

Abstract
Long-term estrogen deprivation (LTED) MCF-7 cells showing estrogen-independent growth, express estrogen receptor (ER) α at a much higher level than wild-type MCF-7 cells. Enhanced expression of ERα associated with partial localization of ERα to the plasma membranes in LTED cells is thought to be an important step for acquisition of estrogen-ablation resistance. In this study, we compared the regulation of ERα gene expression between wild type and LTED cells, examining the usage of the promoters A and C as well as their methylation status. We found that transcription from the promoter C was drastically enhanced in LTED cells, compared with that in wild-type cells. Furthermore, the promoter C region was highly unmethylated in LTED cells, but partially methylated in wild-type cells. Our findings imply that demethylation of promoter C region in the ERα gene is in part responsible for the enhanced expression of ERα gene in LTED cells.
Keywords: Estrogen receptor α, Breast cancer, Methylation
1. Introduction
Experimental, clinical, and epidemiologic data suggest that estrogens contribute to the development of breast cancer. Estrogens bind to ERα or β and stimulate the transcription of target genes involved in cell proliferation. Thus, the anti-estrogen therapy such as tamoxifen has been generally used for ERα-positive breast cancer for several decades. Recently, clinical trials in the adjuvant, neoadjuvant and advanced disease setting have demonstrated a greater clinical efficacy of the aromatase inhibitors aiming to decrease the concentration of estrogen, compared with selective estrogen receptor modulators represented by tamoxifen [1,2]. On the other hand, clinical observations suggested that some human breast cancers adapted to hormone-ablative therapy involving surgically deprivation of estrogen production. Hormone-dependent breast cancers often regress in response to surgical removal of the ovaries, a treatment which lower circulating plasma estradiol (E2) from approximately 200–15 pg/ml [3]. In response to this acute deprivation of E2, tumors regress for 12–18 months on average before they begin to regrow. Second-line therapy with surgical oophorectomy or with aromatase inhibitors can then induce additional tumor regression by lowering E2 concentrations further to 1–5 pg/ml [4]. These observations for the first time demonstrated enhanced sensitivity to circulating E2.
In order to demonstrate the phenomenon of adaptive hypersensitivity and to determine the mechanisms involved, we have established a model system involving MCF-7 human breast cancer cells in vitro. Wild-type MCF-7 cells were cultured over a prolonged period in estrogen-free medium to mimic the effect of ablative endocrine therapy such as induced by surgical oophorectomy [5]. This process involves long-term E2 deprivation; the adapted cells are called LTED cells. When LTED cells were cultured in the presence of E2 for 4 months, the cells showed estrogen-dependent growth as was observed for wild-type MCF-7 cells [5].
Importantly, ERα is expressed at a much higher level in LTED cells than in wild-type MCF-7 cells [6]. In concert with this observation, an elevated basal ER transactivation activity (mean increase; 5-fold) was measured in LTED cells compared with the wild-type cells using pERE-tk-CAT, a reporter gene driven by estrogen responsive element-thymidine kinase promoter [6].
In addition to the established role as a nuclear receptor, ERα may have another function on the plasma membrane. In LTED cells, ERα localizes predominantly to the nuclei and some also present on the plasma membranes [7]. The sub-cellular localization of ERα to the plasma membranes in LTED cells may, at least in part, be due to the enhanced expression of ERα in LTED cells, since plasma membrane-associated ERα can only be observed in ERα-enriched MCF-7 sub-cell lines (mERhigh) but not ERα-depleted ones (mERαlow) [8]. Constitutively activated MAP kinase activity was observed in LTED cells independent of serum factors [9]. A rapid physical interaction of the plasma membrane-associated ERα and an adaptor protein Shc has been observed upon addition of E2 [7]; Shc is subsequently phosphorylated and triggers the MAP kinase signaling pathway [10,11].
Thus, when ERα works as a transcription factor in the nuclei and also as a signal transducer on the plasma membranes, typically in LTED cells, enhanced expression of ERα may be an obligatory step for acquisition of estrogen-ablation resistance. Though, the mechanisms how ERα expresses at high level in LTED cells still remain unknown.
