

Abstract
Primary HIV-1 isolates were evaluated for their sensitivity to inhibition by β-chemokines RANTES (regulated upon activation, normal T-cell expressed and secreted), macrophage inflammatory protein 1α (MIP-1α), and MIP-1β. Virus isolates of both nonsyncytium-inducing (NSI) and syncytium-inducing (SI) biological phenotypes recovered from patients at various stages of HIV-1 infection were assessed, and the results indicated that only the isolates with the NSI phenotype were substantially inhibited by the β-chemokines. More important to note, these data demonstrate that resistance to inhibition by β-chemokines RANTES, MIP-1α, and MIP-1β is not restricted to T cell line-adapted SI isolates but is also a consistent property among primary SI isolates. Analysis of isolates obtained sequentially from infected individuals in whom viruses shifted from NSI to SI phenotype during clinical progression exhibited a parallel loss of sensitivity to β-chemokines. Loss of virus sensitivity to inhibition by β-chemokines RANTES, MIP-1α, and MIP-1β was furthermore associated with changes in the third variable (V3) region amino acid residues previously described to correlate with a shift of virus phenotype from NSI to SI. Of interest, an intermediate V3 genotype correlated with a partial inhibition by the β-chemokines. In addition, we also identified viruses sensitive to RANTES, MIP-1α, and MIP-1β of NSI phenotype that were isolated from individuals with AIDS manifestations, indicating that loss of sensitivity to β-chemokine inhibition and shift in viral phenotype are not necessarily prerequisites for the pathogenesis of HIV-1 infection.
Keywords: nonsyncytium-inducing, syncytium-inducing, RANTES, macrophage inflammatory protein 1α, macrophage inflammatory protein 1β
Biological phenotypes of primary isolates of HIV-1 can be defined according to specific differences in virus–host–cell interactions. The most evident characteristics of these interactions include virus replication rate, syncytia induction, and ability to infect CD4+ cell lines (1–5). Based on an ability to replicate and induce syncytia in transformed T-cell lines, such as the MT-2 cell line, primary HIV-1 isolates can be referred to as either rapid/high syncytium-inducing (SI) or slow/low nonsyncytium-inducing (NSI) (1, 2, 6, 7). Elevated replication rate, tropism for transformed T-cell lines, and SI capacity of the virus appear to correlate with progression to AIDS (1, 3, 4, 8, 9). In contrast, NSI viruses, which lack the ability to infect permanent CD4+ cell lines but readily infect and replicate in primary T cells and monocytes/macrophages, are predominantly harbored by asymptomatic HIV-1-infected individuals (1, 3, 4, 8, 9).
The ability of virus isolates to induce syncytia and to replicate in transformed CD4+ cell lines has been correlated with changes in the charge of certain amino acid positions in the third variable (V3) region of the gp120 envelope protein (10). Viral tropism also appears to shift with variations in the V3 region (11–15). Tropism of different isolates of HIV-1 for distinct cell types has been clarified by the recent findings of coreceptors that, in addition to CD4 molecules, are required for entry of HIV-1 into human cells (16–21). LESTR, a member of the 7-transmembrane G protein-coupled receptor family (22), was described as a coreceptor for HIV-1 isolates with the capacity to infect permanent CD4+ cell lines and was termed “fusin” (16). Subsequent identification of the α-chemokine stromal cell-derived factor 1 as the natural ligand for the coreceptor LESTR/fusin and its ability to block infection in vitro by T-cell line-adapted HIV-1 isolates further substantiated these findings (23, 24).
