

Abstract
Selectins mediate rolling, the initial step of leukocyte adhesion to endothelial cells [Springer, T. A. (1995) Annu. Rev. Physiol. 57, 827–872 and Butcher, E. C. (1991) Cell 67, 1033–1036]. In this study we show that l-selectin triggering of Jurkat cells using different antibodies or glycomimetics resulted in activation of the src-tyrosine kinase p56lck; tyrosine phosphorylation of intracellular proteins, in particular mitogen-activating protein kinase and l-selectin; and association of Grb2/Sos with l-selectin. This association correlated with an activation of p21Ras, mitogen-activating protein kinase, Rac2, and a transient increase of O2− synthesis. Stimulation of the Ras pathway by l-selectin requires functional p56lck, since p56lck-deficient Jurkat cells (JCaM1.6) do not show tyrosine phosphorylation, association of l-selectin with Grb2/Sos, and activation of Ras upon l-selectin triggering. Transfection of JCaM1.6 cells with p56lck reconstitutes the observed signaling events. Genetic inhibition of Ras or Rac2 prevented Rac2 stimulation and O2− synthesis, respectively. The specificity and the physiological significance of the observed signaling cascade is indicated by stimulation of l-selectin-transfected P815, l-selectin-positive CEM or peripheral blood lymphocytes resulting in the same activation events as in Jurkat cells. Our results point to a signaling cascade from l-selectin via p56lck, Grb2/Sos, Ras, and Rac2 to O2− .
Keywords: signal transduction, adhesion proteins, G-proteins, lymphocytes
Adhesion of leukocytes has been shown to be crucial in the regulation of the immune response, during infections, or in recirculation of lymphocytes to lymphatic organs (1–3). Rolling as the first step in this process has been demonstrated to be mediated by transient interactions of selectins with their glycoprotein ligands on endothelial cells and leukocytes, respectively (4, 5). An important function of l-selectin for rolling has been shown in studies employing l-selectin knock-out mice (6), monoclonal antibodies (7, 8), l-selectin-IgG chimera (9), and certain polysaccharides (10–12), demonstrating that the deficiency or inhibition of l-selectin reduces leukocyte adhesion.
However, it is unknown whether l-selectin ligands also induce activation of intracellular signaling events. Several indications point to an active signaling function of the cytoplasmic domain of l-selectin being important for rolling and lymphocyte homing: Partial deletion of the intracellular domain of l-selectin prevents leukocyte rolling without affecting ligand recognition, implying that binding of l-selectin to its ligand is not sufficient for leukocyte rolling (13). Furthermore, triggering neutrophils via l-selectin using various antibodies or sulfatides mobilizes intracellular Ca2+, increases tumor necrosis factor α and interleukin 8–mRNA expression (14), induces O2− generation (15, 16), enhances tyrosine phosphorylation, and activates mitogen-activating protein kinase (MAPK) (17).
In this study we describe intracellular signaling events upon l-selectin triggering in T-lymphocyte lines, peripheral blood lymphocytes (PBL), and l-selectin transfectants. Using different l-selectin antibodies or glycomimetics we provide evidence for a signaling cascade from l-selectin via the tyrosine kinase p56lck, Grb2/Sos, Ras, and Rac2 to the activation of MAPK and the synthesis of O2−. Using genetically p56lck-deficient Jurkat cells, N17Ras transfected Jurkat cells, and Rac2 antisense oligonucleotides a signaling cascade of the activated molecules is suggested.
MATERIALS AND METHODS
Cell Culture and Stimulation.
All reagents were purchased from Sigma, if not otherwise cited. The human T-cell lines Jurkat, p56lck-deficient JCaM1.6 (18, 19), or p56lck-reconstituted JCaM1.6 (19) (a kind gift from A. Weiss, University of California, San Francisco), l-selectin positive or negative CEM cells, and P815 cells were grown in RPMI 1640 medium as described (20). l-selectin-negative CEM cells (CEM−) were obtained from l-selectin-positive CEM cells (CEM+) by spontaneous loss of surface l-selectin after long-term culture and repeated sorting by FACS (Becton Dickinson). PBL were prepared using Ficoll–Histopaque. Transfection of mouse mastocytoma P815 cells with the human l-selectin expression vector pZip-L-sel (P815.L-Sel) or control vector pZip (P815.control) (kindly provided by T. F. Tedder, Duke University) was performed as described (20). For activation, cells (2 × 106/20 μl or 20 × 106/100 μl per sample for cell lysates or immunoprecipitations) were washed twice in sterile Hepes/saline (H/S; 132 mM NaCl/20 mM Hepes/5 mM KCl/1 mM CaCl2/0.7 mM MgCl2/0.8 mM MgSO4) and stimulated at 37°C with NaN3-free, low endotoxin, monoclonal mouse anti-human l-selectin antibody Dreg56 (2 μg/ml, PharMingen), Dreg200 (5 μg/ml, kindly provided by T. K. Kishimoto, Boehringer Ingelheim, Ridgefield, CT), Fucoidan (200 μg/ml), or sialyl LewisX (100 μg/ml; Oxford Glycosystems, Oxford, U.K.). All of these reagents have been shown to recognize l-selectin (14–17, 21–26).
Immunoprecipitation and Immunoblotting.
