

Abstract
The epithelial amiloride-sensitive sodium channel (ENaC) controls transepithelial Na+ movement in Na+-transporting epithelia and is associated with Liddle syndrome, an autosomal dominant form of salt-sensitive hypertension. Detailed analysis of ENaC channel properties and the functional consequences of mutations causing Liddle syndrome has been, so far, limited by lack of a method allowing specific and quantitative detection of cell-surface-expressed ENaC. We have developed a quantitative assay based on the binding of 125I-labeled M2 anti-FLAG monoclonal antibody (M2Ab*) directed against a FLAG reporter epitope introduced in the extracellular loop of each of the α, β, and γ ENaC subunits. Insertion of the FLAG epitope into ENaC sequences did not change its functional and pharmacological properties. The binding specificity and affinity (Kd = 3 nM) allowed us to correlate in individual Xenopus oocytes the macroscopic amiloride-sensitive sodium current (INa) with the number of ENaC wild-type and mutant subunits expressed at the cell surface. These experiments demonstrate that: (i) only heteromultimeric channels made of α, β, and γ ENaC subunits are maximally and efficiently expressed at the cell surface; (ii) the overall ENaC open probability is one order of magnitude lower than previously observed in single-channel recordings; (iii) the mutation causing Liddle syndrome (β R564stop) enhances channel activity by two mechanisms, i.e., by increasing ENaC cell surface expression and by changing channel open probability. This quantitative approach provides new insights on the molecular mechanisms underlying one form of salt-sensitive hypertension.
The amiloride-sensitive epithelial sodium channel (ENaC) is a heteromultimeric protein composed of three homologous subunits, α, β, and γ (1, 2, 4, 5), exhibiting ≈30% identity at the amino acid level. Predicted protein topology reveals a large (≈500 amino acids) extracellular hydrophilic loop with several putative N-linked glycosylation sites flanked by two hydrophobic domains (M1 and M2) that span the membrane.
In aldosterone target epithelia, ENaC represents the rate-limiting step for Na+ reabsorption (6, 7). The control of Na+ movements in these epithelia is critical for the maintenance of extracellular fluid and electrolyte balance, and the central role of ENaC in this regulation is exemplified by the recent discoveries of several heritable human mutations in the genes encoding the ENaC subunits that lead to abnormal regulation of blood pressure and electrolyte balance (8, 9). Therefore, identification of the molecular and cellular mechanisms involved in the regulation of ENaC channel activity at the cell surface is critical for our understanding of the pathogenesis of salt-sensitive hypertension.
ENaC is characterized by its high ionic selectivity for sodium and lithium, by its low single-channel conductance (5 pS in the presence of Na and 8 pS in the presence of Li), by its long open and closed times, and by its high affinity for amiloride. Our knowledge about ENaC biophysical properties comes mainly from patch-clamp studies, allowing recordings of single-channel events (10–12). This technique, however, restricts the study of channel function to conducting channels that may represent only a small fraction of the total number of channels expressed at the cell surface.
In this study, we present a quantitative method to determine the number of ENaC subunits expressed at the cellular surface. This approach is based on binding of an iodinated antibody of known specific activity to an epitope placed in the extracellular part of ENaC. This highly specific binding assay shows that coexpression of the α, β, and γ subunits is required for maximal and efficient cell-surface expression of ENaC channels. In addition, the high level of cell-surface expression relative to the measured amiloride-sensitive current indicates that the overall channel open probability is considerably lower than what is observed in single-channel recordings. The correlation between the number of channel molecules present at the cell surface and the current expressed in individual cells provides evidence that mutations causing Liddle syndrome result in both an increase in channel expression at the cell surface and a predominant change in channel gating. The study provides a new understanding of channel behavior and of the molecular mechanisms underlying gain of function mutations in Liddle syndrome.
MATERIALS AND METHODS
Expression of ENaC in Xenopus Oocytes.
Complementary cRNAs encoding α, β, and γ rat ENaC subunits tagged with FLAG epitope were synthesized in vitro. cRNAs at concentrations ranging from 0.1 to 10 ng total cRNA were injected in Xenopus laevis oocytes. Oocytes were incubated either in a high Na+ Barth’s solution containing 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.8 mM MgSO4, 0.3 mM Ca(NO3)2, 0.4 mM CaCl2, 10 mM Hepes-NaOH (pH 7.2) or in a low Na+ solution containing 10 mM NaCl, 90 mM N-methyl-d-glutamine, 5 mM KCl, 2.4 mM NaHCO3, 0.8 mM MgSO4, 0.3 mM Ca(NO3)2, 0.4 mM CaCl2, 10 mM Hepes-NaOH (pH 7.2). Standard electrophysiological measurements were taken 24 h after injection. Macroscopic amiloride-sensitive Na+ currents (INa) defined as the difference between Na+ currents obtained in the presence (5 μM) and in absence of amiloride in the bath were recorded using the two-electrode voltage clamp. The amiloride-sensitive Na+ current represents a reliable index of the overall channel activity expressed at the cell surface of the oocyte (1). Single- channel recordings were made using the cell-attached configuration of the patch-clamp technique with either Na+ or Li+ ions in the pipette as described (13).