Virus isolates replicating in primary T cells and monocytes/macrophages, also termed “macrophage tropic” or NSI viruses, did not fuse with cells coexpressing CD4 and LESTR/fusin, indicating that such viruses used an alternative coreceptor than was used by T-cell line-adapted viruses (16). This coreceptor was subsequently identified as CC chemokine receptor 5 (CKR-5) (17–21). The natural ligands of the CC CKR-5 receptor are the β-chemokines RANTES (regulated upon activation, normal T-cell expressed and secreted), macrophage inflammatory protein 1α (MIP-1α), and macrophage inflammatory protein 1β (MIP-1β) (25). Chemokines are a group of related peptides that, like other cytokines, mediate signaling between different cells in the immune system. It appears that a more specialized role of the chemokines is to attract inflammatory cells to areas of infection (26). α-Chemokines, also termed CXC chemokines, are primarily chemotactic for neutrophils whereas β-chemokines, or CC chemokines, mainly attract monocytes, lymphocytes, basophils, and eosinophils (27, 28). Recently, it was reported that β-chemokines can inhibit replication of HIV-1 in vitro (29). RANTES, MIP-1α, and MIP-1β, which belong to the β-chemokine family, were identified as the major HIV-1 inhibitory factors produced by CD8+ T cells (29).
In the present study, we evaluated sensitivity of a broad spectrum of primary HIV-1 isolates to β-chemokines RANTES, MIP-1α, and MIP-1β. We demonstrate that, among virus isolates of NSI and SI phenotypes obtained from infected individuals at various stages of infection, NSI viruses were inhibited by these β-chemokines whereas SI viruses were not. When isolates that were obtained sequentially from infected individuals in whom viruses shifted from NSI to SI phenotype were analyzed, the β-chemokines primarily inhibited virus isolates with the NSI phenotype. Thus, a shift of virus phenotype from NSI to SI in infected individuals occurred in parallel with a loss of sensitivity to β-chemokines. In addition, the loss of sensitivity to the β-chemokines correlated with characteristic changes in V3 amino acid residues that were described (10) in the virus phenotype switch from NSI to SI. Furthermore, we also identified individuals with AIDS manifestations with viruses of NSI phenotype shown to be sensitive to RANTES, MIP-1α, and MIP-1β, demonstrating that presence of β-chemokine-sensitive viruses does not necessarily reflect an asymptomatic clinical stage of an HIV-1-infected individual.
MATERIALS AND METHODS
Virus Isolates.
Viruses were isolated as described (30). In brief, isolates were obtained by coculturing 1–2 million peripheral blood mononuclear cells (PBMC) from HIV-infected individuals with phytohemagglutinin (PHA)-stimulated PBMC from two blood donors. The cultures were maintained in complete RPMI 1640 medium (GIBCO) supplemented with 10% fetal calf serum (Flow Laboratories), 5 units/ml recombinant interleukin 2 (Amersham), 2 μg/ml polybrene (Sigma), and antibiotics. Virus antigen production was measured in supernatants twice weekly using an HIV-1 p24 antigen capture ELISA (31). At the time of positive virus cultures, virus stocks were generated by passaging of the virus isolates once or twice in PHA-stimulated blood donor PBMC, after which viruses were aliquotted and cryopreserved at −70°C. Three panels of virus isolates were analyzed for sensitivity to β-chemokines. One panel contained viruses of NSI and SI phenotype obtained from HIV-1-infected individuals at various clinical stages. The two other panels contained isolates obtained sequentially from HIV-1-infected patients with viruses that (i) shifted from NSI to SI phenotype and (ii) maintained NSI virus phenotype despite AIDS. Absolute counts of CD4+ lymphocytes and time span to onset of AIDS and to death in relation to time of virus isolation are depicted in Table 1.
Table 1.