Cell stimulation was terminated by lysis in 25 mM Hepes (pH 7.4), 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 125 mM NaCl, 10 mM each NaF, Na3VO4, and sodium pyrophosphate, and 20 μg/ml of aprotinin/leupeptin (RIPA buffer) for total cell lysates and for immunoprecipitation of l-selectin or Vav. For MAPK tyrosine phosphorylation, kinase activity and co-immunoprecipitation of Sos, Grb2, Shc, and l-selectin, cells were lysed in the same buffer as above with 3% Nonidet P-40 as detergent (TN3 buffer). Agarose-coupled or -uncoupled antibodies to Lck or Fyn, phosphotyrosine 4G10, and Sos were from Upstate Biotechnology, to human β7-integrin from Immunotech (Hamburg, Germany), to Ras (Y13-259) and to a viral epitope (25–9) from the American Type Culture Collection, to Rac2 from Santa Cruz Biotechnology, to CD20 from PharMingen, and to p42/44MAPK from Zymed. All control immunoprecipitates (IPs) were performed with polyclonal or monoclonal rabbit, mouse, or rat Ig. After lysis and a centrifugation step (20,000 × g 15 min) proteins were immunoprecipitated (3 μg antibody). IPs were immobilized by anti-mouse- or anti-rabbit-coupled agarose, washed six times in lysis buffer, and resuspended in SDS sample buffer. Proteins were separated by SDS/PAGE, transferred to Immobilon filters (Millipore), and blotted with the appropriate antibody. Immunoblots were developed by horseradish peroxidase-conjugated protein G (Bio-Rad) and a chemoluminescence kit (Amersham). To test for equal amounts of immunoprecipitated protein blots were stripped (45 min in 20 mM Tris, pH 6.8/2% SDS/70 mM 2-mercaptoethanol at 70°C) and reprobed, or an aliquot of the IPs was analyzed by Western blotting. Association of Sos, Grb2, Shc, or l-selectin was tested by co-immunoprecipitation and blotting the IPs with the corresponding antibody.
Src and MAPK Assays.
The activity of src or MAPK was determined by autophosphorylation or by phosphorylation of myelin basic protein (MBP), respectively. Src kinases p56lck and p59fyn or MAPK were immunoprecipitated, washed four times in TN3 buffer, twice in kinase buffer (25 mM Hepes, pH 7.0/150 mM NaCl/10 mM MnCl2/1 mM Na3VO4/5 mM DTT/0.5% Nonidet P-40) for src kinases or twice in Tris-buffered saline (pH 7.3) and once in kinase buffer (25 mM Hepes, pH 7.4/2 mM DTT/10 mM MgCl2) for MAPK. The reaction was initiated by addition of 10 μCi [32P-γ]-ATP (DuPont/NEN) and ATP (10 μM) in kinase buffer supplemented with MBP (20 μg/ml) for MAPK and incubated at 30°C for 20 min (Lck and Fyn kinases) or 10 min (MAPK). The reaction was stopped by 5× SDS sample buffer. Samples were separated by SDS/PAGE and analyzed by autoradiography.
Ras and Rac2 Activation.
Cells were metabolically labeled for 4 h with 1 mCi/ml [32Pi] (1 Ci = 37 GBq) in phosphate-free DMEM/10% FCS, activated via l-selectin and lysed in 0.1% SDS, 0.5% deoxycholate, 1% Triton X-100, 450 mM NaCl, 10 mM NaF, 25 mM Hepes (pH 7.4), 20 mM MgCl2, and 20 μg/ml aprotinin/leupeptin. Ras or Rac2 were immunoprecipitated by Ras Y13-259 or Rac2 antibodies followed by the addition of anti-rat- or anti-rabbit-coupled agarose. IPs were washed eight times in lysis buffer, resuspended in 20 μl EDTA (1 mM), incubated for 20 min at 68°C, and eluted nucleotides were separated on a thin layer chromatography polyethyleneimine-cellulose plate (Merck) with 0.75 M KH2PO4 (pH 3.5), followed by autoradiography.
Inhibition of Ras or Rac2.
Jurkat cells were transiently transfected with 40 μg transdominant inhibitory N17Ras (pEF-N17ras) or control vector (pEF) and 8 μg of a CD20 expression vector (pRc/CMV-cd20). The ratio of 5:1 (N17ras:cd20) permits expression of N17Ras in all CD20+ cells. Thirty-six hours after transfection, CD20+ cells were sorted by magnetic beads as described (20). Rac2 was inhibited by incubation of the cells for 18 h with 10 μM phosphorothioate antisense oligonucleotides (5′-ACTTGATGGCCTGCA-3′) (27, 28). Control samples were incubated with a random mix of nucleotides (10 μM, 5′-TGGCTATGCCACATG-3′).
O2− Measurement.
Cells (1–2 × 106 cells per 100-μl sample) were stimulated with 2 μg/ml Dreg56 and lysed in 2% Nonidet P-40, 125 mM NaCl, 25 mM Tris (pH 8.0), 10 mM each EDTA, NaF, and sodium pyrophosphate, and 5 μg/ml cytochrome c. Lysates were transferred to cuvettes, covered with mineral oil, and the absorbance at 550 nm was determined. The change of absorption reflects a reduction of cytochrome c by generated O2− ions (29, 30). Lysis of cells permits determination of cellular O2− synthesis, including O2− released to the extracellular space.