Several human ERα gene promoters (A–F) are identified so far [12]. These promoters are differently utilized in a tissue-and cell-dependent manner [13–15]. Among these promoters, we previously demonstrated that the transcript from promoter A was constitutively used in both normal and cancerous mammary tissue, while the transcript from promoter C (formerly called promoter B) showed remarkable correlation to the ERα protein levels in ERα-positive breast cancer [16]. Furthermore, we have identified a cis-acting element, ERBF-1, that plays an important role in the expression of the ERα gene transcribed from promoter C in breast cancer cells [17]. On the other hand, a transcription factor ERF-1, a member of AP2 transcription factor, is important for the transcriptional regulation of promoter A [18,19]. In addition, methylation of the promoter A and C regions was critical for the repression of gene transcription from these promoters [20]. Collectively, methylation of these promoter regions as well as alteration of critical transcription factors are thought to be important for ERα gene expression in breast cancer cells.
In this study, we examined regulation of ERα gene expression in wild-type MCF-7 and LTED cells. We first found that transcription from the promoter C was drastically enhanced in LTED cells, compared with that in the wild-type cells. Transient transfection with a reporter gene driven by the promoter A or promoter C of ERα gene revealed that transcription factors are equally available in these cells. Second, differences in epigenetic alterations of promoter C were found between LTED and wild-type cells: The promoter C region was highly unmethylated in LTED cells, while that in wild-type cells was partially methylated. Our findings imply that demethylation of promoter C region in the ERα gene is in part responsible for the enhanced expression of ERα gene in LTED cells.
2. Materials and methods
2.1. Tissue culture
Human breast cancer cells wild-type MCF-7 were maintained in improved MEM (IMEM) containing 5% dextran-coated charcoal-stripped fetal bovine serum (DCC-FBS) and 10 nM E2. LTED cells were established by long-term culture of wild-type MCF-7 cells in IMEM containing 5% DCC-FBS. The established LTED cells were stored in liquid nitrogen until use. LTED cells were maintained in IMEM containing 5% DCC-FBS.
2.2. Plasmid
Reporter plasmids pGL3-ProA 1.3K and pGL3-ProC (formerly called pGL3-ProB1.4K) were described previously [20]. An internal control pRL-TK was purchased from Promega (Madison, WI).
2.3. RNA extraction and cDNA synthesis
Total RNA was prepared from wild type and LTED cells using RNeasy Mini kit (QIAGEN, Hilden, Germany). One μg of total RNA was reverse transcribed with Quantitect Reverse Transcription (QIAGEN) using RT primer mix as primers in a final volume of 20 μl at 42 °C for 15 min.
2.4. Real-time PCR analysis of ERα mRNA expression
The real-time PCR was performed in triplicate using iCycler iQ (Bio-Rad Laboratories, Hercules, CA). Reaction mixture consisted of 1 μl of cDNA products, 0.2 μM of each primers and 12.5 μl of SYBR Green ROX Mix (ABgene, Epsom, UK) in a total volume of 25 μl. PCR thermal conditions were as following: 95 °C for 15 min for 1 cycle and 95°C for 20 s, 60 °C for 15 s, 72 °C for 10 s, and 86 °C for 15 s (fluorescent signal collection) for 50 cycles for detection of ERα mRNA from promoters A and C; 95 °C for 15 min for 1 cycle and 94 °C for 15 s, 68 °C for 30 s, and 86 °C for 15 s (fluorescent signal collection) for 50 cycles for detection of total ERα mRNA. The following primers were used: PROA1, 5′-ACC TCG GGC TGT GCT CTT-3′ and PRODW, 5′-GAG GGT CAT GGT CAT GGT-3′ for ERα mRNA from promoter A; PROB3, 5′-GCC CAG GAA CAT TTC TGG AA-3′ and PRODW for ERα mRNA from promoter C; EREX1, 5′-AGA ACG AGC CCA GCG GCT AC-3′ and EREX2-R, 5′-CCT TGC AGC CCT CAC AGG AC-3′ for total ERα mRNA. For construction of standard curves, serially diluted plasmids harboring fragment of target gene sequences were used after digestion with Not I restriction enzyme to release the insert fragments. As a control, β-actin mRNA was also measured as described previously [21].
