

Abstract
Serine proteases are secreted from cells as single-chain zymogens, typically having activities orders of magnitude lower than those of the mature two-chain enzymes. Activation occurs by a conformational change initiated by cleavage of a specific peptide bond. We have derived a monoclonal antibody (mAb-112) which binds with subnanomolar affinity to pro-uPA, the zymogen form of urokinase-type plasminogen activator (uPA). We mapped the epitope of the antibody to the autolysis loop, one of the structural elements known to change conformation during zymogen activation. A mechanistic evaluation with biophysical methods elucidated a novel bifunctional inhibitory mechanism whereby mAb-112 not only delays the proteolytic conversion of single-chain pro-uPA into the two-chain form but also subsequently averts the conformational transition to a mature protease by sequestering the two-chain form in a zymogen-like, noncatalytic state. Functional studies employing two variants of human HT-1080 cells, exhibiting high and low levels of dissemination in a chorioallantoic membrane assay, demonstrate that mAb-112 is an effective inhibitor of tumor cell intravasation. These findings show that pharmacological interference with zymogen activation is a plausible and robust means to regulate uPA activity and the downstream effects of plasminogen activation in the spread of cancer and other processes of pathological tissue remodeling. A strategy that targets regions related to pro-enzyme activation likely provide a unique inhibitor-protease interaction surface and is, thus, expected to enhance the chances of engineering high inhibitor specificity. Our results provide new information about the structural flexibility underlying the equilibrium between active and inactive forms of serine proteases.
In nature a key mechanism for regulation of serine proteases with a trypsin-like fold is the activation of secreted zymogens or proenzymes, which typically have activities orders of magnitude lower than the mature enzymes. Zymogen activation is the central step in natural protease cascade regulation, allowing for rapid amplification of the activation signal. The catalytic activity of a zymogen relative to the mature protease can generally be thought of as a problem of equilibrium between active and inactive conformational states of the protease domain. Zymogen activation generally occurs by cleavage of the bond between amino acid residues 15 and 16.2 The liberated amino terminus inserts into a hydrophobic binding cleft of the catalytic domain forming, in addition to hydrophobic interactions, a salt bridge to the side chain of Asp194 which stabilizes the substrate binding pocket and oxyanion hole in a catalytically productive conformation. Conformational changes after cleavage involves four disordered regions of the activation domain, including the activation loop (residues 16-21), the autolysis loop (residues 142-152), the oxyanion stabilizing loop (residues 184-194), and the S1 entrance frame (residues 216-223) (Fig. 1A) (for reviews, see Refs. 1-3).
FIGURE 1.

Three-dimensional structure of uPA. A, overview of the three-dimensional structure of the serine protease domain of active uPA, displayed as ribbons. Depicted as sticks are the residues Ile16, Asp194, and Ser195. The activation domain, i.e. the activation loop (residues 16-21), the autolysis loop (residues 142-152), the oxyanion stabilizing loop (residues 184-193), and the S1 entrance frame (residues 216-223) are colored green. B, the epitope of mAb-112, displayed on a surface presentation of the serine protease domain of active uPA. Alanine substitution of residues depicted in red resulted in a significant change in the affinity to mAb-112, whereas alanine substitution of residues depicted in blue did not. C, a close up view of the autolysis loop (residues Gly141 to Lys156) and residues implicated in the binding of mAb-112. All figures were constructed with Pymol on the basis of the coordinates given in the PDB entry 1C5W.
Several proteases contribute to a variety of pathophysiological states, thus stimulating considerable interest in the design of specific inhibitors for pharmacological intervention. Nonetheless, classical development of small molecule inhibitors of serine proteases has proved a daunting task, with only limited success in engineering inhibitors with high affinity and specificity for related proteases possessing conserved active site architecture and P13 specificity (4, 5). Thus far targeting zymogen activation instead of the mature protease has been a greatly under-exploited strategy in therapeutic regulation of protease activity. This approach provides an opportunity to target more unique interaction surfaces, thereby increasing inhibitor specificity, and ultimately offering novel inhibitory mechanisms. In addition, a more efficient inhibition is expected by targeting enzymes functioning high up in a catalytic cascade.
A serine protease of particular relevance for pursuing therapeutic intervention is urokinase-type plasminogen activator (uPA),4 which catalyzes the conversion of plasminogen to the active protease plasmin, which in turn acts directly on the degradation of extracellular matrix proteins (6). Abnormal expression of uPA is implicated in tissue remodeling in several pathological conditions, including rheumatoid arthritis, allergic vasculitis, and xeroderma pigmentosum. In particular, uPA is central to the invasive capacity of malignant tumors (6). As with all trypsin-like proteases, uPA has a catalytic serine protease domain with surface-exposed loops around residues 37, 60, 97, 110, 170, and 185. Besides the catalytic domain, uPA has an amino-terminal extension consisting of a kringle domain and an epidermal growth factor domain. The latter domain functions in binding to the cell surface-anchored uPA receptor (uPAR) (6). Several proteases including plasmin (6), glandular kallikrein (7), matriptase (8), and hepsin (9) can catalyze the activation of the zymogen, pro-uPA.
A number of inhibitors targeting the proteolytic activity of uPA have been developed, such as small organochemical molecules, peptides, and monoclonal antibodies, with a perspective on their use for elucidating the pathophysiological functions of its various molecular interactions and generating leads during drug development. The most specific inhibitors to date appear to be those derived from antibodies and peptidyl inhibitors, which utilize binding sites involving surface loops of uPA and extended exosite interactions to drive selectivity and specificity (for reviews, see Refs. 4 and 5).
Here we present evidence that targeting zymogen activation is an effective means to regulate protease activity. This conclusion was realized through the development and biochemical analysis of an inhibitory monoclonal antibody, referred to as monoclonal antibody (mAb)-112, which not only delays cleavage of pro-uPA but acts to stabilize the activated two-chain protease in a non-catalytic conformation by restricting the conformational mobility of the activation domain. Characterization of mAb-112 further provides new insights into the flexibility of protease domains and uPA zymogen activation mechanisms. Moreover, mAb-112 was shown to efficiently inhibit human tumor cell intravasation, a step in the metastatic cascade in which activation of pro-uPA was previously implicated as a key event (10).
EXPERIMENTAL PROCEDURES
Buffer Conditions—Unless otherwise indicated, all reactions were carried out in a buffer containing 30 mm HEPES, 135 mm NaCl, 1 mm EDTA, and 0.1% bovine serum albumin, pH 7.4 (HBS-B).
uPA—Human two-chain uPA was purchased from Wakamoto (Tokyo, Japan). Recombinant human pro-uPA was a gift from Abbott Laboratories. uPA-PAI-1 complex and diisopropyl fluorophosphate-uPA were prepared as described (11). Labeling of uPA (25 μm) with EGR-chloromethyl ketone (CMK; 1 mm) or 1,5-dansyl-EGR-CMK (1 mm) was carried out in phosphate-buffered saline at 25 °C for 60 min. Labeling of pro-uPA (12-20 μm) with EGR-CMK or 1,5-dansyl-EGR-CMK (10-20 mm) required the addition of 50 mm Ile-Ile dipeptide and incubation at 37 °C for 4 h. Excess inhibitor was removed by extensive dialysis against 20 mm citric acid, 50 mm NaCl, pH 4.5.