Reporter Epitope Construction.
The α, β, and γ rat ENaC subunits were tagged with the FLAG reporter octapeptide (DYKDDDDK) that is recognized by the anti-FLAG M2 (M2Ab) mouse monoclonal antibody (Kodak). In αENaC, the FLAG sequence replaced the sequence T194RQAGARR201. In βENaC, the FLAG sequence was inserted between T137 and S138. In γENaC, the FLAG sequence replaced the sequence M142PSTLEGT149. The introduction of the epitope was done by PCR and accuracy of constructions was confirmed by DNA sequencing. Immunoprecipitation of the FLAG-tagged ENaC subunits was performed as described (14).
Generation and Purification of the M2Fab Fragment and Iodination Procedure.
Fab fragment of the M2Ab (M2Fab) has been obtained, using the method of papainization described (15). Briefly, M2Ab was incubated for 6 h at 37°C with 2% (wt/wt) papain (Sigma) in 75 mM phosphate buffer (pH 7.0) containing 75 mM NaCl, 10 mM l-cysteine hydrochloride, and 2 mM EDTA. Fab fragment was separated from partially digested M2Ab by filtration on Sephadex G-150 and from Fc fragments by a DE-52 ion exchange chromatography column equilibrated with 0.01 M phosphate buffer (pH 8.0). The M2Fab fragment was eluted with the void volume, whereas the Fc fragment was retained. Purity of the Fab fragment was analyzed by SDS/PAGE (8%) revealing a single band of about 50,000 kDa. Both M2Ab and M2Fab were iodinated using the Iodo-Beads iodination reagent (Pierce) and carrier-free Na125I (Amersham) according to the Pierce protocol. Unincorporated 125I was removed from the iodinated protein by gel filtration on a PD-10 column (Pharmacia). Iodinated protein was collected in either the high or low sodium solutions, and the total quantity of recovered protein was determined by the method of Lowry. The quantity of 125I incorporated into the proteins was determined by TCA precipitation. Iodinated M2Ab and M2Fab had a specific activity of 5–20 × 1017 and of 2–6 × 1017 cpm/mol, respectively, and were used for 2 months after the synthesis.
Binding Assay.
Binding of M2Ab* or M2Fab* to oocytes expressing the FLAG-tagged α, β, and γ subunits of rat ENaC was determined at 20–24 h after the cRNA injection. Twelve oocytes in each experimental group were transferred into a 2 ml Eppendorf tube containing either high or low sodium solutions supplemented with 10% heat-inactivated calf serum and incubated for 30 min on ice. The binding was started upon addition of either 12 nM M2Ab* or 50 nM M2Fab* in a final vol of 100 μl or 50 μl, respectively. After 1 h of incubation on ice, the oocytes were washed eight times with 1 ml of the corresponding sodium solution supplemented with 5% calf serum and then transferred individually into tubes for γ counting containing 250 μl of the same solution. The samples were counted and the same oocytes were reserved for subsequent electrophysiological studies. Nonspecific binding was determined from parallel assays of oocytes injected with nontagged α, β, and γ ENaC cRNAs. Preliminary experiments showed that steady state for antibody binding and INa was achieved as soon as 16 h after cRNA injection. At this time, the rate of internalization and the rate of externalization are equal, and we assume that both rates have the same temperature dependence. M2Ab* binding detected in oocytes coinjected with FLAG-tagged wild-type α, β, and γ subunits varied from 0.3 fmol per oocyte to 0.9 fmol per oocyte. This range of variation corresponds to the day-to-day variation observed for INa in different batches of oocytes. Internal controls were therefore performed for each experiment.
RESULTS
The FLAG Epitope Tagging of the α, β, and γ ENaC Subunits.
The selection of the site to introduce the FLAG epitope was based on current models of ENaC subunits membrane topology in which each homologous subunit contains a large extracellular domain flanked by two transmembrane domains M1 and M2 (16–18). To minimize possible alterations in channel function due to modification of ENaC primary sequence, the epitope was introduced in a region of high degree of sequence divergence between ENaC subunits of human (4, 19), X. laevis (5), and rat (1), located ≈30 residues downstream of the first transmembrane domain M1 (Fig. 1A). In this region, all the eight residues substituted by FLAG sequence differ among the α and γ subunits isoforms. In the β subunit, insertion of the FLAG epitope was performed in a corresponding site where the sequence alignment reveals a gap of 9 amino acids between mammalian and amphibian sequences. The introduction of the FLAG epitope in each subunit did not alter their respective N-linked glycosylation pattern.
Figure 1.