Guanine Nucleotide Exchange and GTPase-Activating Protein (GAP) Assays.
Recombinant Ras (500 fmol) was incubated with 1 pmol GDP for 30 min in 20 mM Tris·HCl (pH 7.4), 1 mM MgCl2, 1 mM DTT, 100 mM NaCl, 50 μg/ml BSA, 1 mM Na3VO4 (Ras loading buffer). Washed l-selectin or Sos IPs, 5 μCi [32P-α]-guanosine 5′-[γ-thio]triphosphate (GTP)/sample (DuPont/NEN) and 1 pmol GTP were added to Ras and incubated at 30°C for 15 min. The agarose was pelleted and an aliquot of the supernatant was filtered through a 0.45-μm nitrocellulose filter (Costar). Filters were washed with of 25 mM Tris·HCl (pH 7.5), 12 mM MgCl2, 100 mM KCl, and 0.5 mM DTT and filter bound counts were determined by liquid scintillation counting. To measure GAP activity, Ras (500 fmol) was preloaded with 5 μCi [32P-γ]-GTP in Ras loading buffer for 30 min, GAP-IPs were added and incubated at 30°C for 15 min, agarose beads were pelleted, an aliquot of the supernatant filtered, washed, and counted as described.
Flow Cytometry.
To analyze l-selectin expression, cells were labeled with Dreg56 and a fluorescein isothiocyanate–anti-mouse Ig. Expression was determined using a FACSort (Becton Dickinson).
RESULTS
l-Selectin Triggers Tyrosine Phosphorylation by Activation of p56lck.
To test whether l-selectin functions as a signaling molecule in lymphocytes we stimulated p56lck-expressing or -deficient Jurkat cells via l-selectin. Similar expression of l-selectin in Jurkat, JCaM1.6, and JCaM1.6+Lck cells was confirmed by flow cytometry. l-selectin stimulation of Jurkat cells resulted in rapid tyrosine phosphorylation of several intracellular proteins (Fig. 1A). Tyrosine phosphorylation was confirmed by immunoprecipitation of several tyrosine phosphorylated proteins with the anti-phosphotyrosine antibody 4G10 after stimulation of Jurkat cells via l-selectin (data not shown). Genetic p56lck deficiency almost completely prevented l-selectin-induced tyrosine phosphorylation (Fig. 1A); reconstitution of p56lck restored tyrosine phosphorylation (Fig. 1A). The specificity of tyrosine phosphorylation was analyzed in CEM+ or CEM− and in P815.L-Sel or P815.control cells (Fig. 1B). Dreg56 treatment resulted in tyrosine phosphorylation in l-selectin-positive cells only (Fig. 1B). l-selectin expression in CEM+ and P815.L-Sel cells was proved by flow cytometry (data not shown). The physiological significance of l-selectin-mediated tyrosine phosphorylation is indicated by the finding that PBL responded to l-selectin triggering with a similar tyrosine phosphorylation. To further investigate l-selectin-induced tyrosine phosphorylation we used Fucoidan (100 μg/ml), sialyl LewisX (100 μg/ml), and Dreg200 (5 μg/ml) as stimuli (Fig. 1C). All stimuli induced a similar tyrosine pattern of proteins in Jurkat and JCaM1.6+Lck cells, whereas JCaM1.6 cells did not respond (Fig. 1C). Crosslinking of Dreg56 or Dreg200 using a secondary antibody did not change tyrosine phosphorylation compared with stimulation without crosslinking (not shown).
Figure 1.

l-selectin triggering induces tyrosine phosphorylation dependent on expression of p56lck. Jurkat, JCaM1.6, JCaM1.6+Lck cells, CEM+, CEM−, P815.L-Sel, P815.control, or PBL cells were stimulated via l-selectin with Dreg56 or as indicated (A–C). l-selectin triggering induced tyrosine phosphorylation of several proteins in Jurkat, JCaM1.6+Lck, CEM+, P815.L-Sel, or PBL cells. No tyrosine phosphorylation was detected in JCaM1.6 cells, CEM−, or P815.control cells (A–C). (D) l-selectin stimulation activates p56lck in Jurkat and JCaM1.6+Lck cells peaking 1 min after stimulation. No activity was seen in JCaM1.6 cells. An aliquot of the IPs was analyzed by Western blotting with anti-p56lck to test for equal amounts of p56lck in all IPs (small panels). Cells for non-specific IPs (n.s.) were stimulated with Dreg56 and incubated with an agarose-coupled rabbit antibody. All experiments were repeated at least three times.
Because tyrosine phosphorylation in lymphocytes can be initiated by src kinases, we tested the activity of p56lck and p59fyn by measuring the autophosphorylation of the kinases (Fig. 1D). We detected an increase in p56lck activity (Fig. 1D), but not of p59fyn (data not shown), upon l-selectin stimulation in Jurkat and JCaM1.6+Lck cells. No signal was obtained in JCaM1.6 cells.
Stimulation via l-Selectin Induces Tyrosine Phosphorylation of l-selectin.