Wild type and mutant recombinant human pro-uPA variants were expressed in HEK 293T cells (12), cultured in the presence of 5 μg/ml aprotinin. The concentrations of wild type and mutant pro-uPA in the conditioned media were determined by surface plasmon resonance analysis using a BIACORE T100 instrument (see below) and were between 80 and 600 nm; however, for the most part variants were consistently expressed at concentrations close to 300 nm. In purified preparations, protease concentrations were determined from the absorbance at 280 nm using an extinction coefficient of 1.36 ml mg-1 cm-1 and an Mr value of 54,000.
Other Proteases—Recombinant human matriptase residues 596-855 was purchased from R&D systems (Wiesbaden-Nordenstadt, Germany). Human Glu-plasminogen was purchased from American Diagnostica (Greenwich, CT).
Antibodies—mAbs were created in-house by immunization of Balb/c mice with recombinant human single chain pro-uPA by intraperitoneal injection. Booster injections of 100 ng of antigen were given on days 35, 65, 100, 130, and 132. Spleen cells were harvested on day 135 and fused with NSO myeloma cells for hybridoma generation (13). Hybridomas were screened for binding to pro-uPA by enzyme-linked immunosorbent assay, yielding 22 clones for further characterization. Conditioned media from these hybridomas were tested for the binding specificity of secreted mAbs toward pro-uPA and uPA using an enzyme-linked immunosorbent assay. Among those tested, one hybridoma produced an antibody, mAb-112, proven to be specific toward pro-uPA. This mAb was determined to belong to subclass IgG1. Antibodies were purified from hybridoma-conditioned medium using protein G-Sepharose 4FF (13). One liter of cell culture supernatant yielded between 15 and 50 mg of purified antibody.
To produce the Fab fragment of mAb-112, the antibody was treated with papain in a 1:100 (w/w) ratio of enzyme to antibody in 15 mm cysteine, 2 mm EDTA for 6 h at 37 °C. Fab-112 was purified from the cleavage reaction mixture by protein A affinity chromatography in phosphate-buffered saline followed by MonoS™ 5/50 GL cation exchange in 25 mm acetate, pH 5.5, using the AmershamÄKTA explorer 100 Air system (GE healthcare).
The following, previously described antibodies were used: anti-uPA mAb-2 and anti-uPA mAb-6 (12); rabbit polyclonal anti-uPA antibodies (14); anti-PAI-1 mAb-2 (15). Anti-uPA mAb-118 was from the same fusion as mAb-112 and found to bind the serine protease domain but did not affect uPA enzyme activity.5
Surface Plasmon Resonance Analysis—Surface plasmon resonance (SPR) analyses were performed on a BIACORE T100 instrument using CM5 sensor chips, flow rates of 30 μl/min, and HBS-B with 0.05% Tween 20. Concentrations of pro-uPA in conditioned media from HEK 293T cells were determined by measuring the initial rate of binding to a chip with 200 response units of anti-uPA mAb-6 (12) using a standard curve of purified pro-uPA. Affinities of mAb-112 for various forms of wild type or mutant pro-uPA and uPA were determined by injecting the proteases (0.5-150 nm), either purified or in conditioned media, over a chip with 200 response units of immobilized mAb-112. The affinity constants kon, koff, and KD were determined by global fitting to a 1:1 binding model with the BIA-CORE evaluation program. In an alternative approach, the binding of mAb-112 to uPA variants was assessed by capturing the variants (300 response units) on a chip with immobilized anti-uPA mAb-6 and subsequently injecting Fab-112 over the chip at a concentration of 100 nm and a flow rate of 10 μl/min.
Amidolytic Assay of uPA Activity—Rates for the uPA-catalyzed hydrolysis of the chromogenic substrate <Glu-Gly-Arg-p-nitroanilide (S-2444; Chromogenix, Mölndal, Sweden) were measured at 37 °C in HBS-B. Reactions of uPA (5 nm) in the presence or absence of mAb-112 (0-500 nm) were acquired over a range of substrate concentrations (0-0.8 mm). Initial rates of S-2444 hydrolysis were monitored at 405 nm.
Plasminogen Activation Assays—Pro-uPA or uPA (0.25 nm) was preincubated with various concentrations of mAb-112 (5-160 nm) in HBS-B at room temperature for 30 min. The addition of 0.5 μm plasminogen and 0.5 mm plasmin substrate H-d-Val-Leu-Lys-p-nitroanilide (S-2251) initiated the reaction at 37 °C. S-2251 hydrolysis was monitored for the parabolic increase in absorbance at 405 nm.
For assaying uPA-catalyzed plasminogen activation activity with uPA bound to uPAR at cell surfaces, U937 cells were cultured as described previously (16), washed, and resuspended in HBS-B at a density of 107 cells/ml. Cells were preincubated for 1 h at 37 °C with 50 nm uPA or pro-uPA and subsequently washed three times with HBS-B to remove unbound uPA or pro-uPA. The cells were then distributed into the wells of non-transparent 96-well microtiter plates (Nunc, Roskilde, Denmark) in a total volume of 200 μl and a cell density of 5 × 106 cells/ml. Cells were preincubated for 30 min with mAb-112 or the unrelated anti-PAI-1 mAb-2 as a negative control before initiating the reactions with plasminogen (270 nm) and the fluorogenic plasmin substrate H-d-Val-Leu-Lys-7-amido-4-methylcoumarine (200 μm). The levels of fluorescence were monitored with 1-min intervals in a Spectromax Gemini fluorescence plate reader (Molecular Devices) using an excitation wavelength of 390 nm and an emission wavelength of 480 nm. uPA-uPAR Binding Analysis—Samples containing 1.0 × 106 U937 cells per ml, 100 pm 125I-pro-uPA or 125I-uPA (1 × 107 cpm/pmol) and varying concentrations of mAb-112 or control amino-terminal fragment were prepared in RPMI medium with 10% fetal bovine serum and incubated overnight at 4 °C with gentle agitation. The following day cells were pelleted, and half of the supernatant was collected for γ-counting. The rest of the supernatant was removed, the cells were washed twice with 1 ml of serum-free RPMI medium and pelleted, and the bound pro-uPA or uPA was quantified by γ-counting plotted as a function of the mAb-112 or amino-terminal fragment concentration.
Analysis of Pro-uPA Cleavage by SDS-PAGE and Immunoblotting Analysis—Samples of pro-uPA (200 nm) were incubated in the presence or absence of 300 nm mAb-112 for 30 min at room temperature in 30 mm HEPES, 135 mm NaCl, 1 mm EDTA, 0.1% polyethylene glycol, pH 7.4. At various times after the addition of plasmin (5 nm) or matriptase (50 nm), samples were removed and analyzed by reducing SDS-PAGE and immunoblotting with rabbit polyclonal anti-uPA antibodies.
Fluorescence Emission Spectroscopy—Fluorescence emission spectra of dansyl-labeled pro-uPA or uPA were collected using a SPEX-3 spectrofluorimeter equipped with a Peltier temperature controller maintaining the measurement and incubation temperatures at 25 °C. Fluorescence experiments were carried out using semi-micro (0.5 × 1.0 cm) quartz cuvettes and in a Hepes-buffered saline reaction buffer containing 0.1% polyethylene glycol 8000 to prevent protein adsorption to the quartz surface. The excitation wavelength used for studying the fluorescence of dansyl-EGR-pro-uPA or dansyl-EGR-uPA was 335 nm, and the emission spectra were scanned from 450 to 625 nm using an excitation bandwidth of 5 or 10 nm and an emission bandwidth of 5 nm. Emission spectra for 200 nm dansyl-EGR-pro-uPA or dansyl-EGR-uPA were recorded before and after the addition of plasmin (10 nm) and with or without 300 nm mAb-112. Results are presented as the averaged spectra of three independent titrations. All individual emission spectra were collected as averages of 3-5 emission scans using a 1.0-s integration over a 1.0-nm step resolution and corrected for background fluorescence and dilution effects, which were typically less than 5%.