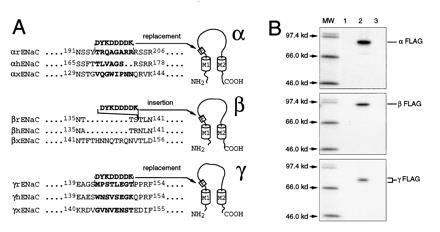
(A) FLAG-epitope placement into the rat ENaC sequence. The alignment of the extracellular loop sequences for the rat, human, and Xenopus ENaC subunits demonstrated the most important divergence in the region near the first transmembrane domain M1. This region was chosen to insert (α and γ subunits) or to replace (β subunit) the FLAG sequence (DYKDDDDK) at the indicated positions. (B) Immunoprecipitation of the FLAG-tagged ENaC subunits, using M2Ab*. Oocytes were injected with an α, β, or γ nontagged subunit alone (lane 1), or with an α, β, or γ FLAG-tagged subunit alone (lanes 2 and 3). Oocytes were metabolically labeled with [35S]methionine for a 4-h pulse followed by a 4-h chase period. Membranes were prepared, and proteins were immunoprecipitated and analyzed on 5–8% gradient SDS/PAGE. Immunoprecipitations of the samples corresponding to lane 3 were performed in the presence of 1000-fold molar FLAG peptide excess over the antibody concentration.
The experiment shown in Fig. 1B demonstrated specific immunoprecipitation of the α, β, and γ FLAG-tagged subunits as proteins of an apparent Mr of 92, 97, and 82, respectively, using the M2Ab. This confirmed the correct protein synthesis (16) with a slight shift of 1.5–2 kDa in the apparent molecular mass due to the presence of the FLAG epitope. To assess the specificity of the recognition of FLAG-tagged ENaC subunits by the M2Ab antibody, immunoprecipitation of the nontagged subunits (lane 1) and of the FLAG-tagged subunits in the presence of 1000-fold molar FLAG peptide excess over the M2Ab antibody concentration (lane 3) were performed. Under both conditions, nonspecific immunoprecipitation was not detected.
M2Ab* and M2Fab* Binding to the ENaC Expressing Xenopus Oocytes.
Quantification of the number of channel molecules expressed at the cell surface requires that one antibody binds specifically to a single subunit. However, antibodies are divalent molecules and the possibility of cross-linking binding interactions exists where one M2Ab antibody binds with two FLAG antigens. To address this possibility, we prepared an univalent Fab fragment of the M2Ab. Fig. 2 A and C shows a linear plot of the saturation binding isotherm for the M2Ab* and M2Fab. Scatchard analysis shown in Fig. 2 B and D revealed an equilibrium dissociation constant (Kd) of 3 nM for the M2Ab* and 31 nM for the M2Fab*, respectively, with a maximal specific binding (Bmax) of 4.9 × 108 molecules M2Ab* bound per oocyte and 5.5 × 108 molecules M2Fab* bound per oocyte. These data indicate different specific binding affinities for M2Ab* or M2Fab* but no differences in the maximum number of binding sites per oocyte for the two antibodies.
Figure 2.
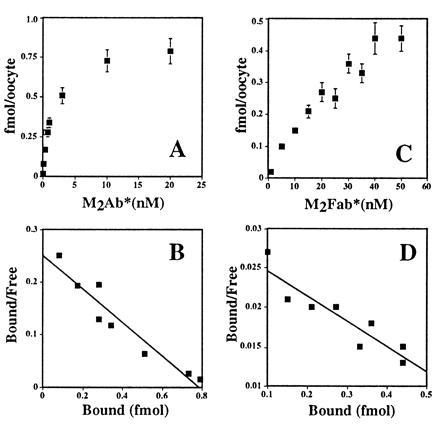
Equilibrium binding of M2Ab* and M2Fab* to the FLAG-tagged ENaC subunits. Stage V oocytes were injected, either with 10 ng of FLAG-tagged or with nontagged ENaC cRNAs. Twenty-four hours later, these oocytes were tested for M2Ab* (A) or M2Fab (C) binding. Twelve oocytes were transferred into 100 μl (M2Ab) or 50 μl (M2Fab) binding buffer and then incubated with increasing concentrations of M2Ab* or M2Fab* for 4 h on ice. The concentration of M2Ab* and M2Fab varied from 0.03–20 nM and 5–50 nM, respectively. Specific binding was calculated as the difference of the binding between the oocytes injected with FLAG-tagged and the oocytes injected with nontagged ENaC cRNAs and transformed into Scatchard plots for M2Ab* (B) or M2Fab* (D). (A and C) The means ± SEM of three independent experiments, each performed on 12 oocytes.
The specificity of M2Ab* and M2Fab* binding interactions with FLAG-tagged ENaC subunits was further evaluated in two independent sets of experiments shown in Fig. 3. These experiments demonstrate that the presence of a 200-fold excess of unlabeled M2Ab completely abolished the specific binding of M2Ab*. In addition, presence of a 1000-molar excess of the FLAG peptide during the binding assay completely abolished both specific bindings of M2Ab* and M2Fab*. Together these experiments establish the specific recognition of the FLAG-tagged ENaC subunits by both M2Ab* and M2Fab*. They also confirm the identical maximal binding obtained with M2Ab* and M2Fab*, strongly suggesting that the stoichiometry of the binding reaction is 1:1 (one M2Ab* or one M2Fab* molecule for one channel subunit molecule). Finally, these observations exclude significant cross-linking interactions between a single M2Ab and multiple binding sites and allowed us to utilize in all subsequent experiments the higher affinity M2Ab* antibody for quantitation.