Using immunoprecipitation and Western blotting two of the tyrosine phosphorylated proteins, were identified as l-selectin (Fig. 2A) and MAPK (Fig. 3A). Tyrosine phosphorylation of l-selectin was dependent on expression of p56lck, since it was absent in JCaM1.6 cells (Fig. 2A). To exclude non-specific immunoprecipitation and tyrosine phosphorylation of l-selectin a “non-specific” IP using a mouse antibody recognizing the human B-lymphocyte antigen CD20 was included. In addition, Jurkat cells were treated with the 25–9 and a human β7-integrin antibody, which recognize antigens not expressed on Jurkat cells, followed by immunoprecipitation with Dreg56. No tyrosine phosphorylation of l-selectin was detected after incubation of Jurkat cells with these antibodies for 1 min or after treatment with antibody storage buffer (Fig. 2A) or sterile H/S (not shown). Tyrosine phosphorylation of l-selectin could also be observed in CEM+ and P815.L-Sel cells and was not detected in CEM− or P815.control cells (Fig. 2B). In addition, triggering of PBL with Dreg56 resulted in tyrosine phosphorylation of l-selectin (Fig. 2C), which could also be induced by triggering Jurkat and JCaM1.6+Lck cells with Fucoidan, sialyl LewisX, and Dreg200 (Fig. 2C) and was absent in JCaM1.6 cells (data not shown).
Figure 2.

(A) l-selectin stimulation induces tyrosine phosphorylation of l-selectin in Jurkat and JCaM1.6+Lck cells only. Cells were stimulated with Dreg56 for the indicated times, l-selectin was immunoprecipitated, and analyzed for tyrosine phosphorylation. Tyrosine phosphorylation of l-selectin was also observed in CEM+ (B), P815.L-Sel (B), or PBL cells (C) stimulated with Dreg56 and after activation of Jurkat cells with Fucoidan, sialyl LewisX, or Dreg200 (C). Stimulation of Jurkat cells with 25–9, anti-human β7-integrin (β7), buffer (b.), CEM−, or P815.control cells with Dreg56 did not result in tyrosine phosphorylation (A and B). Non-specific IPs (n.s.) using an irrelevant mouse anti-CD20 antibody showed no l-selectin IP (A). The small blots display aliquots of the IPs blotted with Dreg56 showing similar amounts of l-selectin in all IPs. These blots confirm expression of l-selectin in CEM+ and P815.L-Sel cells (B). All experiments were repeated three times.
Figure 3.
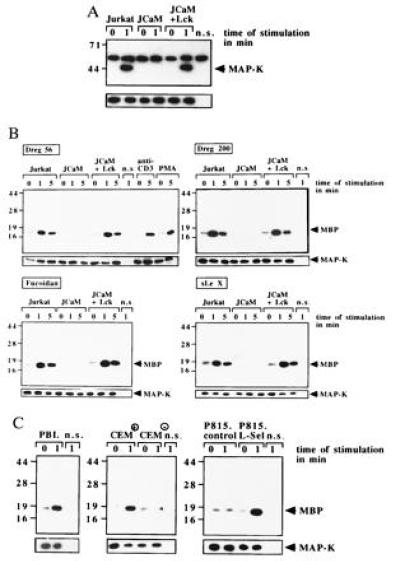
l-selectin triggering using the indicated stimuli induces tyrosine phosphorylation or activation of MAPK in p56lck-positive Jurkat, JCaM+Lck, CEM+, P815.L-Sel, or in PBL cells. Shown is a phosphotyrosine blot of MAPK IPs (B) or autoradiographies of in vitro kinase assays (B and C). The increase in MAPK activity upon stimulation via CD3 (OKT3, 10 μg/ml) or by PMA (10 ng/ml) served as a positive control (B). No increase of MBP phosphorylation is detected in CEM− and in P815.control cells (C). Non-specific controls (n.s.) represent IPs using a mouse anti-CD20 antibody. Equal amounts of MAPK are shown in the lower panels of all parts by Western blotting of aliquots of the IPs with an anti-MAPK antibody.
MAPK Is Tyrosine Phosphorylated and Activated upon l-Selectin Triggering.
l-selectin stimulation resulted in tyrosine phosphorylation of MAPK in Jurkat and JCaM1.6+Lck cells, whereas no tyrosine phosphorylation was detected in JCaM1.6 cells (Fig. 3A). MAPK activity was measured by phosphorylation of MBP and showed an ≈7-fold increase in Jurkat and JCaM1.6+Lck cells after activation with the indicated stimulus (Fig. 3B). No activation was measured in JCaM1.6 cells. Stimulation of Jurkat cells via CD3 or phorbol 12-myristate 13-acetate (PMA) served as positive controls (Fig. 3B). Activation of MAPK was also observed in PBL, CEM+, or P815.L-Sel cells upon stimulation with Dreg56. CEM− or P815.control cells did not respond to Dreg56 (Fig. 3C).
The Ras Pathway Is Stimulated by l-Selectin.
The phosphorylation and activation of MAPK points to a stimulation of the Ras pathway upon l-selectin triggering (31, 32). l-selectin stimulation by Dreg56 induced an ≈6-fold increase in Ras activity in Jurkat and JCaM1.6+Lck cells peaking 1 min after stimulation, whereas JCaM1.6 cells did not show any increase in Ras activity (Fig. 4A), suggesting that the tyrosine kinase p56lck controls Ras activation. Triggering P815.L-Sel or CEM+ cells with Dreg56 elicited an activation of Ras, whereas CEM− or P815.control cells did not respond, indicating the specificity of the activation (Fig. 4B). Stimulation of Ras in a p56lck-dependent manner was also observed upon incubation with Dreg200, Fucoidan, and sialyl LewisX pointing to the physiological significance of Ras activation (Fig. 4C, data not shown).