Clinical data for HIV-1-infected patients
| Patient | Isolate | Virus phenotype* | CD4 count† | AIDS onset, mo‡ | Death, mo‡ |
|---|---|---|---|---|---|
| A | J2195 | NSI | 600 | +55 | +71 |
| J4052 | SI | 260 | +35 | +52 | |
| B | J562 | NSI | 250 | +4 | +25 |
| J975 | SI | 110 | −4 | +17 | |
| C | J669 | NSI | 320 | +11 | +66 |
| J1629 | SI | 210 | −4 | +51 | |
| D | J1874 | NSI | 360 | —§ | —§ |
| J2337 | SI | 410 | —§ | —§ | |
| E | J2090 | NSI | 50 | +10 | +31 |
| J2822 | SI | 20 | +4 | +25 | |
| F | J2234 | NSI | 310 | +24 | +43 |
| J4996 | NSI | 190 | −3 | +16 | |
| G | J1228 | NSI | 320 | +9 | +41 |
| J4482 | NSI | 5 | −26 | +6 | |
| H | J624 | NSI | 290 | +27 | +42 |
| J3899 | NSI | 6¶ | −14 | +1 |
Virus isolates obtained sequentially with (i) switched phenotype from NSI to SI (patients A–E) and (ii) maintained NSI phenotype despite AIDS (patients F–H). Absolute counts of CD4+ lymphocytes were measured at time of virus isolation, and time span to onset of AIDS or death was calculated in relation to the time of virus isolation.
Virus phenotype as classified by MT-2 assay.
CD4+ lymphocyte count in cells/mm3.
Time span in months to the onset of AIDS or to death in relation to the time of the virus isolation; − before isolation; +, after isolation.
Patient D had not developed AIDS and was alive at time of collection of clinical data.
Available CD4 count was measured 6 months before virus isolation.
Virus 50% Infectious Dose (ID50) Determination.
For determination of ID50 of virus stocks, PBMC from one blood donor, BC 550, were used as target cells. The PBMC were activated with PHA at 2.5 μg/ml and cultured for 3 days in RPMI 1640 medium (GIBCO) supplemented with 10% fetal calf serum (Flow Laboratories) and antibiotics. The activated PBMC were thereafter aliquotted, frozen, and stored at −70°C until use. At the time of chemokine inhibition experiments, the activated PBMC were thawed and expanded for 2–3 days in complete RPMI 1640 medium supplemented as described above. CD8+ T cells were depleted from the activated PBMC using Dynabeads M-450 CD8 (Dynal, Oslo) according to the protocol provided by the manufacturer. CD8+ T-cell-depleted PBMC (1 × 105) in 75 μl of complete medium were seeded in each well in microtiter plates (Nunc). Virus stocks were thawed and serially diluted in 5-fold steps starting from a dilution of 1:2. Each dilution of virus in 75 μl of complete medium was added to five parallel wells of cells followed by incubation for 1 h at 37°C. Complete medium (75 μl) was then added per well, and incubation at 37°C was continued. At day 3 postinfection, the old medium was removed, and new complete medium was added. At day 7 postinfection, cell culture medium was harvested, and HIV-1 p24 antigen was detected. The ID50 value was defined as the reciprocal of the virus dilution resulting in 50% positive wells (Reed–Muench calculation).
Determination of Virus Phenotype: MT-2 Assay.
Virus phenotype was determined as described (32). In brief, MT-2 cells and PHA-stimulated PBMC from donors were infected with the primary HIV-1 isolates in parallel. MT-2 cultures were monitored for syncytium formation and HIV-1 p24 antigen production. If syncytia and p24 antigen production were found in the MT-2 culture, the virus was classified as MT-2-positive and with SI phenotype; the virus was classified as MT-2-negative and with NSI phenotype if it replicated in PBMC but showed no syncytia or p24 antigen production in MT-2 culture.
Chemokine Inhibition Assay.
PBMC from one blood donor (BC 550) were used throughout all chemokine inhibition experiments. Preparation of the PBMC was performed as described above. CD8+ T cell-depleted PBMC (1 × 105) in 75 μl of complete medium were seeded in each well in microtiter plates followed by the addition of 75 μl of virus-containing cell culture supernatant. For each separate experiment, ID50 was determined in parallel with the inhibition assay as described above. Plates with cells and virus were incubated for 1 h at 37°C, and then 75 μl of medium with different concentrations of the recombinant chemokines RANTES, MIP-1α, and MIP-1β (PharMingen) was added. Control cultures received 75 μl of complete medium without β-chemokines. At day 3 postinfection, medium was changed, and incubation with chemokines was continued. Supernatants were harvested at day 7, and HIV-1 p24 antigen content was determined using an HIV-1 p24 antigen capture ELISA (31). The mean OD value of control wells was considered as 100% p24 release, and the OD values of sample wells, in which chemokines were added, were calculated as percentage of p24 release compared with control cultures. Chemokine sensitivity of each virus isolate was analyzed at least twice at different virus concentrations.
