenetic factors such as stress or illness. To prevent errors in the assignment of phenotype, progeny testing was used to confirm the phenotype of all critical, recombinant mice. This ability to control for the influence of environmental factors, along with the facility for a large genetic cross and the reduced complexity of genetic background effects in inbred strains, offers power that cannot readily be achieved in human linkage analysis.
The genetic mapping data reported here should facilitate the positional cloning of the Mvwf locus. This gene could exert its marked effect on plasma VWF levels through a number of distinct molecular mechanisms. In addition, the nearly complete dominance of Mvwf (similar phenotypes in heterozygotes and RIIIS/J homozygotes) suggests that the RIIIS/J mutation may confer a unique gain of function. Given the complex multistep biosynthesis and processing of VWF (1), alterations at a number of possible regulatory steps could account for the RIIIS/J low VWF phenotype. Possibilities include defects in endothelial cell-specific synthesis, intracellular targeting or secretion, an abnormality in VWF interaction with platelets or with the subendothelium, or an alteration in plasma VWF clearance. Although Gip is nonrecombinant with Mvwf, this gene is not a likely candidate for Mvwf. Gip functions as a regulator of gastric acid and insulin secretion, and its expression is limited to the small intestine (27, 40).
Few molecular defects have been reported in type 1 VWD patients (41). Genetic analysis of human type 1 VWD performed to date is consistent with linkage to the VWF locus (42–47) although locus heterogeneity has not been excluded. None of the previous studies has individually obtained statistical significance (logarithm of odds score > 3.0). Incomplete penetrance of type 1 VWD complicates linkage analysis by reducing the information available from unaffected family members.
The human homologue of Mvwf would be a strong candidate for an alternative VWD locus. Alterations at such a locus could contribute to the distinct subgroups of type 1 VWD proposed by Weiss et al. (48). Further evidence suggesting the presence of locus heterogeneity for type 1 VWD comes from the analysis of severe (type 3) VWD, which often is assumed to represent homozygous or compound heterozygous type 1 VWD (1). The reported prevalence of type 1 VWD corresponds to an allele frequency of >0.5%, which would predict a prevalence of homozygous patients of >1:40,000. However, the observed frequency of type 3 VWD patients is only ≈1/1 million, which is significantly less frequent than would be predicted if all type 3 VWD patients inherited two type 1 VWD alleles. These data suggest that mechanisms other than simple haploinsufficiency at the VWF locus could account for a significant subset of type 1 VWD.
In addition to its potential role as a second locus for VWD, the human homologue of Mvwf could be an important modifier gene in VWD patients with mutations at the VWF locus. Significant modifying effects on plasma VWF levels already have been demonstrated for the ABO and secretor blood group antigens (5, 6, 49) both in normal and VWD individuals. Allelic heterogeneity at the human homologue of Mvwf could contribute to the wide variability in plasma VWF levels among the normal population and may also account for a significant portion of the variable expression and reduced penetrance observed in human VWD.
Acknowledgments
We thank M. Meisler, M. Burmeister, and D. Burke for critical reading of the manuscript and J. Bonadio for Cola1 primers. D.G. is a Howard Hughes Medical Institute Investigator. This work was supported by National Institutes of Health Grants HL39693 (D.G.), HL31698 (R.T.S.), and HL02839 (K.A.C.).
Footnotes
Abbreviations: VWF, von Willebrand factor; VWD, von Willebrand disease; Mvwf, modifier of VWF; cM, centimorgan.