Figure 3.
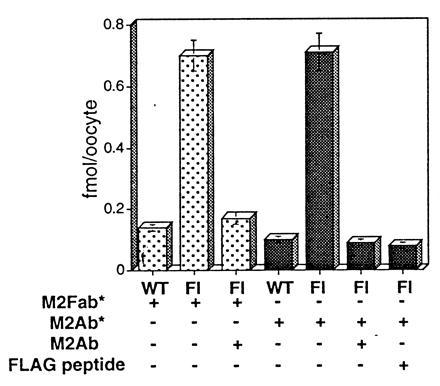
Specific binding of the M2Fab* and M2Ab* to Xenopus oocytes expressing FLAG-tagged ENaC subunits. Binding of the 50 nM M2Fab* (dotted columns) or 12 nM M2Ab* (shaded columns) was measured in parallel on the oocytes expressing either FLAG-tagged (Fl) or nontagged ENaC subunits. Specificity of the binding was tested, using either 1000-molar excess of the FLAG peptide (M2Fab* and M2Ab*) or 200-fold excess of unlabeled M2Ab (M2Ab*). FLAG peptide and unlabeled M2Ab were added in the binding solution simultaneously with the 125I-labeled agents. Shown are means ± SEM from four independent experiments, each performed on 12 oocytes.
The FLAG Epitope in ENaC Subunits Does Not Change the Functional Properties of ENaC.
Introduction of the FLAG epitope in the extracellular loop of ENaC subunits did not change significantly the macroscopic current expressed by ENaC channel in Xenopus oocytes: INa of oocytes expressing the FLAG-tagged ENaC subunits was 6.5 ± 1.1 μA (mean ± SEM, n = 104 oocytes), not different from the currents induced by expression of nontagged ENaC (8.5 ± 3.2 μA, mean ± SEM, n = 106 oocytes). The INa associated with the FLAG-tagged channels was not affected by the presence of M2Fab or M2Ab in the external medium (4.2 ± 0.4 μA and 4.6 ± 0.5 μA with M2Fab or M2Ab, respectively, compared with 5.1 ± 0.5 μA without antibodies, mean ± SEM, n = 37 oocytes for each condition), indicating that the binding interaction between the antibodies and the ENaC channel epitope does not modify the channel functional properties during the binding assay. Finally, the FLAG-tagged ENaC retains its high affinity for amiloride block with an apparent Kd of 64 ± 6 nM (n = 12, mean ± SEM) in the presence of 20 mM Na+.
Patch-clamp studies of oocytes expressing the FLAG-tagged channel revealed active channels characterized by long closing and opening events interrupted by shorter events (Fig. 4). The open probability was quite variable, ranging from 0.09 to 0.81 with an average of 0.42 (n = 20), similar to that previously described (13). Single-channel conductance was 5.5 pS with Na+ and 8.8 pS with Li+ pipette solutions, respectively, indicating that the conductive properties and the ionic selectivity of the FLAG-tagged ENaC remain unchanged with respect to wild-type ENaC.
Figure 4.
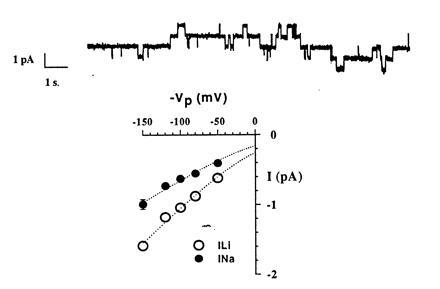
Single-channel properties of FLAG-tagged ENaC channels. (Upper) A single-channel recording performed in cell attached configuration with Na+ ions as major cation in the pipette. Pipette voltage (Vpip) was 80 mV. Downward deflections represent channel openings. The four distinct current levels indicate the presence of at least four conducting channels in the patch. (Lower) Current-voltage relationship in the presence of Na+ or Li+ ions in the pipette. Single-channel conductance estimated from regression analysis between −50 mV and −150 mV was 5.5 pS for Na+ and 8.8 pS for Li+.
Coinjection of All Three α, β, and γ Subunits Determines Cell Surface ENaC Expression.