DNA Sequence Analysis.
DNA for PCR amplification was recovered from virus-infected blood donor PBMC as described (33). In short, 3 days after virus infection, PHA-stimulated blood donor PBMC were lysed at a concentration of 107 cells/ml in lysis buffer containing 10 mM Tris·HCl (pH 8.3), 1 mM EDTA, 0.5% Nonidet P-40, 0.5% Tween 20, and proteinase K at a concentration of 300 μg/ml. Viral DNA was amplified by nested PCR from the cell lysate as described (30, 34) using primer pairs JA9/JA 12 and JA 10/JA 53, which are specific for the HIV-1 gp120 V3 region. Sequencing was performed directly, without cloning, using a solid-phase DNA sequencing protocol (35). An Autoread sequencing kit (Pharmacia) was used for sequencing. The reaction products were separated in a 6% polyacrylamide gel and analyzed with an automated laser fluorescent sequencing apparatus (Pharmacia).
RESULTS
Sensitivity to β-Chemokines of Primary HIV-1 Isolates Correlates with NSI Biological Phenotype.
To examine if different patterns of sensitivity to β-chemokine inhibition could distinguish divergent primary HIV-1 isolates, a panel of isolates obtained from 13 different HIV-1-infected individuals was tested for sensitivity to RANTES, MIP-1α, and MIP-1β. Six (A00IC, A0009, J1496, J8853, J8894, and J9013) were classified as SI viruses, and seven (A00AR, A0022, A0045, A0049, BG178, CLS 1.1, and mp24) were classified as NSI viruses (data not shown). To eliminate possible variations caused by differences in susceptibility of PBMC of different blood donors to HIV-1 infection, target cells for virus infection in the ID50 determinations and chemokine inhibition assays were restricted to PHA-stimulated PBMC from one blood donor throughout the analyses. To test virus sensitivity to inhibition by β-chemokines, HIV-1 isolates were cultured in the presence of 3-fold serially diluted β-chemokines, RANTES, MIP-1α, and MIP-1β, starting at a concentration of 200 ng/ml of each chemokine. After 7 days of culture in the presence of the highest concentration of β-chemokines, the levels of p24 antigen produced by all seven NSI viruses were <10% of those produced by the same viruses in the absence of β-chemokines (Fig. 1a). Six NSI viruses were inhibited to >90% when exposed to 67 ng/ml of each chemokine. In contrast, all of the SI viruses, except J1496, replicated to the same levels independently of the absence or presence of the β-chemokines (Fig. 1b). At the highest concentration of chemokines (200 ng/ml), a less than 2-fold inhibitory effect was observed with SI isolate J1496. These findings demonstrated that primary virus isolates with an SI phenotype had no or limited sensitivity to the β-chemokines RANTES, MIP-1α and MIP-1β and that replication of viruses with an NSI phenotype was readily inhibited after exposure to the β-chemokines.
Figure 1.
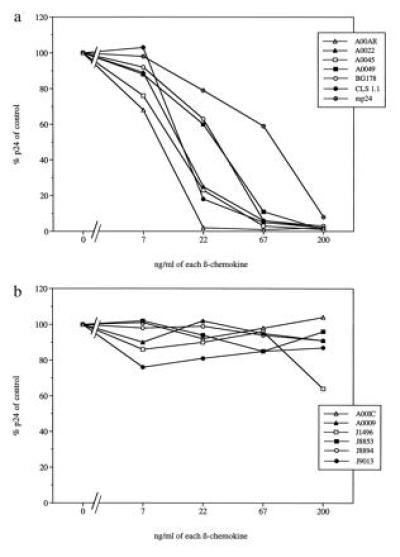
Sensitivity to β-chemokines of primary HIV-1 isolates with either NSI or SI biological phenotype. Percentage of HIV-1 p24 release, in relation to controls, of β-chemokine-exposed virus cultures of (a) NSI phenotype or (b) SI phenotype.