Human Tumor/Chick Embryo Chorioallantoic Membrane (CAM) Assay—HT-hi/diss and HT-lo/diss variants, previously selected in vivo from HT-1080 human fibrosarcoma (ATCC, Manassas, VA) for a 50-100-fold differential in their ability to intravasate (10), were cultured under standard conditions. The day before experiments, cells were passaged and, after an overnight incubation, detached with trypsin/EDTA, washed, and resuspended in serum-free Dulbecco's modified Eagle's medium. Fertilized SPAFAS White Leghorn eggs (Charles River, North Franklin, CT) were incubated in a humidified rotary incubator at 38 °C. On day 10 of incubation, 2 × 105 HT-hi/diss cells or 4 × 105 HT-lo/diss cells were grafted in 25 μl of serum-free medium on the CAM. The developing HT-hi/diss tumors were treated on days 1 and 3 with 0.1 ml of 1 mm aprotinin or 25 μg of normal mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), mAb-118, mAb-2, or mAb-112. A group of embryos was treated with mAb-112 on day 3 only. Control embryos were treated with 0.1 ml of vehicle (30 mm HEPES, pH 7.4, 135 mm NaCl, 1 mm EDTA, 0.1% w/v polyethylene glycol 8000). On day 5, primary tumors were excised and weighed. Portions of the distal CAM were harvested and frozen on dry ice. Levels of human cell intravasation in the chick embryo were determined by quantitative PCR of the Alu repeats (10). The actual numbers of tumor cells in each tissue sample were determined using a standard curve generated by a serial dilution of HT-1080 cells within a constant number (106) of chick embryo fibroblasts. To determine statistical significance, the data sets were analyzed by Fisher or Mann-Whitney two-tailed test for p < 0.05.
RESULTS
Discovery of an Inhibitory Antibody to Pro-uPA—In hope of producing mAbs with preferential affinity to pro-uPA, mice were immunized with human pro-uPA. Hybridomas were generated by standard techniques and screened by enzyme-linked immunosorbent assay for binding to pro-uPA and uPA. Of the 22 hybridoma clones found to bind pro-uPA, seven produced antibodies that bound to the catalytic domain, whereas the remaining clones were specific for the amino-terminal fragment of uPA. A single hybridoma clone produced an antibody, mAb-112, which bound to the catalytic domain and demonstrated a clear preference for binding to pro-uPA over active uPA as determined by enzyme-linked immunosorbent assay (data not shown).
mAb-112 Inhibits Pro-uPA Activation—The functional effect of mAb-112 on pro-uPA activation was investigated in a coupled plasminogen activation assay in which pro-uPA was incubated with plasminogen and a plasmin substrate. The assay is designed so that trace amounts of plasmin in the plasminogen preparation will initiate pro-uPA activation and the further generation of plasmin, which in turn activates additional pro-uPA creating a reciprocal activation cycle stimulating plasmin generation (17). Preincubation of pro-uPA with mAb-112 inhibited the generation of plasmin with an estimated IC50 value in the 5 nm range (Fig. 2A). Plasminogen activation catalyzed directly by active two-chain uPA was also reduced by mAb-112, however at much higher effective concentrations of mAb-112, suggesting that mAb-112 preferentially targets pro-uPA activation (Fig. 2B). In contrast to mAb-112, no inhibitory effects on plasminogen activation by either pro-uPA or uPA were observed in the presence of a control antibody, anti-PAI-1 mAb-2 (data not shown).
FIGURE 2.

mAb-112 inhibits in vitro plasminogen activation initiated by Pro-uPA or uPA. 0.25 nm pro-uPA (panel A) or 0.25 nm uPA (panel B) was incubated without (black dots) or with 5 nm (red dots), 10 nm (green dots), 20 nm (yellow dots), 40 nm (dark blue dots), 80 nm (pink dots), and 160 nm (light blue dots) of mAb-112. After incubation for 30 min at room temperature, plasminogen was added to 0.5 μm and S-2251 to 0.5 mm. Substrate hydrolysis was followed at 37 °C by measuring the absorbance at 405 nm. Pro-uPA or uPA was omitted in controls (gray curves).
We further evaluated the effect of mAb-112 on the activation of pro-uPA by both plasmin and matriptase in a more direct way. To this end, pro-uPA was incubated with plasmin or matriptase for various time periods after which the activation reactions were quenched by the addition of aprotinin. The amount of generated active uPA was estimated from the rate of hydrolysis of the uPA amidolytic substrate S-2444. As shown in Fig. 3, the presence of mAb-112 in this assay resulted in a total inhibition of pro-uPA activation by both plasmin and matriptase.
FIGURE 3.

mAb-112 inhibits the activation of Pro-uPA. Ten nm pro-uPA was incubated with 100 nm mAb-112 (black dots), 100 nm anti-PAI-1 mAb-2 (open circles), or buffer (black triangles). At time 0, 0.5 nm plasmin or 5 nm matriptase was added. Then the mixtures were incubated at room temperature for the indicated time periods, after which the activity of plasmin and matriptase was quenched by the addition of 1 μm aprotinin. S-2444 was added to a final concentration of 0.5 mm, and the amount of active uPA formed was estimated by the rate of hydrolysis and comparison to a standard curve of active two-chain uPA.
The ability of mAb-112 to inhibit uPAR-bound pro-uPA activation and, thus, downstream uPA activity on the cell surface was further examined using U937 cells, which up-regulates uPAR expression and surface display after stimulation with phorbol esters (18). As shown in Fig. 4, cell surface plasminogen activation initiated by pro-uPA bound to uPAR at the surface of U937 cells was also dose-dependently inhibited. Inhibition of plasmin generation was considered to be in direct response to mAb-112 acting on cell surface-associated pro-uPA activity rather than an effect on the binding of pro-uPA to uPAR. This conclusion was supported by experiments demonstrating that mAb-112 was unable to compete 125I-pro-uPA or 125I-uPA binding to uPAR on the surface of U937 cells. One million cells/ml were incubated with 100 pm 125I-pro-uPA with or without mAb-112. In the absence of mAb-112, 4.1 ± 0.2% of the added tracer bound to the cells. In the presence of 150 nm mAb-112, 4.5 ± 0.2% bound. The amino-terminal fragment of uPA effectively competed for binding of the labeled proteases in control experiments.
FIGURE 4.

mAb-112 inhibits cell surface-associated plasminogen activation initiated by pro-uPA. U937 cells (5 × 106 cells/ml) that had been saturated with pro-uPA were incubated at 37 °C in the absence (red dots) or in the presence of 5 nm (light green dots), 10 nm (yellow dots), 20 nm (blue dots), 40 nm (violet dots), 80 nm (cyan dots), or 160 nm of mAb-112 (gray dots). Cells incubated without pro-uPA (black dots) or with anti-PAI-1 mAb-2 (not shown) served as negative controls. After 30 min of incubation, the addition of plasminogen (270 nm) and the fluorogenic plasmin substrate H-d-Val-Leu-Lys-7-amido-4-methylcoumarine (200 μm) started the reaction, which was recorded by reading the fluorescence at regular time intervals for 120 min. The experiment shown is a representative from a total of three independent experiments.