2.5. Transient transfection assays
A transient transfection of the plasmids was performed in triplicate using SuperFect Transfection Reagent (QIAGEN) under manufacturer’s instruction. Briefly, 1 × 105 cells were plated onto 24 wells plastic dish. After over night culture, 1 μg of pGL3-ProA1.3K or pGL3-ProC and 0.1 μg of pRL-TK were mixed with 5 μl of the SuperFect Transfection Reagent in 350 μl of the medium with 5% DCC-FBS and subjected to transfection. After 2 h incubation, the medium was replaced with a fresh medium and cells were incubated for 48 h. Then, cells were collected and lysed using Passive Lysis Buffer (Promega). Luciferase assays were performed using Dual-Luciferase Reporter Assay System (Promega) and TD-20/20 Luminometer (TURNER DESIGNS, CA).
2.6. Bisulfite modification and methylation-specific PCR
Total DNA was extracted from wild type and LTED cells using NucleoSpin Tissue (MACHEREY-NAGEL, Düren, Germany). DNA was subjected to bisulfite conversion using EZ DNA Methylation kit (ZYMO Research, Orange, CA) according to the manufacture’s instruction, essentially based on the report by Herman et al. [22]. Briefly, after denaturation with NaOH, 500 ng of DNA was incubated in a buffer containing sodium bisulfite at 50 °C for 16 h, followed by purification using ZYMO-Spin I column. Bisulfite-converted DNA was eluted in 10 μl of M-Elution buffer. First PCR amplification aiming to amplify bisulfite-converted DNA fragments was performed in 20 μl of reaction mixture containing 1 μl of bisulfite-treated genomic DNA, 0.5 μM each primers, 0.2 mM dNTPs, 2.5 mM MgCl2 and 1.25 units of AmpliTaq Gold DNA polymerase (Applied Biosystem, Foster, CA) under the following conditions: 95 °C for 5 min for 1 cycle and 95 °C for 15 s, 54 °C for 15 s, and 72 °C for 1 min for 35 cycles. Second PCR amplification using methylation-or unmethylation-specific primers was performed in 20 μl of reaction mixture containing 1 μl of first PCR product (1/100 diluted with MilliQ water), 0.5 μM each primers, 0.2 mM dNTPs, 2.5 mM MgCl2 and 1.25 units of AmpliTaq Gold DNA polymerase under the following conditions: 95 °C for 5 min for 1 cycle and 95 °C for 15 s, 57 °C (methylation) or 54 °C (unmethylation) for 30 s, and 72 °C for 20 s for 35 cycles. The amplified fragments were electrophoresed to 8% polyacrylamide gel. All the primers used for this methylation analysis were designed using MethPrimer [23]. Primers used for the first PCR and second PCR are summarized in Table 1.
Table 1.
Primers used for the methylation-specific PCR
| Promoter | PCR | Specification | Primer name | Primer sequence | Annealing temperature (°C) |
|---|---|---|---|---|---|
| A | 1st | Bisulfite conversion specific | ERPROABSF | 5′-TTAATGTTAGGGTAAGGTAATAGTTTT-3′ | 54 |
| ERPROABSR | 5′-AACCACCTAAAAAAAAAACACAA-3′ | ||||
| 2nd | Methylation specific | ERPROAM1F | 5′-AGTTTAGGAGTTGGCGGAGGGC-3′ | 60 | |
| ERPROAM1R | 5′-CCGAAATTAAAAACGACGCAACG-3′ | ||||
| Unmethylation specific | ERPROAU1F | 5′-GGGAGTTTAGGAGTTGGTGGAGGGT-3′ | 60 | ||
| ERPROAU1R | 5′-ACCCAAAATTAAAAACAACACAACACA-3′ | ||||
| C | 1st | Bisulfite conversion specific | ERPROCBSF | 5′-AGTAGATAGTAAGTTTTTTTTTATTTTTT-3′ | 54 |
| ERPROCBSR | 5′-AAAAACAACCAATAAACAAAA-3′ | ||||
| 2nd | Methylation specific | ERPROCM1F | 5′-TTTTTTATTGTTATTTATTTAGCGT-3′ | 57 | |
| ERPROCM1R | 5′-AAAACACTTAACAACCCCTCCCGAC-3′ | ||||
| Unmethylation specific | ERPROCU1F | 5′-TATTTTTTATTGTTATTTATTTAGTGT-3′ | 54 | ||
| ERPROCU1R | 5′-AAACACTTAACAACCCCTCCCAAC-3′ |
For quantification, real-time PCR amplification was performed in 25 μl of reaction mixture containing 1 μl of the first PCR product (1/100 diluted with MilliQ water), 0.2 μM each primers and 12.5 μl of SYBR Green Rox Mix according to the manufacturer’s protocol using iCycler iQ under the following conditions: 95 °C for 15 min for 1 cycle and 95 °C for 15 s, 57 °C (methyl) or 54 °C (unmethyl) for 15 s, 72 °C for 15 s, and fluorescent signal collection at 77 °C (methyl) or 76 °C (unmethyl) for 20 s for 40 cycles. The assay was conducted in triplicate and repeated for three times. For construction of a standard curve, serially diluted control PCR fragments were used.