Concurrent Loss of Sensitivity to β-Chemokines and Shift in Virus Phenotype of Sequentially Obtained Primary Virus Isolates.
To further examine the association between virus sensitivity to β-chemokines and biological phenotype, we examined primary virus isolates obtained sequentially from five HIV-1-infected individuals. During the course of infection, the phenotype of the virus present in these individuals switched from the NSI to the SI phenotype (Table 1). The time span between the isolation of the last NSI virus and the isolation of the first SI virus from the same individual ranged from 6 to 20 months. To eliminate any influence of variation in virus input in the inhibition analysis, virus isolates from individual patients were assayed at equivalent concentrations of virus as determined by ID50 titration before, as well as in parallel with, the β-chemokine inhibition assay. All early isolates of NSI type from the five HIV-1-infected patients were sensitive to β-chemokines (Fig. 2a). On the contrary, none of the later SI isolates was substantially inhibited after exposure to the same concentration of β-chemokines (Fig. 2b). A partial block (51% and 73% of p24 release compared with control cultures, respectively) was observed at the highest concentration of β-chemokines using SI isolates from patients B and C. Despite the short interval of 6 months between the isolation of the NSI and SI isolates from patients D and E, a complete block of p24 antigen release was observed after exposure of the NSI virus-infected cells to 67 or 22 ng/ml of each chemokine, respectively. In contrast, the later SI isolates from the same individuals were unaffected by the presence of β-chemokines in the cell culture medium. These results demonstrate that the sensitivity to β-chemokines RANTES, MIP-1α, and MIP-1β is lost in parallel with a switch in phenotype from NSI to SI of HIV-1 isolates obtained sequentially from single individuals.
Figure 2.
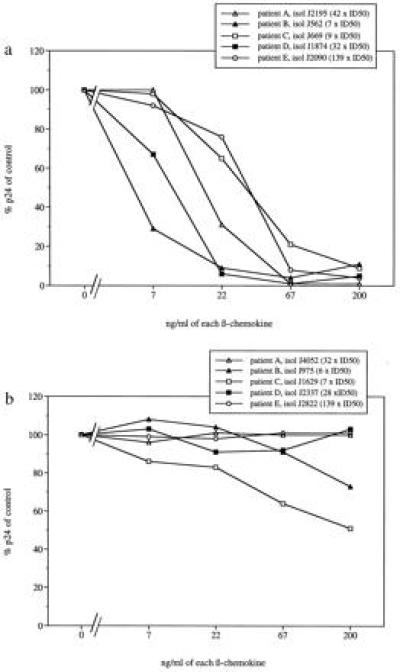
Sensitivity to β-chemokine-mediated replication inhibition of primary HIV-1 isolates with different biological phenotypes obtained sequentially from individual patients. Percentage of p24 release, in relation to controls, of β-chemokine-exposed virus cultures with (a) NSI phenotype or after switch with (b) SI phenotype.
V3 Sequence Change Parallels Loss of Sensitivity to β-Chemokine.