To directly evaluate whether mAb-112 influenced the rate of cleavage of single-chain pro-uPA, pro-uPA was incubated with plasmin or matriptase in the presence or absence of mAb-112, and the reaction products were analyzed by reducing SDS-PAGE and immunoblotting. The proteolytic cleavage of pro-uPA is marked by the conversion of a single band at Mr ∼ 55,000 to two bands representing the serine protease domain and amino-terminal fragment that migrate with Mr ∼ 30,000 and Mr ∼ 20,000, respectively. In the presence of excess mAb-112, the plasmin or matriptase-catalyzed proteolytic cleavage of pro-uPA is significantly hindered, reflecting a delay in activation that is in agreement with the delay in appearance of catalytic activity (Fig. 5).
FIGURE 5.

mAb-112 hinders the cleavage event in plasmin-mediated pro-uPA activation. Aliquots of pro-uPA (200 nm) were incubated with plasmin (5 nm, panel A) or matriptase (50 nm, panel B) in the absence or presence of mAb-112 (300 nm). After the indicated incubation periods, the reaction products were analyzed by reducing SDS-PAGE and immunoblotting with a polyclonal anti-uPA antibody. The cleavage was observed as the conversion of the Mr ∼ 54,000 band of single-chain pro-uPA to the Mr ∼ 30,000 catalytic domainand Mr ∼ 20,000 amino-terminal fragment. tc, two chain.
Stabilization of a Zymogen-like Conformation of the Protease by mAb-112—Although single-chain pro-uPA was ultimately cleaved into a two-chain enzyme by plasmin, the presence of a molar excess of mAb-112 during the cleavage reaction completely abrogated all amidolytic activity even at time points after which complete cleavage was shown to have occurred (Fig. 3 and data not shown). This finding suggested that the activated protease may be trapped in a non-catalytic or zymogen-like conformation such that mAb-112 also interferes with the concerted conformational change within the activation domain of uPA after cleavage of the activation loop.
To substantiate the aforementioned hypothesis and directly observe the conformational change associated with pro-uPA activation, we developed a novel method for specific labeling of the active site cleft in pro-uPA with a fluorescent probe by means of covalently modifying Ser195 and His57 with a dansyl-labeled chloromethyl ketone inhibitor. We took advantage of the fact that a dipeptide mimic of the amino-terminal residues (Ile16-Ile17), which normally inserts into the activation domain after proteolytic cleavage, shifts the equilibrium from an inactive toward an active protease conformation in the absence of proteolytic cleavage (19). In the presence of Ile-Ile dipeptide, we were able to label the Ser195 and His57 residues within the active site cleft of pro-uPA with the fluorescent and irreversible inhibitor, dansyl-EGR-CMK. Upon removal of the dipeptide by extensive dialysis, the enzyme reverts to the inactive but now dansyl-labeled zymogen conformation. Based on the absence of amidolytic activity subsequent to plasmin cleavage of the dan-syl-EGR-CMK-labeled pro-uPA, the efficiency of labeling was at least 99% (data not shown). This zymogen labeling technique was adopted from procedures previously developed for labeling plasminogen and prothrombin in the presence of streptokinase or staphylocoagulase, which induced activity of these zymogens by a mechanism similar to dipeptides corresponding to the amino-terminal residues of the mature enzymes (20, 21).
The fluorescence spectra of dansyl-EGR-CMK labeled pro-uPA and uPA are markedly dissimilar with dansyl-EGR-pro-uPA showing a broad fluorescence maximum at 543 ± 5 (n = 3) nm, whereas that of dansyl-EGR-uPA is red-shifted to 568 ± 3 (n = 3) nm with a prominent quench in fluorescence (Fig. 6A). Plasmin cleavage of dansyl-EGR-CMK-labeled pro-uPA resulted in a time-dependent quench and red shift of the fluorescence emission spectrum from 543 to 568 nm, consistent with the expected fluorescence profile for active, two-chain dansyl-EGR-CMK-labeled uPA (Fig. 6B). When the cleavage was performed in the presence of mAb-112, we observed negligible changes in the fluorescence emission spectrum even after 120 min of incubation with plasmin (Fig. 6C). In a parallel experiment, SDS-PAGE visualization of the reaction products of dansyl-EGR-pro-uPA demonstrated full cleavage to the two-chain enzyme within 60 min (Fig. 6D). Thus, despite the presence of a cleaved product, a zymogen-like conformation persisted in the presence of mAb-112.
FIGURE 6.

mAb-112 prevents the conformational change associated with Pro-uPA activation. Emission spectra of 100 nm dansyl-EGR-labeled pro-uPA (black) or 100 nm dansyl-EGR-labeled uPA (red) were collected with an excitation wavelength of 335 nm, and emission spectra were scanned from 450 to 625 nm (panel A). Dansyl-EGR-labeled pro-uPA (200 nm) was incubated with plasmin (20 nm) for 0 (black), 15 (red), 30 (yellow), 60 (green), or 120 min (blue) in the absence (panel B) or presence (panel C) of mAb-112 (300 nm). The black dashed and fully drawn lines in panel B represent the spectrum of pro-uPA with and without mAb-112, respectively. In parallel, samples of the dansyl-EGR-pro-uPA incubation with mAb-112 were removed at the indicated time points for analysis by reducing SDS-PAGE. After electrophoresis, dansyl-labeled pro-uPA and uPA bands were visualized using a Gel-Doc imaging system (Bio-Rad). Dansyl-EGR-uPA was co-electrophoresed as a standard (first lane)(panel D).
Inhibition of uPA Activity by mAb-112—Given that mAb-112
displayed inhibitory activity against active two-chain uPA, we decided to
further characterize the mechanism of mAb-112 inhibition of uPA-catalyzed
hydrolysis of a tripeptide substrate (Fig.
7). The apparent Km and
Vmax values (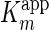 and
and 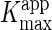 ) were determined
under steady state conditions and fitting of the experimental data to Equation
1,
) were determined
under steady state conditions and fitting of the experimental data to Equation
1,
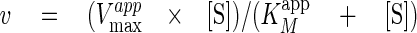 |
(Eq.1) |
The mode of inhibition was purely non-competitive, as the
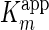 was invariable with increasing
mAb-112 concentrations, whereas the
was invariable with increasing
mAb-112 concentrations, whereas the
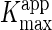 decreased severalfold,
reflecting a reduction in kcat
(Fig. 7A). The
Ki was determined to be 164 nm by plotting the
decreased severalfold,
reflecting a reduction in kcat
(Fig. 7A). The
Ki was determined to be 164 nm by plotting the
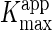 values against mAb-112
concentration (Fig.
7B) and evaluation of the data with Equation 2 describing
a non-competitive mode of inhibition
values against mAb-112
concentration (Fig.
7B) and evaluation of the data with Equation 2 describing
a non-competitive mode of inhibition
 |
(Eq.2) |
FIGURE 7.

mAb-112 is a non-competitive inhibitor of active two-chain uPA. The
dependence of the amidolytic activity of uPA on the concentration of the
substrate S-2444 in the presence of mAb-112 at concentrations of 0
nm (black dots), 10 nm (red dots), 40
nm (green dots), 160 nm (blue dots),
320 nm (purple dots), or 500 nm (cyan
dots) is shown (panel A). For each mAb-112 concentration, the
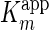 and
and
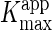 was estimated by
fitting the data to Equation 1. The
was estimated by
fitting the data to Equation 1. The
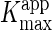 values were plotted
against the mAb-112 concentration, and the data were fit to Equation 2,
expected to apply for non-competitive inhibition. The fit yielded a
Ki value of 163.8 ± 0.2 nm (panel
B).
values were plotted
against the mAb-112 concentration, and the data were fit to Equation 2,
expected to apply for non-competitive inhibition. The fit yielded a
Ki value of 163.8 ± 0.2 nm (panel
B).