References
- 1.Ginsburg D, Bowie E J W. Blood. 1992;79:2507–2519. [PubMed] [Google Scholar]
- 2.Rodeghiero F, Castaman G, Dini E. Blood. 1987;69:454–459. [PubMed] [Google Scholar]
- 3.Werner E J, Broxson E H, Tucker E L, Giroux D S, Shults J, Abshire T C. J Pediatr. 1993;123:893–898. doi: 10.1016/s0022-3476(05)80384-1. [DOI] [PubMed] [Google Scholar]
- 4.Miller C H, Graham J B, Goldin L R, Elston R C. Blood. 1979;54:117–145. [PubMed] [Google Scholar]
- 5.Gill J C, Endres-Brooks J, Bauer P J, Marks W J, Montgomery R R. Blood. 1987;69:1691–1695. [PubMed] [Google Scholar]
- 6.Orstavik K H, Kornstad L, Reisner H, Berg K. Blood. 1989;73:990–993. [PubMed] [Google Scholar]
- 7.Harrison R L, McKee P A. Blood. 1984;63:657–665. [PubMed] [Google Scholar]
- 8.Alperin J B. Am J Med. 1982;73:367–371. [PubMed] [Google Scholar]
- 9.Triplett D A. Mayo Clin Proc. 1991;66:832–840. doi: 10.1016/s0025-6196(12)61202-6. [DOI] [PubMed] [Google Scholar]
- 10.Rogers J S, Shane S R, Jencks F S. Ann Intern Med. 1982;97:713–716. doi: 10.7326/0003-4819-97-5-713. [DOI] [PubMed] [Google Scholar]
- 11.Dalton, R. G., Dewar, M. S., Savidge, G. F., Kernoff, P. B., Matthews, K. B., Greaves, M. & Preston, F. E. (1987) Lancet i, 1007–1009. [DOI] [PubMed]
- 12.Conlan M G, Folsom A R, Finch A, Davis C E, Sorlie P, Marcucci G, Wu K K. Thromb Haemostasis. 1993;70:380–385. [PubMed] [Google Scholar]
- 13.Bloom A L. Mayo Clin Proc. 1991;66:743–751. doi: 10.1016/s0025-6196(12)62088-6. [DOI] [PubMed] [Google Scholar]
- 14.Rickles F R, Hoyer L W, Rick M E, Ahr D J. J Clin Invest. 1976;57:1618–1625. doi: 10.1172/JCI108432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeuchi M, Nagura H, Kaneda T. Blood. 1988;72:850–854. [PubMed] [Google Scholar]
- 16.Hampton K K, Grant P J, Primrose J, Dean H G, Davies J A, Prentice C R. Clin Sci. 1991;81:257–260. doi: 10.1042/cs0810257. [DOI] [PubMed] [Google Scholar]
- 17.Sweeney J D, Novak E K, Reddington M, Takeuchi K H, Swank R T. Blood. 1990;76:2258–2265. [PubMed] [Google Scholar]
- 18.Nichols W C, Cooney K A, Mohlke K L, Ballew J D, Yang A, Bruck M E, Reddington M, Novak E K, Swank R T, Ginsburg D. Blood. 1994;83:3225–3231. [PubMed] [Google Scholar]
- 19.Miller S A, Dykes D D, Polesky H F. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietrich W F, Katz H, Lincoln S E, Shin H-S, Friedman J, Dracopoli N, Lander E S. Genetics. 1992;131:423–447. doi: 10.1093/genetics/131.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor B A, Navin A, Phillips S J. Genomics. 1994;21:626–632. doi: 10.1006/geno.1994.1323. [DOI] [PubMed] [Google Scholar]
- 22.Dietrich, W. F., Miller, J. C., Steen, F. G., Merchant, M., Damron, D., Nahf, R., Gross, A., Joyce, D. C., Wessel, M., Dredge, R. D., Marquis, A., Stein, L. D., Goodman, N., Page, D. C. & Lander, E. S. (1994) Nat. Genet. 7, Suppl., 220–245. [DOI] [PubMed]
- 23.Copeland N G, Jenkins N A, Gilbert D J, Eppig J T, Maltais L J, Miller J C, Dietrich W F, Weaver A, Lincoln S E, Steen R G, Stein L D, Nadeau J H, Lander E S. Science. 1993;262:57–66. doi: 10.1126/science.8211130. [DOI] [PubMed] [Google Scholar]
- 24.Takada Y, Murphy E, Pil P, Chen C, Ginsberg M H, Hemler M E. J Cell Biol. 1991;115:257–266. doi: 10.1083/jcb.115.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamura R N, Cooper H M, Collo G, Quaranta V. Proc Natl Acad Sci USA. 1991;88:10183–10187. doi: 10.1073/pnas.88.22.10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radeke M J, Misko T P, Hsu C, Herzenberg L A, Shooter E M. Nature (London) 1987;325:593–597. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- 27.Sharma S K, Austin C, Howard A, Lo G, Nicholl C G, Legon S. J Mol Endocrinol. 1992;9:265–272. doi: 10.1677/jme.0.0090265. [DOI] [PubMed] [Google Scholar]