Switches in virus phenotype have been associated with changes of certain amino acids inside the HIV-1 gp120 V3 loop sequence (10), so we examined if changes of the same amino acids correlated with loss of virus sensitivity to β-chemokines. Viral DNA of the V3 region were sequenced from the sequential isolates of the five individuals described above. V3 amino acid residues were numbered according to the HIV-1MN sequence presented in the Los Alamos data base (36). Results of the deduced amino acids indicated that changes in charge of the V3 amino acid residues at positions 311 and 325 were associated with changes in sensitivity to β-chemokines. In patients A, D, and E, the amino acid in position 311 changed from Ser to Arg in parallel with the loss of virus sensitivity to β-chemokines (Fig. 3). Amino acid position 325 in isolates from these patients did not change in charge between the two time points. In contrast, sequence analysis of NSI and SI isolates obtained from patients B and C indicated that an acidic amino acid, Glu, was replaced by a basic amino acid, Arg or Lys, at amino acid position 325. The charge of the V3 amino acid residue in position 311 did not change between the NSI and SI isolates of these patients; in patient B, a Ser was substituted by a Gly, and, in patient C, a Gly residue was preserved in both isolates (Fig. 3). Of interest, SI isolates of patients B and C were partially sensitive to the β-chemokine mixture at the highest concentration (200 ng/ml) whereas the isolates of SI type from patients A, D, and E were completely insensitive to inhibition when exposed to this concentration of β-chemokines (Fig. 2b). In V3 loop position 311 of NSI isolates from four of the five patients, a Ser residue was found, and a Gly residue was detected in the corresponding V3 position of NSI virus from patient C (Fig. 3). This isolate of patient C was also determined to be one of the least sensitive NSI isolates to inhibition by the β-chemokines (Fig. 2a). Taken together, these findings suggest that virus sensitivity to inhibition by β-chemokines RANTES, MIP-1α, and MIP-1β is associated with changes in V3 amino acid residues positions 311 and 325. These changes also correlate with NSI or SI virus phenotype. Our results also indicate that SI viruses with a Gly amino acid in V3 position 311 together with a positively charged amino acid in position 325 display intermediate sensitivities to β-chemokines.
Figure 3.

V3 region amino acid sequences obtained from viruses losing β-chemokine sensitivity simultaneously with a switch from NSI to SI phenotype in infected individuals. V3 amino acid residues are numbered according to the HIV-1MN sequence presented in the Los Alamos data base (35). A dash (-) indicates an amino acid identical to that present in the V3 sequence of the early isolate obtained from a corresponding individual, and a gap was introduced to align the sequences and is pointed out by a dot (.). Sequence heterogeneity is indicated by assigning two amino acids at the same position.
Viruses with NSI Phenotype Obtained from Patients with AIDS Maintain Sensitivity to β-Chemokines.
Although progression to AIDS is usually accompanied by a switch of the virus phenotype from NSI to SI, there are instances of individuals who develop AIDS in the absence of detectable virus of SI phenotype (32, 37, 38). This suggests that disease progression may not only depend on the presence of SI virus. We therefore wished to investigate whether virus isolates of NSI phenotype derived from HIV-1-infected individuals with AIDS were sensitive or resistant to inhibition by β-chemokines. Virus isolates were obtained from three patients with manifestation of AIDS who still only had detectable NSI virus when they were close to death (Table 1). For comparison, additional NSI isolates from the same patients, obtained before the onset of AIDS, were included in the β-chemokine inhibition assay. The results revealed that NSI isolates obtained from patients F and H after progression to AIDS remained equally sensitive to β-chemokines as virus obtained before the manifestation of AIDS. (Fig. 4a and c). In the case of patient G, replication of the early NSI isolate was completely inhibited after exposure to 200 or 67 ng/ml of the chemokine mixture whereas replication of the late virus was only partially blocked (69% and 67% p24 antigen release compared with control, respectively) (Fig. 4b). The durations of AIDS manifestation in patients F, H and G at the time of the late NSI virus isolation were 3, 14, and 26 months, respectively, and p24 releases in the presence of 200 ng/ml of each chemokine were 2%, 35%, and 69%, respectively (Fig. 4). Taken together, our results demonstrate that NSI isolates obtained from HIV-1-infected individuals with AIDS are sensitive to β-chemokine inhibition.
Figure 4.