Kinetics of pro-uPA and uPA Binding to mAb-112—Using SPR analyses, the KD value for pro-uPA was found to be >100-fold lower than that for active uPA, with the difference being predominantly attributed to changes in kon. Furthermore, the calculated KD value for active uPA was in close agreement with the experimentally determined Ki value (Fig. 7). Although EGR-CMK-modified pro-uPA exhibited the same affinity to mAb-112 as pro-uPA, EGR-uPA, diisopropyl fluorophosphate-modified uPA, or the uPA-PAI-1 complex had a more than 3-fold lower affinity than uPA. We cannot exclude that minor conformational differences may exist between the individual active uPA derivatives, accounting for the observed differences between EGR-CMK and diisopropyl fluorophosphate-labeled uPA derivatives (Table 1).
TABLE 1.
Kinetic analysis of the binding of mAb-112 to Pro-uPA and active uPA
Association (kon) and dissociation (koff) rate constants and the equilibrium dissociation constant (KD) were determined by global fitting of the SPR data to a 1:1 binding model.
| uPA variant | kon | koff | KD | KD-Variant/KD-pro-uPA | n |
|---|---|---|---|---|---|
| m−1s−1 | s−1 | nm | |||
| Pro-uPA | (1.2 ± 0.4) × 105 | (4.4 ± 1.5) × 10−5 | 0.39 ± 0.16 | 1.0 | 12 |
| Active uPA | (1.4 ± 0.5) × 103a | (2.0 ± 1.2) × 10−4a | 141 ± 47a | 362 | 6 |
| EGR-pro-uPA | (1.3 ± 0.4) × 105 | (8.1 ± 4.7) × 10−5 | 0.63 ± 0.32 | 1.6 | 4 |
| EGR-uPA | (6.9 ± 3.0) × 102 | (2.8 ± 0.2) × 10−4a | 420 ± 111b | 1017 | 3 |
| DFP-uPA | NBc | NBc | NBc | ||
| uPA-PAI-1 | NBc | NBc | NBc |
Significantly different from corresponding value for pro-uPA (p < 0.01).
Significantly different from corresponding value for uPA (p < 0.01).
NB, no measurable binding at 150 nm protease.
The Epitope of mAb-112 Is Localized to the Autolysis Loop—The epitope of mAb-112 was determined by alanine scanning mutagenesis utilizing variants of uPA expressed recombinantly as the zymogen from HEK 293T cells. The primary focus was residues in the activation domain (22), although residues within adjacent secondary structural elements or residues previously implicated in stabilizing the zymogen structure were also analyzed (1, 19). Alanine substitutions of Glu144, Tyr149, Tyr151, Lys156, or Asp194 significantly affected mAb-112 binding in SPR analyses; likewise, triple alanine mutations at Phe141-Gly142-Lys143 reduced mAb-112 binding (Table 2) (Fig. 1B). These results were confirmed by an alternative BIACORE format, in which the pro-uPA mutants were captured on a chip with anti-uPA mAb-6, and the binding of a Fab-fragment of mAb-112 to the immobilized pro-uPA was measured (data not shown).
TABLE 2.
Mapping the epitope for mAb-112 by alanine scanning mutagenesis
Association (kon) and dissociation (koff) rate constants and the equilibrium dissociation constant (KD) were determined by global fitting of the SPR data to a 1:1 binding model. The following pro-uPA variants demonstrated binding kinetics indistinguishable from wild type: T9A, K15A, I16A, I17A, E20A, F21A, Y34A, R35A, R36A, H37A, R37aA, S37dA, T39A, Y40A, V41A, P60cA, K61A, K62A, E62aA, D63A, I65A, Y67A, R70A, R72A, L73A, N75A, N76A, Q78A, K82A, E84A, K92A, K110aA, E110bA, R110dA, K143A, N145A, S146A, T147A, D148A, L150A, P152A, E153A, Q154A,, D185A, Q185bA, W186A, K187A, D189A, S190A, Q192A, R217A, K223A, K225A, H241A, K243A, E244A.
| Pro-uPA variant | kon | koff | KD | KD-Variant/KD-wt | n |
|---|---|---|---|---|---|
| m−1s−1 | s−1 | nm | |||
| Wild type | (7.1 ± 2.8) × 104 | (6.1 ± 4.0) × 10−5 | 0.80 ± 0.29 | 1.0 | 5 |
| E144A | (1.3 ± 0.9) × 105 | (3.9 ± 1.8) × 10−4a | 3.1 ± 0.7a | 3.9 | 2 |
| Y149A | (1.2 ± 0.7) × 105 | (9.1 ± 0.3) × 10−3a | 93 ± 50a | 116 | 3 |
| Y151A | (1.9 ± 0.3) × 105 | (2.4 ± 0.6) × 10−2a | 124 ± 12a | 155 | 3 |
| K156A | (2.5 ± 0.5) × 105a | (1.3 ± 0.3) × 10−2 | 56 ± 19a | 70 | 4 |
| D194A | (6.4 ± 0.3) × 104 | (1.4 ± 0.1) × 10−3 | 22 ± 8a | 28 | 3 |
| F141A-G142A-K143A | (1.2 ± 0.2) × 105 | (7.1 ± 0.9) × 10−3 | 60 ± 5a | 75 | 2 |
Significantly different from corresponding value for wild type (p < 0.01).
Unfortunately, the three-dimensional structure of pro-uPA remains to be reported. The best possible information about the localization of the residues implicated in the epitope could, thus, only be obtained from the published structure of active uPA (22). Analysis of this structure predicted that Glu144, Tyr149, and Tyr151, all localized in the autolysis loop, are surface-exposed, whereas Lys156 is only partly exposed, and Phe141, Gly142, Lys143, and Asp194 are buried and localized to the autolysis loop or near the active site, respectively. For all variants, the observed effects could be attributed to increased dissociation rates (koff) (Table 2). The identified surface-exposed residues likely form the epitope, whereas the buried residues are expected to affect mAb-112 binding by conformational effects on the autolysis loop (Fig. 1). Although it is expected that the conformation of pro-uPA will differ in the region of the autolysis loop, the changes in solvent exposure for most residues is generally not expected to be significant.
Effect of mAb-112 on Human Tumor Cell Intravasation—We investigated the potential for mAb-112 to inhibit tumor cell dissemination in the chicken embryo model where human tumor cells are grafted on the CAM, providing the vasculature readily available for tumor cell intravasation (23). We used a high disseminating (HT-hi/diss) variant of the human HT-1080 fibrosarcoma cell line generated by in vivo selection (24). The low disseminating (HT-lo/diss) counter-part served as a negative control as these cells, although generating primary tumors, exhibit very low levels of intravasation. Previous studies have demonstrated that pro-uPA activation is a key step in HT-hi/diss intravasation (10).