We previously showed that expression of β and γ subunits individually does not induce an amiloride-sensitive current (1). When β or γ subunits were coexpressed with α subunit, a 3- to 5-fold potentiation of the current induced by α alone was observed. Coexpression of α, β, and γ subunits led to maximal channel activity, characterized by a 25- to 50-fold increase over the current due to the expression of either αβ or αγ. It is not known whether β and γ subunits can reach the plasma membrane as nonconductive channels without α or whether assembly and oligomerization of the three subunits in the endoplasmic reticulum compartment is a prerequisite for export of conductive heteromultimeric channels to the membrane. As shown in Fig. 5, M2Ab* binding (0.92 fmol per oocyte) detected in oocytes coinjected with α, β, and γ subunits was more than 20-fold greater (P < 0.001) than the binding measured in αγ- (0.04 fmol per oocyte) or αβ- (0.06 fmol per oocyte) expressing oocytes. Due to the low level of counts, significant binding for mono- or dimeric-channels over the level of water-injected oocytes was not observed (P > 0.05). Overall, our binding data fits exactly the pattern of functional expression. It is consistent with the idea that assembly of α, β, and γ and their oligomerization in the endoplasmic reticulum compartment is required before active channels can be exported to the plasma membrane. Only the α subunit alone or αβ or αγ channels can be targeted and constitute active channel at the plasma membrane, though very inefficiently (0.5 to 2–5% of maximum). Our binding assay will thus preferentially measure a major and homogenous pool of αβγ heteromultimeric channel. Only around 5% of the total binding could be accounted for by the presence of α, αβ, or αγ complexes.
Figure 5.
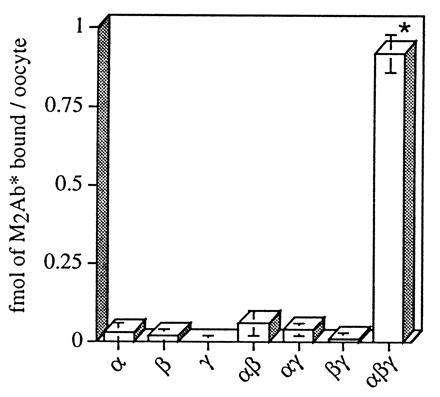
Specific M2Ab* binding to the oocytes injected with different combinations of ENaC subunits. X. laevis oocytes were injected with FLAG-tagged cRNAs (3 ng) of α, β, and γ ENaC, or combinations of αβ, αγ, βγ, αβγ, or nontagged αβγ as control. Binding analysis was performed with 12 nM M2Ab*, and the specific binding has been determined as difference between the oocytes injected with the FLAG-tagged ENaC subunits and the oocytes injected with nontagged α, β, and γ ENaC subunits. The data are means ± SEM of three independent experiments, each performed on 12 oocytes. ∗, P < 0.001.
Quantitative Correlation of the Macroscopic Amiloride-Sensitive Na+ Current with the Number of ENaC Subunits at the Cell Surface.
Our binding assay allowed us to quantitatively correlate the level of amiloride-sensitive Na+ current expressed by individual oocytes with the number of ENaC subunits present at the cell surface. Oocytes were first assayed individually for the number of specific M2Ab binding sites, then the same oocytes were taken for measurements of the expressed amiloride-sensitive Na+ current.
Graphs in Fig. 6 plot the specific binding of M2Ab* versus the macroscopic INa for individual oocytes incubated either in a low (Fig. 6A) or a high (Fig. 6B) sodium medium. Under each conditions, we observed a significant correlation (r2 = 0.83) between M2Ab* binding and INa, indicating that the expressed INa increases proportionally with the number of channel molecules present at the cell surface, as shown by the regression lines that cross the axes at values not significantly different from zero (−0.023 ± 0.157). We have, therefore, no evidence for the presence of “truly silent” or nonconductive channels in the oocyte expression system. Comparison of Fig. 6 A and B shows that INa for a given number of M2Ab* binding sites varied with external sodium concentration. With 10 mM Na, a current-binding relationship of 4.8 μA/fmol M2Ab* bound was determined. In other words, the M2Ab recognizes 125 binding sites for every pAmps of INa measured in oocytes. By contrast, in the high-sodium medium, INa was ≈4-fold lower per M2Ab* binding sites. Alternatively, the slope of the current-binding relationship was 1.1 μA/fmol M2Ab*, which corresponds to 550 M2Ab binding sites per pAmps of INa. This 4-fold change in amiloride-sensitive conductance relative to specific FLAG binding at the cell surface is entirely consistent with previous studies showing that increasing extracellular Na decreases open probability of the channel.
Figure 6.
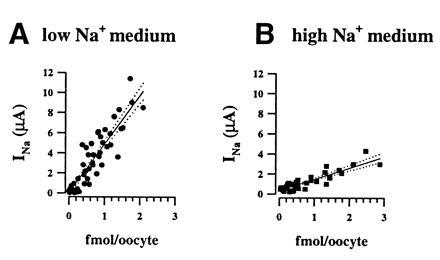
Relationships between macroscopic amiloride-sensitive Na+ current (INa) and surface expression of ENaC channel subunits. Specific M2Ab* binding is expressed as a function of INa measured at −100 mV. Each symbol represent one oocyte, the type of symbol refers to different batches of oocytes. (A) Experiments (n = 4) performed with oocytes incubated in the low sodium solution. Regression analysis on all the data points gave a linear correlation (r2= 0.85) with a slope (solid line; dotted lines are 95% confidence limit) of 4.82 μA/fmol crossing the origin at 0.09 μA. (B) Oocytes incubated in the high sodium medium. Regression analysis of the correlation between INa and the bound M2Ab* molecules gave a slope of 1.13 μA/fmol (r2 = 0.83) and a y value at the origin of −0.13.