2.7. Bisulfite-sequencing and combined bisulfite restriction analysis (COBRA)
PCR amplification of ERα promoter C region using bisulfite-converted DNA fragments was performed as described above except for the cycle numbers to be 50. The amplified fragments were purified using NucleoSpin Extract II kit (MACHEREY-NAGEL). Direct sequencing of the fragments was conducted using ERPROCBSF as a primer, BigDye Terminators v1.1 Cycle Sequencing Kit and ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). For COBRA, 5 μl of purified DNA fragment was digested in a total volume of 20 μl using 5 units of Hpy188III restriction enzyme (New England BioLabs, Ipswich, MA) that cleave CpG sites retained because of methylation at 37 °C for 4 h. The resultant DNA fragments together with undigested ones were electrophoresed onto 8% polyacrylamide gel. After staining with ethidium bromide, the image was visualized under UV illumination. Intensity of the fluorescence of each band was quantified using ChemImager 5500 (Alpha Innotech, San Leandro, CA).
2.8. Statistical analysis
Student’s t-test was conducted for statistical analysis. When necessary, the t-test was modified to all for unequal variances. P-values less than 0.05 were considered as significant.
3. Results
3.1. Expression levels of ERα mRNA in wild-type and LTED cells
We previously demonstrated that ERα protein and mRNA were expressed at higher levels in LTED cells than in wild-type cells using Western and Northern blot analyses [6]. As a first step, we conducted real-time PCR analysis to quantitatively evaluate ERα mRNA expression in wild-type and LTED cells: LTED cells showed 27-fold total ERα mRNA expression levels as compared with wild-type cells did (P = 0.004) (Fig. 1).
Fig. 1.
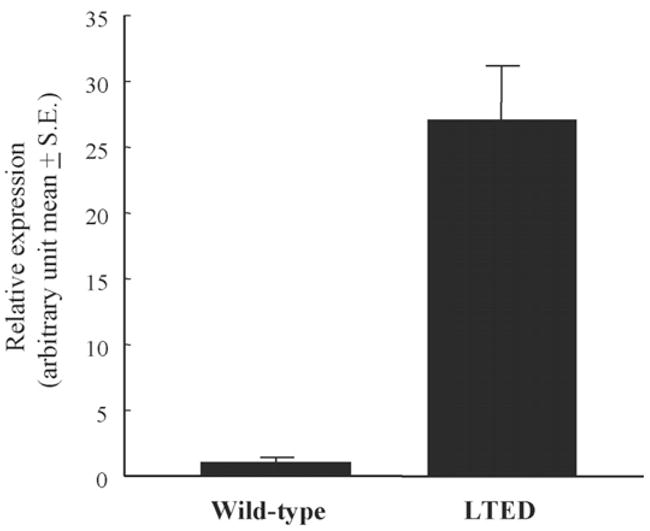
Relative expression of total ERα mRNA in wild type and LTED cells. Real-time RT-PCR was conducted for quantification of total ERα and β-actin mRNA as described in Section 2. Expression levels of total ERα mRNA were normalized using β-actin mRNA.
Since an increase of ERα mRNA from promoter C was responsible for the enhanced expression of ERα protein in ERα-positive primary breast cancer and since promoter A was constitutively utilized in both normal and cancerous mammary tissue [16], we next compared ERα mRNA expression levels transcribed from these promoters A and C between wild-type and LTED cells using real-time PCR (Fig. 2). ERα mRNA from promoter A in LTED cells was 35-fold higher than that in wild-type cells (P = 0.0007) (Fig. 2). Furthermore, ERα mRNA expression level from promoter C in LTED was 149-fold higher than that in wild-type cells (P = 0.01) (Fig. 2). In wild-type cells, promoter A was dominant while promoters A and C were equally utilized in LTED cells (Fig. 2). These results indicate that reinforced utilization of promoter C in LTED cells as compared with wild-type cells may be important for the enhanced expression of ERα in LTED cells.