HT-hi/diss and HT-lo/diss cells were grafted on the CAM of chicken embryos. On days one and three, the developing primary tumors were treated with anti-uPA mAb-112. A group of embryos was treated with mAb-112 on day 3 only. In parallel, embryos were treated with a serine protease inhibitor, aprotinin, used as a positive control for inhibition of plasmin activity and previously demonstrated to effectively inhibit HT-hi/diss intravasation (10). As shown in Fig. 8, the expected low numbers of HT-lo/diss cells were detected in the CAM by quantitative PCR of the Alu repeats. In contrast, HT-hi/diss cells exhibited high levels of intravasation. The addition of mAb-112 caused a substantial reduction in the number of intravasated HT-hi/diss cells, whereas control mAb had no effect. Importantly, the addition of mAb-112 on days 1 and 3 after cell grafting resulted in a significant, 66 ± 8% inhibition of intravasation (p < 0.001). A similar application of mAb-112 performed on day 3 only was less effective (p < 0.05), thus implicating pro-uPA activation and/or uPA activity in the early stages of HT-hi/diss dissemination. Aprotinin caused an ∼50% decrease in HT-hi/diss intravasation as compared with a vehicle control (p < 0.05), confirming the dependence of HT-hi/diss dissemination on the activity of serine proteases. We have attempted to analyze the effects of mAb-112 and aprotinin on the uPA cleavage status in the lysates of CAM tumors. However, uPA was not detected by Western blotting, most probably due to the highly labile status of this secreted protease in the tissues in vivo.
FIGURE 8.

Inhibitory effects of anti-uPA mAb-112 and mAb-2 on tumor cell intravasation. HT-hi/diss and HT-lo/diss cells were grafted on the CAM of chick embryos on day 10 of incubation. The developing CAM tumors were treated topically with buffer, aprotinin, control mAb-118, or anti-uPA antibodies mAb-112 and mAb-2 on days 1 and 3 after cell grafting. A group of embryos bearing HT-hi/diss tumors was treated with mAb-112 on day 3 only (d3). On day 5 the primary tumors were excised and weighed. Portions of the distal CAM were harvested and analyzed by quantitative PCR of the Alu repeats to determine levels of intravasation (number of human cells detected in the CAM tissue per 106 chick embryo cells). Presented are normalized data from 4 independent experiments involving from 7 to 41 embryos per cell variant or treatment condition. The data are the means ± S.E. of percentage of intravasation (A) and tumor growth (B) determined by comparison with control conditions set as 100% (buffer for aprotinin and HT-lo/diss and control mAb for anti-uPA mAbs). *, p < 0.05 in Fisher test; p < 0.05 (**) and p < 0.001 (***) in Mann-Whitney tests, respectively.
We also tested the effect of anti-uPA mAb-2, which inhibits uPA-catalyzed plasminogen activation in vitro with a Ki around 0.5 nm, whereas it does not inhibit pro-uPA activation. It has the same affinity to pro-uPA and active uPA and has an epitope adjacent to the uPA active site and presumably sterically hinders the access of plasminogen to the active site.6 When added in the CAM assay at the same dose as mAb-112, mAb-2 caused a 32% reduction in HT-hi/diss intravasation, indicating that tumor cell dissemination can also be inhibited by mAb-mediated blocking of plasminogen activation, although less efficiently than by mAb-112. In contrast, there was no effect of mAb-118, which inhibits neither pro-uPA activation nor plasminogen activation catalyzed by two-chain uPA.5 Likewise, there was no effect of IgG purified from mouse plasma (data not shown). Importantly, none of these treatments affected primary tumor growth.
DISCUSSION
The in vitro and in vivo data presented in this report provide evidence that targeting the zymogen activation step is a realistic approach to intervention with uPA activity. We generated and characterized an anti-uPA monoclonal antibody, mAb-112, exhibiting more than 100-fold higher affinity for pro-uPA than for active uPA. The mAb-112 epitope is localized to the autolysis loop and comprised of the surface-exposed residues Glu144, Tyr149, and Tyr151, which are close to the Lys15-Ile16 bond that is cleaved upon conversion of single-chain pro-uPA to two-chain active uPA. From a functional standpoint, the antibody targets pro-uPA at two distinct points in the activation mechanism. Initially, mAb-112 delays conversion of the zymogen to the two-chain protease, most likely through sterically occluding access of the activation loop to activating proteases to obstruct the proteolytic cleavage of the Lys15-Ile16 bond. Nonetheless, the primary mechanism of action appears to be prevention of the conformational change associated with pro-uPA activation by maintaining the autolysis loop in a zymogen-like conformation and/or sterically hindering the interaction between Ile16 and Asp194 after cleavage of the activation loop, thus preventing stabilization of the oxyanion hole in an active conformation. This observation is in excellent agreement with the autolysis loop being among the key structural elements altering conformation during zymogen activation and contributing to the stability of the activation domain to provide transition state stabilization for enzyme activity (1, 2, 25).
In addition to inhibiting the activation of pro-uPA, we demonstrate that the antibody is a non-competitive inhibitor of two-chain uPA, albeit at a significantly reduced affinity. Binding of mAb-112 was found to destabilize the activation domain by ∼1 kcal/mol as determined by calculating ΔΔGT from the ratios of kcat/Km in the presence and absence of mAb-112 at close to 90% inhibition. This is much less than the 8 kcal/mol of stabilization energy reported for the trypsinogen to trypsin conversion (25). Thus, either a small active site destabilization has relatively profound effects, on catalysis or the pro-uPA to active uPA transition may require much less stabilization energy. The latter explanation is consistent with the findings that the zymogen pro-uPA is only 250-fold less active than active uPA, whereas trypsinogen is 107-fold less active than trypsin (25).
Given that derivatization of uPA with covalent inhibitors, i.e. transition state analogues (26) or the serpin PAI-1, causes a severalfold reduction in its affinity to mAb-112, we propose that the antibody locks two-chain uPA in a conformation that does not permit stabilization of the transition state and, thus, effective catalysis. The notion of mAb-112 stabilizing an otherwise sparsely populated conformational state of two-chain uPA is supported by the fact that the difference in mAb-112 affinity to pro-uPA and active uPA is largely due to a difference in the association rate constant. Although the derivatization of active uPA with a chloromethyl ketone inhibitor further reduces the affinity of mAb-112, derivatization of pro-uPA with the same inhibitor has no effect on the high affinity of mAb-112. A ready explanation of this contrasting observation is that although pro-uPA was labeled with a transition state inhibitor, cleavage of the activation loop at the Lys15-Ile16 bond is a necessary requirement for maintaining the full organization of the mature active site architecture. Support for this view is illustrated by Fig. 6, A and B, which clearly shows a distinct fluorescence spectrum for dansyl-EGR-chloromethyl ketone-labeled pro-uPA relative to that of the labeled two-chain enzyme after proteolysis and activation by plasmin.
When viewed in its entirety, our data suggest that the catalytic domain of uPA is flexible, existing in conformational equilibrium between inactive and active states and that mAb-112 binds to and sequesters uPA in a catalytically deficient conformation. Fig. 9 provides a graphical representation of the proposed inhibitory mechanism of mAb-112.
FIGURE 9.

Schematic summarization of the inhibitory mechanism of mAb-112. Both the zymogen form of uPA (pro-uPA) and active two-chain uPA are expected to exist in conformational equilibrium between active and inactive states. With pro-uPA the equilibrium is predominantly shifted toward the inactive conformation of the active site, whereas the opposite is true for two-chain uPA where the equilibrium is shifted toward an active conformation. When bound to single-chain pro-uPA, mAb-112 acts to stabilize the conformation toward the inactive one. In two-chain uPA, mAb-112 stabilizes the protein in an inactive conformation, which is unable to stabilize the transition state. mAb-112 also sterically hinders the access of proteases to the activation bond at Lys15-Ile16 and thereby delays the proteolytic conversion of one-chain pro-uPA to two-chain uPA. SPD, serine protease domain; K, Kringle domain; G, growth factor Domain.