Figure 4.
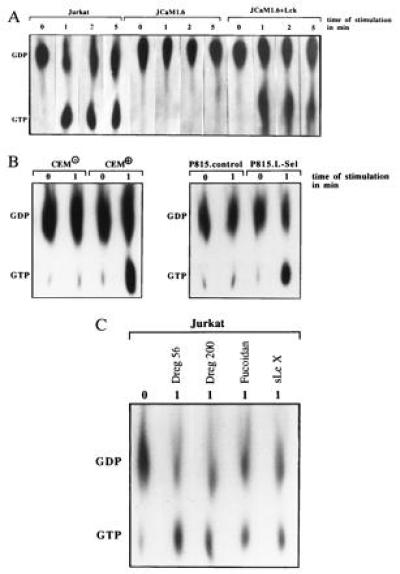
l-selectin triggering with Dreg56 or the indicated stimulus activates Ras in Jurkat, JCaM1.6+Lck, CEM+, and P815.L-Sel cells. Deficiency of p56lck in JCaM1.6 cells (A) or of l-selectin (B) prevent Ras activation after l-selectin triggering. Results are representative of three experiments.
Ras and Sos Associate upon l-Selectin Stimulation.
Because Ras is activated by guanine nucleotide exchange factors (GEF) (33), we tested in co-immunoprecipitation experiments the physical association of Sos, Grb2, or l-selectin. l-selectin associates with Sos and Grb2 upon activation (Fig. 5 A and B). The formation of this complex seems to be dependent on the function of p56lck, since it could not be detected in JCaM1.6 cells (Fig. 5 A and B). The association of l-selectin and Grb2/Sos was confirmed by an increase of GDP/GTP exchange activity in l-selectin-IPs by a functional assay (Fig. 5C). We were unable to detect any significant association of the Shc protein with l-selectin IPs (data not shown). Ras can be either activated by increased activity of guanine nucleotide exchange factors or by a decrease of p120GAP protein activity (33). However, l-selectin triggering did not change the high basal GAP activity measured by hydrolysis of [32P-γ]-GTP.Ras, whereas PMA induced a 60% reduction of GAP activity as described (34). Thus, l-selectin triggering seems to translocate Grb2/Sos from the cytoplasm to the cell membrane bringing Sos in close contact to Ras resulting in Ras activation.
Figure 5.
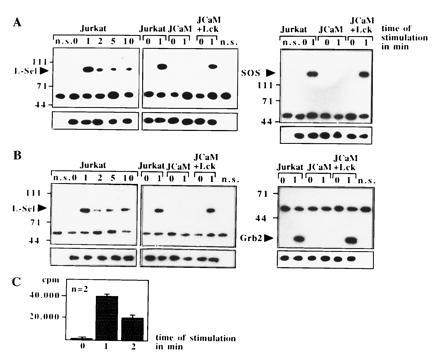
(A and B) The GEF Sos, the adapter protein Grb2, and l-selectin associate upon stimulation via l-selectin in Jurkat or JCaM1.6+Lck cells. Association was revealed by co-immunoprecipitations and blotting Sos- or Grb2-IPs with anti-l-selectin or vice versa. No association of Sos, Grb2, and l-selectin was detected in JCaM1.6 cells. All blots were reprobed with the immunoprecipitating antibody (small panels). Non-specific IPs (n.s.) were performed with anti-CD20 as above. The blots are representative of three experiments. (C) Exchange activity in l-selectin IPs increases upon l-selectin triggering. l-selectin IPs were incubated with recombinant GDP. Ras and [32P-α]-GTP. Increase in binding of [32P-α]-GTP to Ras reflects exchange activity.
l-Selectin Triggering Activates Rac2 and Synthesis of O2− via Ras.
Because Rac2 is a downstream target of Ras (35), Rac2 activity was determined. GTP binding to Rac2, representing Rac2 activation, increased ≈8-fold after l-selectin triggering (Fig. 6A). Activation of Rac2 was inhibited by transient transfection of N17Ras (Fig. 6B Left), which has been shown by us and others to block endogenous Ras (20, 36). Controls showed that N17Ras expression efficiently prevented stimulation of endogenous Ras by l-selectin (Fig. 6B Right). Rac2 has been shown in lymphocytes (27) to regulate the synthesis of O2−. l-selectin induced a rapid and transient O2− synthesis peaking 1 min after stimulation in Jurkat and JCaM1.6+Lck cells, which was completely absent in JCaM1.6 cells (Fig. 6C). Likewise, inhibition of Ras by transient transfection of N17Ras prevented l-selectin stimulated O2− synthesis (Fig. 6C). To test the involvement of Rac2 in l-selectin mediated O2− release, Rac2 protein expression was inhibited by more than 90% using antisense oligonucleotides (Fig. 6C Inset). Stimulation of these Rac2-depleted Jurkat cells via l-selectin failed to induce a release of O2−, whereas treatment of cells with a control oligonucleotide did not affect Rac2 protein expression and O2− synthesis after l-selectin triggering (Fig. 6C). Co-incubation of cell lysates with superoxide dismutase (200 units/ml), which specifically degrades O2− (29), prevented l-selectin induced cytochrome c reduction demonstrating the specificity of O2− synthesis.