Fig. 2.

Expression of ERα mRNA transcribed from promoters A and C in wild-type and LTED cells. Schematic representation of a part of ERα gene organization is shown above. The transcription start site of promoter A is defined as +1. Relative expression of ERα mRNA from promoters A and C in wild-type and LTED cells is shown. Expression levels of ERα mRNA from promoters A and C were quantified by real-time RT-PCR as described in Section 2 being normalized by β-actin mRNA.
3.2. Transient transfection of reporter gene constructs with ERα gene promoters
The cis- and trans-acting factors are thought to generate differences in transcription activity on various promoters. We first tested the possibility that alterations of transcription factors may generate an increased level of ERα expression in LTED cells, compared with wild-type cells. We then measured the promoter activities in wild type and LTED cells using reporter gene constructs driven by ERα promoters A and C. No significant differences in promoter activities between wild-type and LTED cells were observed for these two promoters (P = 0.3 and 0.1 for promoters A and C, respectively, Fig. 3), suggesting that trans-acting factors specific to promoters A and C are not responsible for the different utilization of ERα promoters A and C between wild-type and LTED cells.
Fig. 3.

Reporter activities of promoters A and C in wild-type and LTED cells. Wild-type and LTED cells were transiently transfected with a reporter gene construct pGL3-ProA 1.3K or pGL3-ProC together with control vector pRL-TK as described in Section 2. The measured luciferase activities were normalized using the control Renilla luciferase activity.
3.3. Different methylation status of ERα gene promoters in wild-type and LTED cells
We next hypothesized that alternations of higher order chromatin structure caused by DNA methylation may contribute to the expression level of ERα in these cells. Then, we compared methylation status of ERα gene promoters A and C in wild-type and LTED cells by methylation-specific PCR. Promoter A of ERα gene was unmethylated in both wild-type and LTED cells (Fig. 4). On the other hand, promoter C of ERα gene showed partial methylation in wild-type cells (Fig. 4), in good agreement with our previous report [20]. In LTED cells, the unmethylated band for promoter C was clearly observed, while the methylated one was considerably weaker (Fig. 4), in accordance with our observation that promoter C was actively utilized in LTED cells.
Fig. 4.

Methylation-specific PCR analysis of ERα gene promoters A and C in wild-type and LTED cells. Location of primers used in methylation-specific PCR for promoters A and C is shown in the left. Photos of electrophoresis of PCR products of methylated (M) or unmethylated (UM) DNA fragments are shown in the right.
In order to confirm the difference of methylation status of the promoter C between wild-type and LTED cells, we next conducted direct sequencing of the PCR fragment amplified from bisulfite-converted DNA. Cytosines were predominantly observed at nucleotides −2103, −2082, and −2073 within CpG dinucleotides, while thymines are faintly detected at these sites in wild-type cells (Fig. 5). On the other hand, both thymines and cytosines were clearly observed at these sites in LTED cells (Fig. 5), confirming the difference of methylation status between wild-type and LTED cells.
Fig. 5.

Direct sequencing of PCR products of ERα gene promoter C using bisulfite-converted DNA from wild-type and LTED cells. First PCR products of bisulfite-converted DNA from wild-type or LTED cells using primers ERPROCBSF and ERPROCBSR were subjected to direct sequencing with ERPROCBSF primer as described in Section 2. Three CpG sites within the amplified region are indicated by rectangles with numbering over the rectangle. Cytosine residues that will be shown as thymine in the sequence of PCR product of bisulfite converted DNA are marked with vertical arrows in black. Thick arrows indicate the positions of three CpG sites.
3.4. Quantitative analysis of methylation status of ERα gene promoter C in wild-type and LTED cells
In order to quantitatively analyze the methylation of the promoter C region, we next performed two different analyses, i.e. COBRA assay and bisulfite-real-time PCR. In COBRA assay, the fragment amplified from methylated DNA can be identified as digestible bands with restriction enzyme Hpy188III, because of the retention of methyl-cytosine residue at −2073 even after bisulfite-treatment (Fig. 6). Quantification of the fragments revealed that 54% of the fragments were methylated in wild-type cells, while only 7.7% were methylated in LTED cells (Fig. 6).