The D194A variant has a reduced binding to mAb-112, indicating that interaction with the buried side chain of Asp194 likely contributes to stabilization of the autolysis loop in a zymogen-like conformation. The observation provides a ready explanation of the effect of mAb-112 on the active site, as the orientation of the side chain of Asp194 is decisive for the oxyanion hole being in an active or an inactive conformation. This finding is also rather consistent with the work of Pasternak et al. (27), demonstrating that trypsinogen variants of Asp194 gain enzymatic activity without proteolytic processing. Due to the lack of knowledge of the three-dimensional structure of pro-uPA, we do not know exactly how Asp194 links to the autolysis loop. In active uPA, however, the γ-carboxylate of Asp194, besides salt bridging to the amino group of Ile16, also participates in a hydrogen bonding network involving the main chain amino group of Gly142 and the main chain carbonyl group of Lys143. The strong effect on binding of mAb-112 to the pro-uPA variants with the K156A and F141A-G142A-K143A mutations, implicating buried residues, is also likely to be caused by conformational effects on the surface presentation of the epitope. This conclusion is also consistent with Lys156 forming a salt bridge to Glu144 (22).
Dennis et al. (28) described the isolation of a disulfide-bridged constrained peptide from a phage-displayed peptide library targeting the serine protease FVIIa by a non-competitive mechanism. Crystallographic analysis demonstrated that peptide binding induced a structural alteration within the autolysis loop, involving disruption of a hydrogen bond between Gln143 and Lys192, probably leading to deformation of the oxyanion hole. Such an inhibition mechanism bears resemblance to the present effect of mAb-112 on active uPA. Also, our finding is another example of a substrate-targeting protease inhibitor, recently described in the case of γ-secretase and its substrate the β-amyloid precursor protein (29). In our case the proteases are plasmin or matriptase, and the substrate is pro-uPA. Small molecule inhibitors of the cysteine proteases caspase-1, -3, and -7 have been identified that bind a common allosteric site and prevent the conformational change associated with zymogen activation; however, these function by a mechanism distinct from mAb-112 (30, 31).
The efficacy of mAb-112 at regulating uPA-dependent plasminogen activation in vivo was investigated in the CAM model. A critical role for pro-uPA activation in tumor cell intravasation was established by exploiting high and low disseminating variants of HT-1080 fibrosarcoma cells. Whereas both cell types produced comparable amounts of pro-uPA, only the HT-hi/diss cells converted the inactive zymogen to the active two-chain protease, and activation of pro-uPA appeared to be the rate-limiting step during spontaneous tumor cell dissemination in the chick embryo (10). Our present study demonstrates that mAb-112 is able to efficiently inhibit tumor cell intravasation in vivo. The finding that the efficacy of intravasation inhibition by mAb-112 depended on the time of application indicates that uPA is functionally involved starting at early stages of tumor cell dissemination, which includes the intravasation step. Anti-uPA mAb-2 was also found to inhibit intravasation of HT-hi/diss cells although less effectively than mAb-112. Anti-uPA mAb-2 inhibits plasminogen activation by two-chain uPA with a Ki very similar to the KD for binding of mAb-112 to pro-uPA, both being in the subnanomolar range, but does not inhibit pro-uPA activation. The fact that anti-uPA mAb-2 is even slightly less efficient in inhibiting intravasation than mAb-112 makes it highly unlikely that the direct effect of mAb-112 on plasminogen activation, having a Ki of 164 nm, could contribute to the effect of this antibody on intravasation. However, the overall outcome of mAb-112, pronouncedly inhibiting activation of pro-uPA, in fact prevents plasminogen conversion to plasmin, the latter being possibly the actual effector serine protease in the intravasation step. The involvement of plasmin would be consistent with moderate inhibition of tumor cell intravasation by aprotinin along with partial inhibition by mAb-2 and substantial inhibition by mAb-112, as this antibody would block the initial step in the serine protease cascade. It is also unlikely that the observed inhibition of intravasation by mAb-112 was caused by inhibition of activation of chicken pro-uPA, as only one of the residues in the epitope, Tyr151, is conserved in chicken uPA. It should be noted, however, that in the in vivo CAM model, active human uPA is capable of activating chicken plasminogen, which would be readily available due to significant plasma leakage from the angiogenic CAM vasculature induced within HT-hi/diss primary tumors.
Initially, this model was used to demonstrate the functional role of uPA in tumor cell invasion of the CAM and metastasis to the lungs (32). In a later work, Ossowski (33) demonstrated that the uPA needs to be bound to its cell surface receptor uPAR to be able to stimulate invasion of human tumor cells into the CAM stroma. However, binding of uPA to uPAR may not only support the plasminogen activation activity of uPA but also have another effect on human cancer cells grafted on the CAM. Binding of the proteolytically inactive pro-uPA to uPAR stimulated growth of human tumors developing on the CAM through uPAR-mediated mechanisms independent of enzymatic activity of uPA (34, 35). The functional role of uPA in vivo, therefore, could be dual, i.e. manifested both in promoting tumor cell proliferation and in stimulating intravasation and metastatic spread. It is noteworthy that anti-uPA mAb-112 and mAb-2 did not inhibit primary tumor growth, which is consistent with the fact that none of these antibodies affected the binding of uPA to uPAR on the cell surface. The uPA produced by the HT-hi/diss primary tumors in the presence of these mAbs can, therefore, bind to uPAR and stimulate tumor cell growth, but its proteolytic activity and, thus, intravasation, will be inhibited. In conclusion, our data effectively demonstrate the in vivo efficacy of mAb-112 in the inhibition of tumor cell functions and support our initial hypothesis that regulation of pro-uPA activation provides a robust strategy for pharmacological intervention in the early steps of cancer dissemination.
While this report was being considered for publication, Lund et al. (36) reported that a monoclonal antibody against murine uPA was able to block the activation of murine pro-uPA in vitro and to block uPA-dependent activation of an engineered anthrax toxin and uPA-dependent fibrinolysis in mice in vivo, whereas an antibody inhibiting only the enzyme activity of uPA was much less effective. It will be interesting to investigate if that antibody acts by a mechanism similar to that of mAb-112. This report independently corroborates our present conclusion that inhibition of pro-uPA activation is a feasible and effective means of inhibiting uPA-dependent processes in vivo.
Acknowledgments
The technical assistance of Anni Christensen and Signe Kristiansen is gratefully acknowledged.
This work was supported, in whole or in part, by National Institutes of Health Grants CA55852 and CA105412 (to J. P. Q.). This work was also supported by grants from the Danish Cancer Society, the Danish Cancer Research Foundation, the Danish Research Agency, the Carlsberg Foundation, the Novo-Nordisk Foundation, European Union FP6 Contract LSHC-CT-2003 h503297 (the Cancer Degradome) (to P. A. A.), and the Danish Heart Association (to G. E. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Amino acid numbering used in the text and figures refers to the standard chymotrypsinogen template numbering.
P1, P2, P3, P4, etc. and P1′,P2′,P3′,P4′, etc. are of the nomenclature of Schechter and Berger (37) and denote those residues on the amino-terminal and carboxyl sides of the scissile bond (P1-P1′), respectively.
The abbreviations used are: uPA, urokinase-type plasminogen activator; uPAR, uPA receptor; CAM, chorioallantoic membrane; CMK, chloromethylketone; mAb, monoclonal antibody; PAI-1, plasminogen activator inhibitor type-1; SPR, surface plasmon resonance; EGR, Glu-Gly-Arg.