Cell Surface Expression of ENaC Mutant Causing Liddle Syndrome.
We used our binding assay to see whether a gain of function mutations causing Liddle syndrome results in a higher number of ENaC molecules expressed at the cell surface or in an increase in channel open probability. The nonsense mutation at Arg codon 564 in the C terminus of ENaC β subunit (β R564 stop) identified in the original Liddle’s pedigree was introduced in the FLAG-tagged β subunit and coexpressed with the other FLAG-tagged subunits. The mean increase in INa caused by the βR564stop mutation was 5.6 ± 1.6-fold (P < 0.05), whereas the increase in the number of specific M2Ab* binding sites was 2.0- ± 0.3-fold (P < 0.05) (Fig. 7A). Thus, although the βR564stop mutation doubles the number of ENaC subunits on the cell surface, this 2-fold higher ENaC surface expression cannot account for the measured increase in INa. Fig. 7B shows the current-binding relationship of the ENaC wild type and the Liddle mutant. For the wild type, regression analysis of individual values gave a mean slope of 1.8 μA/fmol, consistent with the results in Fig. 6B obtained under the same conditions. Oocytes injected with the βR564stop mutant display a higher ratio of current over specific M2Ab* binding with a mean of 9 μA/fmol. These data indicate that Liddle’s mutation affects both the expression of ENaC at the cell surface and the open probability of the channel. Furthermore, our data predict that the change in channel gating is the main determinant underlying the gain of function of channel mutant since it accounts for more than 65% of the stimulation of the macroscopic INa induced by the βR564stop mutation.
Figure 7.
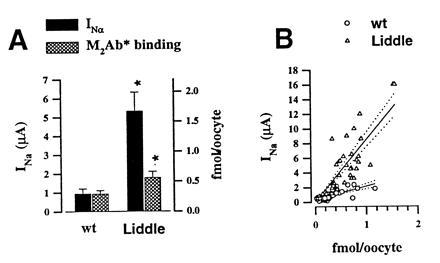
Effects of βR564stop mutation on INa and channel expression at the cell surface. Five independent experiments were performed in which 10 to 12 oocytes were injected with either β subunit or βR564stop mutant, together with α and γ ENaC wild type. (A) Mean INa and M2Ab* binding was, respectively, 0.94 ± 0.27 μA and 0.29 ± 0.06 fmol for wt and 5.31 ± 1.01 μA and 0.56 ± 0.1 fmol for the β564R stop. ∗, A statistical significance P < 0.05. (B) Correlation between INa measured at −100 mV and specific M2Ab* binding in individual oocytes. Regression analysis (regression line, straight line; dotted lines are 95% confidence limit) gave a linear correlation with a slope of 1.8 μA/fmol M2Ab* (r2 = 0.6) for the wt and 9.1 μA/fmol M2Ab* (r2 = 0.77) for βR564stop.
DISCUSSION
This study describes the relationship existing between the number of ENaC channels present at the cell surface and the corresponding sodium current. The experimental approach was based on a specific and quantitative binding assay, using an iodinated antibody against a FLAG epitope introduced into the α, β, and γ ENaC subunits.
This study first shows that α, β, and γ subunit assembly is required for maximal efficient expression of the ENaC at the cell surface. α, β, and γ subunits were translated in oocytes injected with α, β, or γ cRNA, but no surface labeling was detected, consistent with the absence or small measurable currents (in the nAmp range). For other heteromultimeric plasma membrane proteins, assembly, folding, and oligomerization of subunits take place in the endoplasmic reticulum compartment and have been shown to be a prerequisite for exit to the Golgi compartment and targeting to the plasma membrane (20). According to the present study, we propose similar requirements for ENaC to exit the endoplasmic reticulum compartment.
The Open Probability (Po) of ENaC Is Considerably Lower than Previously Thought.
A striking observation is the high level of specific M2Ab* binding sites relative to the amiloride-sensitive current measured in individual oocytes. Depending on the incubation conditions, the ratio of specific M2Ab* binding sites expressed at the cell surface over amiloride-sensitive current carried ranged from 125 to 550 sites per pA. Assuming that these specific binding sites represent one channel subunit, and that ENaC is made of three subunits, 1 pA of amiloride-sensitive inward current would correspond to the activity of 42 to 167 channels. From the single-channel conductance (0.6 pA at −100 mV), our results predict that the mean open probability of all the ENaC channels expressed at the cell surface range from 0.014 to 0.004. Even if ENaC is made of nine subunits, the mean open probability would increase by a factor of 3 (0.042 to 0.012), a figure still one order of magnitude lower than that reported previously in the literature (21). In native tissues or cultured cells, the open probability of ENaC is subject to a great variability from channel to channel (21). In cortical-collecting duct of salt-depleted rats with high plasma aldosterone levels Po ranged from 0.05 to 0.9 with an average value for mean Po of 0.5 (21). Similar ENaC open probabilities were observed in frog skin (22) and in the A6 kidney cell line. However, due to the wide distribution of spontaneous Po and the slow transitions between open and closed states, the estimation of the number of active channels present in the membrane patch is usually difficult, leading to overestimation of Po. Therefore, the characteristic ENaC channel behavior described in patch clamp experiments exhibiting long opening events and Po ≥ 0.5 likely concerns only a minority of ENaC channels present at the cell surface. The majority of the channel expressed are either nonconducting channels or channels with a extremely low Po. The linearity of the relationship between INa and the number of channel subunits present at the cell surface suggests that the increasing level of INa in individual oocytes is not due to the recruitment of nonconducting channels from a pool of inactive channels, but rather to the insertion of new channel molecules at the plasma membrane.