Fig. 6.

COBRA assay of ERα gene promoter C in wild-type and LTED cells. First PCR products of bisulfite converted DNA from wild-type or LTED cells using primers ERPROCBSF and ERPROCBSR were digested with restriction enzyme Hpy188III as described in Section 2. Rectangle indicates the position of the CpG site to be tested. Underline indicates the recognition site of Hpy188III that will be generated by bisulfite conversion of methylated DNA, but not unmethylated one. An image of the polyacrylamide gel electrophoresis is shown below. U, untreated. D, digested with Hpy188III.
This was also examined with bisulfite-real-time PCR analysis using methylation- or unmethylation-specific primers as described above. Amount of methylated DNA in the tested sample from LTED cells was 42% of that from wild-type cells, though this was not statistically significant (P = 0.063) (Fig. 7). On the other hand, unmethylated DNA amount was 14.2-fold in LTED cells as compared with wild-type cells (P < 0.001) (Fig. 7). These results demonstrated that the promoter C region was highly unmethylated in LTED cells, but partially methylated in wild-type cells.
Fig. 7.

Bisulfite-real-time PCR analysis of ERα gene promoter C in wild-type and LTED cells. Bisulfite-real-time PCR analysis based on SYBR-Green chemistry was conducted using methylation-specific primers ERPROCM1F and ERPROCM1R or unmethylation-specific primers ERPROCUM1F and ERPROCUM1R as described in Section 2. Representative results of bisulfite-real-time PCR are shown above. Comparison of methylated or unmethylated DNA amounts in tested samples from wild-type and LTED cells are shown below. S.D.: standard deviation.
4. Discussion
Clinical observations suggest that human breast tumors can adapt to endocrine therapy by developing hypersensitivity to estrogen. To understand the mechanisms underlying this, we have previously examined estrogenic stimulation of cell proliferation in a model system and provided in vitro and in vivo evidence that long-term E2 ablation causes adaptive hypersensitivity. Importantly, LTED cells express ERα at a much higher level than wild cells and consequently show sub-cellular localization of ERα to the plasma membrane in addition to the nucleus. LTED cells showed higher basal estrogen responsive transcription activity as compared with wild-type cells. In addition, LTED cells activate MAP kinase signaling pathway mediated by plasma membrane-bound ERα. In both roles of ERα in the nuclei and on the plasma membranes of LTED cells, enhanced expression of ERα protein may be an obligatory step for acquisition of estrogen-ablation resistance.
Previously, the expression level of ERα transcript from promoter A was reported to be several folds higher than that from promoter C in MCF-7 cells [24,25], in good agreement with our current study for wild-type MCF-7 cells (Fig. 2). On the other hand, in this study, we for the first time demonstrated that ERα transcripts from promoters A and C expressed at high levels in LTED cells as compared with wild-type cells using real-time PCR (Fig. 2). Specifically, transcripts from promoter C in LTED cells were 149-fold higher than those in wild-type cells and the transcripts from promoter A and C were equally expressed in LTED cells (Fig. 2), indicating that enhanced transcription of ERα gene from promoter C may in part be responsible for the enhanced expression of ERα protein in LTED cells.
Our transient transfection experiments indicated that alterations of transcription factors may not be a major cause for the enhanced expression of ERα mRNA from promoter C in LTED cells (Fig. 3). On the other hand, methylation status of the promoter C region of ERα gene was drastically changed in LTED cells as compared with wild-type cells (Figs. 4–7). We have previously reported that specific in vitro methylation of promoter C region of ERα gene significantly reduced its promoter activity in transient transfection experiment [20]. In addition, methylation of promoter C was inversely associated with expression of transcripts from this promoter as well as ERα protein in primary breast cancer [20]. Taken together, hypomethylation of promoter C region in LTED cells can be one of the mechanisms responsible for the enhanced transcription of ERα gene from promoter C.