- 28.Bogarad L D, Utset M F, Awgulewitsch A, Miki T, Hart C P, Ruddle F H. Dev Biol. 1989;133:537–549. doi: 10.1016/0012-1606(89)90056-0. [DOI] [PubMed] [Google Scholar]
- 29.Frohman M A, Boyle M, Martin G R. Development (Cambridge, UK) 1990;110:589–607. doi: 10.1242/dev.110.2.589. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen P J, Ayane M, Kohler G. Nucleic Acids Res. 1989;17:6723. doi: 10.1093/nar/17.16.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fitzgerald L A, Steiner B, Rall S C, Jr, Lo S S, Phillips D R. J Biol Chem. 1987;262:3936–3939. [PubMed] [Google Scholar]
- 32.Lossie A C, MacPhee M, Buchberg A M, Camper S A. Mamm Genome. 1994;5:S164–S180. [PubMed] [Google Scholar]
- 33.Barrow L L, Simin K, Mohlke K, Nichols W C, Ginsburg D, Meisler M H. Mamm Genome. 1993;4:343–346. doi: 10.1007/BF00357095. [DOI] [PubMed] [Google Scholar]
- 34.Ruggeri Z M, De Marco L, Gatti L, Bader R, Montgomery R R. J Clin Invest. 1983;72:1–12. doi: 10.1172/JCI110946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pareti F I, Niiya K, McPherson J M, Ruggeri Z M. J Biol Chem. 1987;262:13835–13841. [PubMed] [Google Scholar]
- 36.Johnson D, Lanahan A, Buck C R, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao M. Cell. 1986;47:545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- 37.Bentley K L, Bradshaw M S, Ruddle F H. Genomics. 1993;18:43–53. doi: 10.1006/geno.1993.1425. [DOI] [PubMed] [Google Scholar]
- 38.Anderson L A, Friedman L, Osborne-Lawrence S, Lynch E, Weissenbach J, Bowcock A, King M C. Genomics. 1993;17:618–623. doi: 10.1006/geno.1993.1381. [DOI] [PubMed] [Google Scholar]
- 39.Lewis T B, Saenz M, O’Connell P, Leach R J. Genomics. 1994;19:589–591. doi: 10.1006/geno.1994.1114. [DOI] [PubMed] [Google Scholar]
- 40.Inagaki N, Seino Y, Takeda J, Yano H, Yamada Y, Bell G I, Eddy R L, Fukushima Y, Byers M G, Shows T B, Imura H. Mol Endocrinol. 1989;3:1014–1021. doi: 10.1210/mend-3-6-1014. [DOI] [PubMed] [Google Scholar]
- 41.Ginsburg D, Sadler J E. Thromb Haemostasis. 1993;69:177–184. [PubMed] [Google Scholar]
- 42.Bernardi F, Guerra S, Patracchini P, Volinia S, Buzzoni D, Ballerini G, Casonato A, Marchetti G. Br J Haematol. 1988;68:243–248. doi: 10.1111/j.1365-2141.1988.tb06196.x. [DOI] [PubMed] [Google Scholar]
- 43.Bahnak B R, Lavergne J-M, Verweij C L, Rothschild C, Pannekoek H, Larrieu M-J, Meyer D. Thromb Haemostasis. 1988;60:178–181. [PubMed] [Google Scholar]
- 44.Bernardi F, Marchetti G, Casonato A, Gemmati D, Patracchini P, Legnani C, DeRosa V, Girolami A, Conconi F. Br J Haematol. 1990;74:282–289. doi: 10.1111/j.1365-2141.1990.tb02584.x. [DOI] [PubMed] [Google Scholar]
- 45.Bignell P, Standen G R, Bowen D J, Peake I R, Bloom A L. Lancet. 1990;336:638–639. doi: 10.1016/0140-6736(90)93445-u. [DOI] [PubMed] [Google Scholar]
- 46.Standen G R, Bignell P, Bowen D J, Peake I R, Bloom A L. Br J Haematol. 1990;76:242–249. doi: 10.1111/j.1365-2141.1990.tb07879.x. [DOI] [PubMed] [Google Scholar]
- 47.Inbal A, Kornbrot N, Zivelin A, Shaklai M, Seligsohn U. Blood Coagulation Fibrinolysis. 1992;3:167–177. [PubMed] [Google Scholar]
- 48.Weiss H J, Piétu G, Rabinowitz R, Girma J-P, Rogers J, Meyer D. J Lab Clin Med. 1983;101:411–425. [PubMed] [Google Scholar]
- 49.Orstavik K H, Magnus P, Reisner H, Berg K, Graham J B, Nance W. Am J Hum Genet. 1985;37:89–101. [PMC free article] [PubMed] [Google Scholar]