Extra- and/or Intracellular Sodium Controls Po Without Changing the Number of ENaC Molecules.
In epithelial cells expressing ENaC channels, the rate of Na+ entry is controlled to maintain intracellular homeostasis. This process involves different regulatory mechanisms dependent on extracellular Na+, membrane voltage, intracellular Ca2+, or protein kinases (21, 23, 24). During the present study, we observed that oocytes expressing identical number of ENaC subunits exhibited lower INa when incubated in a high Na+ medium, compared with oocytes incubated in a low Na+ medium. Incubation of oocytes expressing ENaC in a high Na+ medium leads to intracellular Na+ load, as shown by a depolarizing shift in the Nernst equilibrium potential for Na+ (1). Our observation suggests that raising of the intracellular Na+ content decreases channel’s open probability without affecting surface expression of ENaC. This down regulation of channel activity may prevent intracellular Na+ overload. The precise mechanism involved in the down-regulation of the channel by Na+ remains to be determined.
Dual Effects of the Liddle’s Mutation on Po and the Number of Channels Expressed at the Cell Surface.
The recent identification of mutations in ENaC subunits, causing diseases such as the Liddle syndrome or pseudohypoaldosteronism type 1, has stressed the critical role of ENaC in the maintenance of salt and water homeostasis (25). Mutations of a conserved PY motif of the C terminus in the β or γ subunits result in a channel gain of function, leading to hypertension and hypokalemia (26, 27). We have postulated that the increased sodium conductance caused by these mutations could potentially involve channel gating, surface expression of the channel, or both (13). A recent study by Snyder et al. (26) addressing this question found a 2-fold increase in macroscopic INa on oocytes expressing the Liddle mutation, with no evidence for a change in Po, reported to be around 0.4–0.5. Snyder et al. (26) reported, however, a relative 40% increase in surface expression in COS cells of an HLA chimeric protein expressing the mutant C terminus of the β subunit. Our present results show that in Liddle syndrome, the number of channel subunits expressed at the cell surface cannot completely account for the increase in the macroscopic current and that a change in channel gating, leading to a significantly higher open probability represents an equally important factor in increasing channel activity at the cell surface.
All missense or nonsense mutations causing Liddle syndrome target a conserved PY motif in the C terminus of β and γ ENaC subunits. This PY motif is involved in binding interactions with specific domains of a Nedd4 protein called WW domains, as recently shown by Staub et al. (3). Interestingly, the Nedd4 protein binding to the PY motif of ENaC subunits contains a ubiquitin-ligase homology domain and may be involved in the ubiquitination of the channel that tags the channel for degradation. Although ubiquitinization of the ENaC channel remains to be demonstrated, the binding interaction between Nedd4 and the PY motif suggests how Liddle’s mutations could impair channel retrieval from the plasma membrane and lead to an increased number of active channels at the cell-surface as also observed in the present study. The increase in channel open probability that we observed with the Liddle’s mutant is not incompatible with this model. Regulation of channel Po at the cell surface and channel retrieval could be mediated by two distinct functional domains of Nedd-4. Alternatively, regulation of Po could be mediated by distinct proteins and/or factors.
Acknowledgments
We thank Nicole Skarda-Coderey for secretarial assistance and Hans-Peter Gaeggeler for technical help. We would like to thank Jean-Pierre Mach (Institut de Biochimie, Lausanne) for technical help and helpful comments, as well as Dirk Claeys for comments and suggestions. This work was supported by the Swiss National Fund for Scientific Research Grants 31–43384.95 (to B.C.R.) and 31–39435.93 (to L.S.).
Footnotes
Abbreviations: ENaC, amiloride-sensitive epithelial Na+ channel; M2Ab, anti-FLAG M2 mouse monoclonal antibody; M2Fab, Fab fragment of the M2Ab; M2Ab*, 125I-iodinated M2Ab; M2Fab*, 125I-iodinated M2Fab.