In this study, we demonstrated that ERα transcript from promoter A also expressed at high levels in LTED cells as compared with wild-type cells (Fig. 2). Since the promoter A was highly unmethylated in both LTED and wild-type MCF-7 cells (Fig. 4), some mechanisms other than DNA methylation should be involved in the regulation of promoter A in LTED cells. Our transient transfection experiments did not show any difference of the reporter gene activity of promoter A constructs in between LTED and wild-type MCF-7 cells (Fig. 3). One possible explanation for the enhanced expression of the transcript from promoter A in LTED cells could be alteration of transcription factors that bind to downstream sequences including intron of the ERα gene that are not present in our tested construct; AP2 transcription factor that bind to the ERF-1 cis-acting element located downstream of the transcription start site of promoter A may be one of the candidates [18,19].
Transcripts from promoter A and C share an identical coding sequence for ERα protein. Then, is there any functional difference between these transcripts in relation to the protein level of ERα? Importantly, it has recently been shown that 5′ upstream open reading frame (ORF) that is specific to the transcript A has inhibitory effects on the translation of ERα protein, while the 5′ upstream ORF of transcript C did not show the effect [26]. Notably, the inhibitory effect of the ORF of transcript A was most remarkable in MCF-7 cells among the tested cell lines, indicating that there are differences in the translational potential of transcripts from these two major promoters [26]. It would be interesting to test whether such regulatory effects of upstream ORF specific to transcript A is also present in MCF-7 LTED cells.
In conclusion, we found that transcription from promoters A and C were highly activated in LTED cells. Specifically, hypomethylation of promoter C correlated well with its drastically enhanced expression in LTED cells, suggesting that epigenetic alterations may play some roles in enhanced expression of ERα important for the acquisition of estrogen-ablation resistance of breast cancer cells.
Acknowledgments
We thank Ms. Shoko Hirano for her excellent technical assistance. This work was supported in part by a Grant-in-Aid for Science, Ministry of Education, Culture, Sports, Science, and Technology, Japan.
References
- 1.Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 2004;5(3):207–213. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 2.Yue W, Wang JP, Li Y, Bocchinfuso WP, Korach KS, Devanesan PD, Rogan E, Cavalieri E, Santen RJ. Tamoxifen versus aromatase inhibitors for breast cancer prevention. Clin Cancer Res. 2005;11(2):925–930. [PubMed] [Google Scholar]
- 3.Santen RJ, Harvey HA. Use of aromatase inhibitors in breast carcinoma. Endocr Relat Cancer. 1999;6(1):75–92. doi: 10.1677/erc.0.0060075. [DOI] [PubMed] [Google Scholar]
- 4.Santen RJ, Manni A, Harvey HA, Redmond C. Endocrine treatment of breast cancer in woman. Endocr Rev. 1990;11(2):221–265. doi: 10.1210/edrv-11-2-221. [DOI] [PubMed] [Google Scholar]
- 5.Masamura S, Santner SJ, Heitjan DF, Santen RJ. Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 1995;80(10):2918–2925. doi: 10.1210/jcem.80.10.7559875. [DOI] [PubMed] [Google Scholar]
- 6.Jeng MH, Shupnik MA, Bender TP, Westin EH, Bandyopadhyay D, Kumar R, Masamura S, Santen RJ. Estrogen receptor expression and function in long-term estrogen-deprived human breast cancer cells. Endocrinology. 1998;139(10):4164–4174. doi: 10.1210/endo.139.10.6229. [DOI] [PubMed] [Google Scholar]
- 7.Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, Santen RJ. Linkage of rapid estrogen action to MAPK activation by ERα-Shc association and Shc pathway activation. Mol Endocrinol. 2002;16(1):116–127. doi: 10.1210/mend.16.1.0748. [DOI] [PubMed] [Google Scholar]