G. E. Blouse and K. A. Bøtkjær, unpublished information.
P. A. Andreasen, unpublished data.
References
- 1.Hedstrom, L. (2002) Chem. Rev. 102 4501-4524 [DOI] [PubMed] [Google Scholar]
- 2.Huber, R., and Bode, W. (1978) Acc. Chem. Res. 11 114-122 [Google Scholar]
- 3.Madison, E. L., Kobe, A., Gething, M. J., Sambrook, J. F., and Goldsmith, E. J. (1993) Science 262 419-421 [DOI] [PubMed] [Google Scholar]
- 4.Rockway, T. W., Nienaber, V., and Giranda, V. L. (2002) Curr. Pharm. Des. 8 2541-2558 [DOI] [PubMed] [Google Scholar]
- 5.Stoppelli, M. P., Andersen, L. M., Votta, G., and Andreasen, P. A. (2008) in The Cancer Degradome-Proteases and Cancer Biology (Edwards, D. R., Høyer-Hansen, G., Blase, F., and Sloane, B. F., eds) pp. 721-758, Springer-Verlag New York Inc., New York
- 6.Andreasen, P. A., Egelund, R., and Petersen, H. H. (2000) Cell. Mol. Life Sci. 57 25-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.List, K., Jensen, O. N., Bugge, T. H., Lund, L. R., Ploug, M., Dano, K., and Behrendt, N. (2000) Biochemistry 39 508-515 [DOI] [PubMed] [Google Scholar]
- 8.Kilpatrick, L. M., Harris, R. L., Owen, K. A., Bass, R., Ghorayeb, C., Bar-Or, A., and Ellis, V. (2006) Blood 108 2616-2623 [DOI] [PubMed] [Google Scholar]
- 9.Moran, P., Li, W., Fan, B., Vij, R., Eigenbrot, C., and Kirchhofer, D. (2006) J. Biol. Chem. 281 30439-30446 [DOI] [PubMed] [Google Scholar]
- 10.Madsen, M. A., Deryugina, E. I., Niessen, S., Cravatt, B. F., and Quigley, J. P. (2006) J. Biol. Chem. 281 15997-16005 [DOI] [PubMed] [Google Scholar]
- 11.Egelund, R., Petersen, T. E., and Andreasen, P. A. (2001) Eur. J. Biochem. 268 673-685 [DOI] [PubMed] [Google Scholar]
- 12.Petersen, H. H., Hansen, M., Schousboe, S. L., and Andreasen, P. A. (2001) Eur. J. Biochem. 268 4430-4439 [DOI] [PubMed] [Google Scholar]
- 13.Jespersen, M. H., Jensen, J., Rasmussen, L. H., Ejlersen, E., Moller-Petersen, J., and Sperling-Petersen, H. U. (1993) Scand. J. Clin. Lab. Investig. 53 639-648 [DOI] [PubMed] [Google Scholar]
- 14.Knoop, A., Andreasen, P. A., Andersen, J. A., Hansen, S., Laenkholm, A. V., Simonsen, A. C., Andersen, J., Overgaard, J., and Rose, C. (1998) Br. J. Cancer 77 932-940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen, L. S., Andreasen, P. A., Grondahl-Hansen, J., Huang, J. Y., Kristensen, P., and Dano, K. (1986) Thromb. Haemostasis 55 206-212 [PubMed] [Google Scholar]
- 16.Lund, L. R., Georg, B., Nielsen, L. S., Mayer, M., Dano, K., and Andreasen, P. A. (1988) Mol. Cell. Endocrinol. 60 43-53 [DOI] [PubMed] [Google Scholar]
- 17.Behrendt, N., List, K., Andreasen, P. A., and Dano, K. (2003) Biochem. J. 371 277-287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picone, R., Kajtaniak, E. L., Nielsen, L. S., Behrendt, N., Mastronicola, M. R., Cubellis, M. V., Stoppelli, M. P., Pedersen, S., Dano, K., and Blasi, F. (1989) J. Cell Biol. 108 693-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bode, W., and Huber, R. (1976) FEBS Lett. 68 231-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bock, P. E., Day, D. E., Verhamme, I. M., Bernardo, M. M., Olson, S. T., and Shore, J. D. (1996) J. Biol. Chem. 271 1072-1080 [DOI] [PubMed] [Google Scholar]
- 21.Panizzi, P., Friedrich, R., Fuentes-Prior, P., Kroh, H. K., Briggs, J., Tans, G., Bode, W., and Bock, P. E. (2006) J. Biol. Chem. 281 1169-1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spraggon, G., Phillips, C., Nowak, U. K., Ponting, C. P., Saunders, D., Dobson, C. M., Stuart, D. I., and Jones, E. Y. (1995) Structure 3 681-691 [DOI] [PubMed] [Google Scholar]
- 23.Ossowski, L., and Reich, E. (1980) Cancer Res. 40 2300-2309 [PubMed] [Google Scholar]
- 24.Deryugina, E. I., Zijlstra, A., Partridge, J. J., Kupriyanova, T. A., Madsen, M. A., Papagiannakopoulos, T., and Quigley, J. P. (2005) Cancer Res. 65 10959-10969 [DOI] [PubMed] [Google Scholar]
- 25.Hedstrom, L., Lin, T. Y., and Fast, W. (1996) Biochemistry 35 4515-4523 [DOI] [PubMed] [Google Scholar]
- 26.Creigthon, T. E. (1993) Proteins: Structures and Molecular Properties pp. 419-428, W. H. Freeman and Co., New York
- 27.Pasternak, A., Liu, X., Lin, T. Y., and Hedstrom, L. (1998) Biochemistry 37 16201-16210 [DOI] [PubMed] [Google Scholar]
- 28.Dennis, M. S., Eigenbrot, C., Skelton, N. J., Ultsch, M. H., Santell, L., Dwyer, M. A., O'Connell, M. P., and Lazarus, R. A. (2000) Nature 404 465-470 [DOI] [PubMed] [Google Scholar]
- 29.Kukar, T. L., Ladd, T. B., Bann, M. A., Fraering, P. C., Narlawar, R., Maharvi, G. M., Healy, B., Chapman, R., Welzel, A. T., Price, R. W., Moore, B., Rangachari, V., Cusack, B., Eriksen, J., Jansen-West, K., Verbeeck, C., Yager, D., Eckman, C., Ye, W., Sagi, S., Cottrell, B. A., Torpey, J., Rosenberry, T. L., Fauq, A., Wolfe, M. S., Schmidt, B., Walsh, D. M., Koo, E. H., and Golde, T. E. (2008) Nature 453 925-929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy, J. A., Lam, J., Nguyen, J. T., O'Brien, T., and Wells, J. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 12461-12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheer, J. M., Romanowski, M. J., and Wells, J. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 7595-7600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ossowski, L., and Reich, E. (1983) Cell 35 611-619 [DOI] [PubMed] [Google Scholar]
- 33.Ossowski, L. (1988) J. Cell Biol. 107 2437-2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aguirre Ghiso, J. A., Kovalski, K., and Ossowski, L. (1999) J. Cell Biol. 147 89-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu, D., Aguirre Ghiso, J., Estrada, Y., and Ossowski, L. (2002) Cancer Cell 1 445-457 [DOI] [PubMed] [Google Scholar]
- 36.Lund, I. K., Jogi, A., Rono, B., Rasch, M. G., Lund, L. R., Almholt, K., Gardsvoll, H., Behrendt, N., Romer, J., and Hoyer-Hansen, G. (2008) J. Biol. Chem. 283 32506-32515 [DOI] [PubMed] [Google Scholar]
- 37.Abramowitz, N., Schechter, I., and Berger, A. (1967) Biochem. Biophys. Res. Commun. 29 862-867 [DOI] [PubMed] [Google Scholar]