Figure 6.
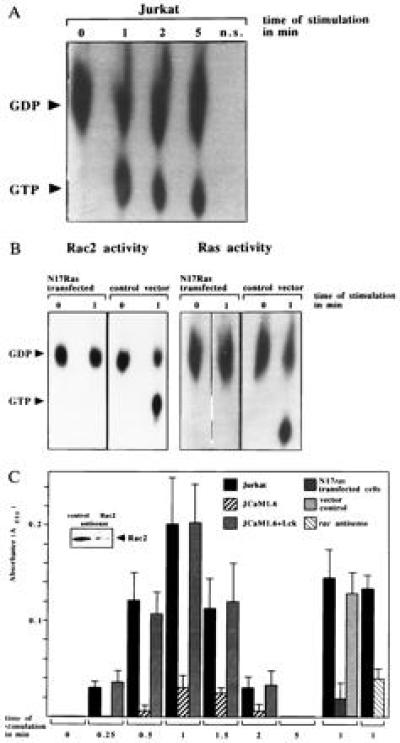
(A) l-selectin triggering stimulates Rac2. Rac2 was immunoprecipitated from [32Pi]-labeled, Dreg56-treated Jurkat cells and the increase of GTP binding was analyzed by autoradiography. (B) Inhibition of endogenous Ras by N17Ras prevents Rac2 and Ras activation upon l-selectin triggering. Transfection of the vector did not affect l-selectin-induced Rac2 or Ras stimulation. Rac2 or Ras activity in CD20+ sorted cells was determined as above. (C) l-selectin triggering leads to synthesis of O2− in Jurkat and in JCaM1.6+Lck cells. Genetic deficiency of p56lck, inhibition of Ras with N17Ras or of Rac2 by antisense oligonucleotides prevents O2− synthesis. Mean values are ± SD.
DISCUSSION
The interaction of l-selectin with its endothelial ligands has been shown to play a fundamental role in immune homeostasis by recruiting leukocytes to inflammatory sites (1, 2, 5) and mediating lymphocyte homing (3, 4).
We describe a signaling cascade in lymphocytes upon l-selectin triggering via activation of p56lck, tyrosine phosphorylation of the l-selectin molecule, physical association of l-selectin with Sos/Grb2, and stimulation of Ras, MAPK, and Rac2 to O2− synthesis. Using different l-selectin positive and negative cell lines, l-selectin transfectants, and control antibodies, we confirm the specificity of the observed signaling events initiated by l-selectin. The physiological significance is indicated by experiments applying PBL and different ligands of l-selectin. A signaling cascade is demonstrated using genetically deficient cells, transdominant inhibitory mutants, and antisense oligonucleotides.
To establish the specificity of cellular activation via l-selectin triggering we first transfected the mouse mastocytoma cell line P815 with an expression vector of human l-selectin or a control vector and stimulated the cells with the human-l-selectin antibody Dreg56. Second, we compared signal transduction events in l-selectin negative and l-selectin positive CEM cells upon l-selectin stimulation. These experiments show the activation of the same signaling molecules in P815.L-Sel or in CEM+ as in Jurkat cells or PBLs, whereas the negative controls did not respond to l-selectin triggering.
Furthermore, treatment of Jurkat cells with two antibodies recognizing epitopes not expressed in Jurkat cells (anti-25-9 and anti-human-β7-integrin), with antibody storage buffer or H/S, did not induce tyrosine phosphorylation of l-selectin indicating a specific stimulation of Jurkat cells by Dreg56.
In conclusion, the experiments using l-selectin transfectants, l-selectin positive/negative CEM cells, and the control antibodies show a specific activation after stimulation with Dreg56.
To show that activation of intracellular signaling molecules by l-selectin is not restricted to cell lines, we stimulated PBL via l-selectin and detected tyrosine phosphorylation or activation of l-selectin or MAPK similar to Jurkat, CEM+, or P815.L-Sel cells.
To test whether Dreg56 induced signaling events can also be initiated by other stimuli of l-selectin we used the l-selectin-antibody Dreg200 and the glycomimetics Fucoidan or sialyl LewisX, which have been shown to interact with l-selectin (12, 23–25). All stimuli resulted in similar tyrosine phosphorylation or activation of proteins, in particular l-selectin, MAPK, and Ras. Because the four stimuli activate the same signaling molecules, our results suggest a physiological function of the observed signaling cascade initiated by l-selectin. Furthermore, the results also underline the specificity of the activation via l-selectin, because the four stimuli are different in molecular structure and binding characteristics.
To show the activation of an intracellular signaling cascade by l-selectin we applied JCaM1.6 cells, transdominant inhibitory N17Ras, and Rac2 antisense oligonucleotides. The data suggest first, that p56lck functions upstream of Ras and Rac 2, second, that Ras is upstream of Rac2 and O2− synthesis and third, that O2− synthesis is regulated by Rac2 upon l-selectin triggering. The failure to show tyrosine phosphorylation in JCaM1.6 cells is due to a signaling defect and not due to a lower surface expression of the l-selectin molecule as tested by flow cytometry.