- 8.Zivadinovic D, Watson CS. Membrane estrogen receptor-α levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005;7(1):130–144. doi: 10.1186/bcr959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yue W, Wang JP, Conaway M, Masamura S, Li Y, Santen RJ. Activation of the MAPK pathway enhances sensitivity of MCF-7 breast cancer cells to the mitogenic effort of estradiol. Endocrinology. 2002;143(9):3221–3229. doi: 10.1210/en.2002-220186. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Z, Kumar R, Santen RJ, Song RX. The role of adapter protein Shc in estrogen non-genomic action. Steroids. 2004;69(89):523–529. doi: 10.1016/j.steroids.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 11.Santen RJ, Song RX, Zhang Z, Kumar R, Jeng MH, Masamura S, Lawrence J, Berstein L, Yue W. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer. 2005;12(Suppl 1):61–73. doi: 10.1677/erc.1.01018. [DOI] [PubMed] [Google Scholar]
- 12.Koš M, Reid G, Denger S, Gannon F. Minireview: genomic organization of the human ERα gene promoter region. Mol Endocrinol. 2001;15(12):2057–2063. doi: 10.1210/mend.15.12.0731. [DOI] [PubMed] [Google Scholar]
- 13.Flouriot G, Griffin C, Kenealy M, Sonntag-Buck V, Gannon F. Differentially expressed messenger RNA isoforms of the human estrogen receptor-α gene are generated by alternative splicing and promoter usage. Mol Endocrinol. 1998;12(12):1939–1954. doi: 10.1210/mend.12.12.0209. [DOI] [PubMed] [Google Scholar]
- 14.Malyala A, Kelly MJ, Rønnekleiv OK. Estrogen modulation of hypothalamic neurons: activation of multiple signaling pathways and gene expression changes. Steroids. 2005;70(5–7):397–406. doi: 10.1016/j.steroids.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Österlund KM, Grandied K, Keller E, Hurd YL. The human brain has distinct regional expression patterns of estrogen receptor α mRNA isoforms derived from alternative promoters. J Neurochem. 2000;75(4):1390–1397. doi: 10.1046/j.1471-4159.2000.0751390.x. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi S, Imai K, Suga K, Kurihara T, Higashi Y, Nakachi K. Two promoters in expression of estrogen receptor messenger RNA in human breast cancer. Carcinogenesis. 1997;18(3):459–464. doi: 10.1093/carcin/18.3.459. [DOI] [PubMed] [Google Scholar]
- 17.Tanimoto K, Eguchi H, Yoshida T, Hajiro-Nakanishi K, Hayashi S. Regulation of estrogen receptor α gene mediated by promoter B responsible for its enhanced expression in human breast cancer. Nucl Acids Res. 1999;27(3):903–909. doi: 10.1093/nar/27.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.deConinck EC, McPherson LA, Weigel RJ. Transcriptional regulation of estrogen receptor in breast carcinomas. Mol Cell Biol. 1995;15(4):2191–2196. doi: 10.1128/mcb.15.4.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPherson LA, Baichwal VR, Weigel RJ. Identification of ERF-1 as a member of the AP2 transcription factor family. Proc Natl Acad Sci USA. 1997;94(9):4342–4347. doi: 10.1073/pnas.94.9.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida T, Eguchi H, Nakachi K, Tanimoto K, Higashi Y, Suemasu K, Iino Y, Morishita Y, Hayashi S. Distinct mechanisms of loss of estrogen receptor α gene expression in human breast cancer: methylation of the gene and alteration of trans-acting factor. Carcinogenesis. 2000;21(12):2193–2201. doi: 10.1093/carcin/21.12.2193. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida N, Omoto Y, Inoue A, Eguchi H, Kobayashi Y, Kurosumi M, Saji S, Suemasu K, Okazaki T, Nakachi K, Fujita T, Hayashi S. Prediction of prognosis of estrogen receptor-positive breast cancer with combination of selective estrogen-regulated genes. Cancer Sci. 2004;95(6):496–502. doi: 10.1111/j.1349-7006.2004.tb03239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman GJ, Graff RJ, MyöHänen S, Nelkin DB, Baylim BS. Methylation-specific, PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93(18):9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinfomatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 24.Fasco MJ. Estrogen receptor mRNA splice variants produced from the distal and proximal promoter transcripts. Mol Cell Endocrinol. 1998;138:51–59. doi: 10.1016/s0303-7207(98)00048-3. [DOI] [PubMed] [Google Scholar]
- 25.Weigel RJ, Crooks DL, Iglehart JD, deConinck EC. Quantitative analysis of the transcriptional start sites of estrogen receptors in breast carcinoma. Cell Growth Differ. 1995;6:707–711. [PubMed] [Google Scholar]
- 26.Pentecost BT, Song R, Luo M, DePasquale JA, Fasco MJ. Upstream regions of the estrogen receptor alpha proximal promoter transcript regulate ER protein expression through a translational mechanism. Mol Cell Endocrinol. 2005;229:83–94. doi: 10.1016/j.mce.2004.09.002. [DOI] [PubMed] [Google Scholar]