References
- 1.Canessa C M, Schild L, Buell G, Thorens B, Gautschi I, Horisberger J-D, Rossier B C. Nature (London) 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 2.Lingueglia E, Renard S, Voilley N, Waldmann R, Chassande O, Lazdunski M, Barbry P. Eur J Biochem. 1993;216:679–687. doi: 10.1111/j.1432-1033.1993.tb18188.x. [DOI] [PubMed] [Google Scholar]
- 3.Staub O, Dho S, Henry P C, Correa J, Ishikawa T, McGlade J, Rotin D. EMBO J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- 4.Mcdonald F J, Price M P, Snyder P M, Welsh M J. Am J Physiol. 1995;268:C1157–C1163. doi: 10.1152/ajpcell.1995.268.5.C1157. [DOI] [PubMed] [Google Scholar]
- 5.Puoti A, May A, Canessa C M, Horisberger J D, Schild L, Rossier B C. Am J Physiol. 1995;38:C188–C197. doi: 10.1152/ajpcell.1995.269.1.C188. [DOI] [PubMed] [Google Scholar]
- 6.Rossier B C, Canessa C M, Schild L, Horisberger J-D. Curr Opin Nephrol Hypertens. 1994;3:487–496. doi: 10.1097/00041552-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Rossier B C, Palmer L G. In: Mechanisms of Aldosterone Action on Sodium and Potassium Transport. Seldin D W, Giebiesch G, editors. New York: Raven; 1992. pp. 1373–1409. [Google Scholar]
- 8.Shimkets R A, Warnock D G, Bositis C M, Nelsonwilliams C, Hansson J H, Schambelan M, Gill J R, Ulick S, Milora R V, Findling J W, Canessa C M, Rossier B C, Lifton R P. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 9.Chang S S, Grunder S, Hanukoglu A, Rösler A, Mathew P M, Hanukoglu I, Schild L, Lu Y, Shimkets R A, Nelson-Williams C, Rossier B C, Lifton R P. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 10.Palmer L G, Frindt G. Fed Proc Fed Am Soc Exp Biol. 1986;45:2708–2712. [PubMed] [Google Scholar]
- 11.Palmer L G, Frindt G. Proc Natl Acad Sci USA. 1986;83:2767–2770. doi: 10.1073/pnas.83.8.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamilton K L, Eaton D C. Am J Physiol. 1985;249:C200–C207. doi: 10.1152/ajpcell.1985.249.3.C200. [DOI] [PubMed] [Google Scholar]
- 13.Schild L, Canessa C M, Shimkets R A, Gautschi I, Lifton R P, Rossier B C. Proc Natl Acad Sci USA. 1995;92:5699–5703. doi: 10.1073/pnas.92.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geering K, Theulaz I, Verrey F, Hauptle M T, Rossier B C. Am J Physiol. 1989;257:C851–C858. doi: 10.1152/ajpcell.1989.257.5.C851. [DOI] [PubMed] [Google Scholar]
- 15.Buchegger F, Haskell C M, Schreyer M, Scazziga B R, Randin S, Carrel S, Mach J-P. J Exp Med. 1983;158:413–427. doi: 10.1084/jem.158.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canessa C M, Merillat A-M, Rossier B C. Am J Physiol. 1994;267:C1682–C1690. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 17.Snyder P M, Mcdonald F J, Stokes J B, Welsh M J. J Biol Chem. 1994;269:24379–24383. [PubMed] [Google Scholar]
- 18.Renard S, Lingueglia E, Voilley N, Lazdunski M, Barbry P. J Biol Chem. 1994;269:12981–12986. [PubMed] [Google Scholar]
- 19.Voilley N, Lingueglia E, Champigny G, Mattei M G, Waldmann R, Lazdunski M, Barbry P. Proc Natl Acad Sci USA. 1994;91:247–251. doi: 10.1073/pnas.91.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geering K. In: Oligomerization and Maturation of Eukaryotic Membrane Proteins. Heijne G v., editor. Austin, TX: Landes; 1996. pp. 173–188. [Google Scholar]
- 21.Palmer L G, Frindt G. J Gen Physiol. 1996;107:35–45. doi: 10.1085/jgp.107.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helman S I, Baxendale L M. J Gen Physiol. 1990;95:647–678. doi: 10.1085/jgp.95.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frindt G, Silver R B, Windhager E E, Palmer L G. Am J Physiol. 1995;37:F480–F489. doi: 10.1152/ajprenal.1995.268.3.F480. [DOI] [PubMed] [Google Scholar]
- 24.Palmer L G, Frindt G. Am J Physiol. 1987;253:F333–F339. doi: 10.1152/ajprenal.1987.253.2.F333. [DOI] [PubMed] [Google Scholar]
- 25.Lifton R P. Science. 1996;272:676–680. doi: 10.1126/science.272.5262.676. [DOI] [PubMed] [Google Scholar]
- 26.Snyder P M, Price M P, McDonald F J, Adams C M, Volk K A, Zeiher B G, Stokes J B, Welsh M J. Cell. 1995;83:969–978. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 27.Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton R P, Rossier B C. EMBO J. 1996;15:2381–2387. [PMC free article] [PubMed] [Google Scholar]