The efficiency of N17Ras transfection and the purification process is shown by the almost complete suppression of Ras activation in N17Ras-transfected cells upon l-selectin triggering. The specificity of the inhibition of endogenous Ras by N17Ras has been previously shown by us (20). In conclusion, these experiments point to a signaling cascade from l-selectin via p56lck, Ras, and Rac2 to the synthesis of O2−.
Since Ras can be activated by translocation of Sos to the membrane bringing the protein in close contact with Ras (37, 38), the observed association of Sos and l-selectin after l-selectin triggering may enable Sos to stimulate Ras. The association of the proteins correlates with the tyrosine phosphorylation of l-selectin. l-selectin contains only one tyrosine residue in the intracellular domain without a typical Src homology 2 domain-binding motif (39); thus, the mechanism of Sos and l-selectin association remains to be elucidated. Because no association of l-selectin and Sos could be detected in p56lck-deficient JCaM1.6 cells, the expression of p56lck seems to be required for the translocation of Sos upon l-selectin triggering. However, the exact function of p56lck for the association of Grb2/Sos with l-selectin has to be determined.
An active signaling function of the l-selectin molecule described in this study is supported by experiments demonstrating that stimulation of neutrophils with anti-l-selectin antibodies or sulfatides increases cytosolic free calcium and tumor necrosis factor α or interleukin 8–mRNA expression (14, 16). In accordance with our results in lymphocytes, activation of neutrophils via l-selectin by F(ab′)2 of Dreg56, Dreg200, Dreg55, or sulfatides induced tyrosine phosphorylation of cellular proteins, an activation of MAPK and a transient O2− generation (15–17).
Our results show that l-selectin activates an intracellular signaling cascade, which may mediate several potential biological functions of the l-selectin molecule. The activation events may be linked to rolling of leukocytes, which is supported by the finding that partial deletion of the intracellular receptor domain or cellular treatment with cytochalasin B abrogates leukocyte rolling in vivo without affecting ligand recognition (13). A function of p56lck in rolling of leukocytes is supported by our preliminary experiments showing an almost complete absence of rolling in JCaM1.6 cells in a Fucoidan-coated flow channel, whereas Jurkat and JCaM1.6+Lck cells exhibited similar rolling on Fucoidan in vitro. However, a requirement of the identified intracellular signaling events for the ability of leukocytes to roll on endothelial cells in vivo has to be tested.
Some of the observed signaling events after triggering of Jurkat cells via l-selectin resemble signaling pathways involved in T-cell antigen receptor/CD3 signaling (33, 34, 40). l-selectin may therefore function as costimulus for antigen-mediated cell proliferation or differentiation. This hypothesis is supported by experiments showing an association of l-selectin with the CD3-zeta-chain and a synergistic effect of T-cell antigen receptor-induced cell proliferation by l-selectin triggering (41).
In summary, our results show a signaling cascade in lymphocytes upon l-selectin triggering from l-selectin via the tyrosine kinase p56lck to Sos, Ras, MAPK, and Rac2 resulting in synthesis of O2−. This signaling cascade might be important for the biological functions of the l-selectin molecule.
Acknowledgments
B.B. and E.G. contributed equally to this work and share first authorship. We would like to thank Drs. A. Weiss (University of California, San Francisco) for Lck-transfected JCaM1.6 cells, T. F. Tedder (Duke University) for the l-selectin-expression construct, and T. K. Kishimoto (Boehringer Ingelheim) for Dreg200. This work was supported by grants from the University of Heidelberg (134\N96) to B.B., from the Deutsche Forschungsgemeinschaft (Gu335/2-1, La315/5-1, Schl390/1-1), and the Sandoz Foundation (96\N18) to E.G., and from National Science Foundation (MCB9317027) to K.M.C.
Footnotes
Abbreviations: MAPK, mitogen-activating protein kinase; PBL, peripheral blood lymphocyte; IP, immunoprecipitate; GAP, GTPase-activating protein; MBP, myelin basic protein; H/S, Hepes/saline.
References
- 1.Bevilacqua M P. Annu Rev Immunol. 1993;11:767–804. doi: 10.1146/annurev.iy.11.040193.004003. [DOI] [PubMed] [Google Scholar]
- 2.Lasky L A. Science. 1992;258:964–969. doi: 10.1126/science.1439808. [DOI] [PubMed] [Google Scholar]
- 3.Butcher E C, Picker L J. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 4.Springer T A. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 5.Butcher E C. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 6.Arbonés M L, Ord D C, Ley K, Ratech H, Maynard-Curry C, Oten G, Capon D J, Tedder T F. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 7.Kishimoto T K, Warnock R A, Jutila M A, Butcher E C, Lane C, Anderson D C, Smith C W. Blood. 1991;78:805–811. [PubMed] [Google Scholar]
- 8.Ley K, Tedder T F, Kansas G S. Blood. 1993;82:1632–1638. [PubMed] [Google Scholar]
- 9.Watson S R, Fennie C, Lasky L A. Nature (London) 1991;349:164–167. doi: 10.1038/349164a0. [DOI] [PubMed] [Google Scholar]
- 10.Tangelder G J, Arfors K E. Blood. 1991;77:1565–1571. [PubMed] [Google Scholar]
- 11.Nelson R M, Cecconi O, Roberts W G, Aruffo A, Linhardt R J, Bevilacqua M P. Blood. 1993;82:3253–3258. [PubMed] [Google Scholar]
- 12.Ley K, Linnemann G, Meinen M, Stoolman L M, Gaehtgens P. Blood. 1993;81:177–185. [PubMed] [Google Scholar]
- 13.Kansas G S, Ley K, Munro J M, Tedder T F. J Exp Med. 1993;177:833–838. doi: 10.1084/jem.177.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laudanna C, Constantin G, Baron P, Scarpini E, Scarlato G, Cabrini G, Dechecchi C, Rossi F, Cassatella M A, Berton G. J Biol Chem. 1994;269:4021–4026. [PubMed] [Google Scholar]
- 15.Waddell T K, Fialkow L, Chan C K, Kishimoto T K, Downey G P. J Biol Chem. 1994;269:18485–18491. [PubMed] [Google Scholar]
- 16.Crockett-Torabi E, Sulenbarger B, Smith C W, Fantone J C. J Immunol. 1995;154:2291–2302. [PubMed] [Google Scholar]
- 17.Waddell T K, Fialkow L, Chan C K, Kishimoto T K, Downey G P. J Biol Chem. 1995;270:15403–15411. doi: 10.1074/jbc.270.25.15403. [DOI] [PubMed] [Google Scholar]
- 18.Goldsmith M A, Weiss A. Proc Natl Acad Sci USA. 1987;84:6879–6883. doi: 10.1073/pnas.84.19.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strauss D B, Weiss A. Cell. 1992;70:585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 20.Gulbins E, Bissonette R, Mahboubi A, Martin S, Nishioka W, Brunner T, Baier G, Baier-Bitterlich G, Byrd C, Lang F, Kolesnick R, Altman A, Green D. Immunity. 1995;2:341–351. doi: 10.1016/1074-7613(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 21.Rosen S O, Chi S I, True D D, Singer M S, Yednock T A. J Immunol. 1989;142:1895–1902. [PubMed] [Google Scholar]
- 22.Jutila M A, Kishimoto T K, Butcher E C. Blood. 1990;76:178–183. [PubMed] [Google Scholar]
- 23.Kishimoto T K, Jutila M, Butcher E C. Proc Natl Acad Sci USA. 1990;87:2244–2248. doi: 10.1073/pnas.87.6.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imai Y, True D D, Singer M S, Rosen S D. J Cell Biol. 1990;111:1225–1232. doi: 10.1083/jcb.111.3.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kansas G S, Spertini O, Stoolman L M, Tedder T F. J Cell Biol. 1991;114:351–358. doi: 10.1083/jcb.114.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berg E L, Robinson M K, Warnock R A, Butcher E C. J Cell Biol. 1991;114:343–349. doi: 10.1083/jcb.114.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorseuil O, Vasquez A, Lang P, Bertoglio J, Gacon G, Leca G. J Biol Chem. 1992;267:20540–20542. [PubMed] [Google Scholar]
- 28.Gulbins E, Coggeshall K M, Brenner B, Schlottmann K, Linderkamp O, Lang F. J Biol Chem. 1996;271:26389–26394. doi: 10.1074/jbc.271.42.26389. [DOI] [PubMed] [Google Scholar]
- 29.Mayo L A, Curnutte J T. Methods Enzymol. 1990;186:567–575. doi: 10.1016/0076-6879(90)86151-k. [DOI] [PubMed] [Google Scholar]
- 30.Gulbins E, Brenner B, Schlottmann K, Welsch J, Heinle H, Linderkamp O, Coggeshall K M, Lang F. Immunology. 1996;89:205–212. doi: 10.1046/j.1365-2567.1996.d01-743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelech S L, Sanghera J S. Science. 1992;257:1355–1356. doi: 10.1126/science.1382311. [DOI] [PubMed] [Google Scholar]
- 32.deVries-Smits A M M, Burgering B M T, Leevers S J, Marshall C J, Bos J L. Nature (London) 1992;357:602–604. doi: 10.1038/357602a0. [DOI] [PubMed] [Google Scholar]
- 33.Boguski M S, McCormick F. Nature (London) 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 34.Downward J, Graves J D, Warne P H, Rayter S, Cantrell D. Nature (London) 1990;346:719–723. doi: 10.1038/346719a0. [DOI] [PubMed] [Google Scholar]
- 35.Ridley A J, Paterson H F, Johnston C L, Diekmann D, Hall A. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 36.Feig L A, Cooper G M. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aronheim A, Engelberg D, Li N, al-Alawi N, Schlessinger J, Karin M. Cell. 1994;78:949–961. doi: 10.1016/0092-8674(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 38.Quilliam L A, Huff S Y, Rabun K M, Wei W, Park W, Broek D, Der C J. Proc Natl Acad Sci USA. 1994;91:8512–8516. doi: 10.1073/pnas.91.18.8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch C A, Anderson D, Moran M F, Ellis C, Pawson T. Science. 1991;252:668–674. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- 40.Weiss A, Littman D R. Cell. 1994;76:263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 41.Murakawa Y, Minami Y, Strober W, James S P. J Immunol. 1992;148:1771–1776. [PubMed] [Google Scholar]