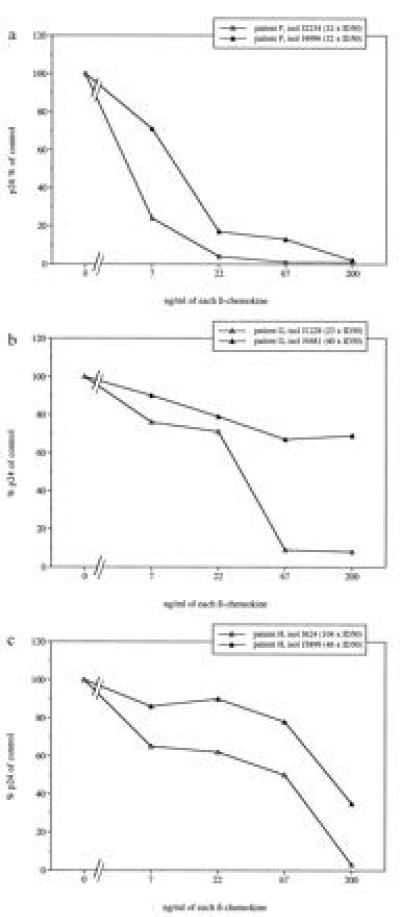
Sensitivity to chemokine-mediated replication inhibition of primary HIV-1 isolates with NSI phenotype obtained sequentially from three individual patients with AIDS. Percentage of p24 release, in relation to controls, of chemokine-exposed virus isolates obtained from patients F (a), G (b), and H (c).
DISCUSSION
CD8+ T cells were described earlier to mediate suppression of HIV-1 replication in a noncytotoxic and major histocompatibility complex-unrestricted manner (39–41). It is known that activated CD8+ T cells from asymptomatic HIV-1-infected individuals secrete one or more factors that can control HIV-1 replication in vitro, but it was not until recently that soluble factors with HIV-1 inhibitory activity were identified. The β-chemokines RANTES, MIP-1α, and MIP-1β were reported to be the major factors with HIV-1 inhibitory properties produced by CD8+ T cells (29). The T-cell line-adapted HIV-1 isolate IIIB, in contrast to the primary macrophage tropic virus isolate BaL, was reported to lack sensitivity to inhibition by the β-chemokines. The present study was initiated with the aim of examining a wide range of primary HIV-1 isolates and their sensitivities to β-chemokines RANTES, MIP-1α, and MIP-1β. Our results demonstrate that primary HIV-1 isolates can be divided into different groups according to their sensitivity to inhibition by these β-chemokines. When we analyzed a panel of unrelated, primary HIV-1 isolates containing viruses of both NSI and SI phenotypes, it became evident that viruses of NSI phenotype were sensitive to β-chemokines because HIV-1 p24 antigen production was undetectable in all of the NSI virus cultures in the presence of 200 ng/ml of each β-chemokine. In contrast, primary viruses of SI phenotype were resistant to inhibition by β-chemokines; no substantial reduction in p24 antigen release was observed in these cultures. It was recently described (42) that CD4+ T cells obtained from HIV-1-exposed but uninfected individuals were resistant to HIV-1 infection and that addition of β-chemokines RANTES, MIP-1α, and MIP-1β to CD4+ T cells of unexposed individuals mimicked the resistance to infection with the NSI isolate SF162. In contrast, no resistance to infection was observed when lymphocytes from exposed but uninfected individuals were infected with the T-cell line-adapted variant SF162 R3H, even with the addition of β-chemokines (42). Recent studies have described the β-chemokine receptor CC CKR5 is a coreceptor that, in addition to CD4, is required for entry of NSI macrophage-tropic HIV-1 isolates into human cells (17–21). It also was reported that recombinant viruses expressing envelope proteins of macrophage-tropic isolates such as JR-FL, ADA, and BaL were sensitive to inhibition by β-chemokines RANTES, MIP-1α, and MIP-1β whereas the laboratory- and T cell line-adapted viruses HXB2 and NL4/3 were resistant (17–19). Our results confirm that sensitivity of primary HIV-1 isolates to β-chemokines is associated with an NSI phenotype of the virus isolate, supporting the proposal that viruses of NSI type are using the CC CKR-5 as a coreceptor to enter cells (17–21). Furthermore, we extend previous observations by demonstrating that resistance to inhibition by β-chemokines RANTES, MIP-1α, and MIP-1β is not restricted to T cell line-adapted SI isolates but is also a general property of primary SI isolates, presumably due to a switch of virus coreceptor usage. The present analysis of primary virus isolates obtained sequentially from single HIV-1-infected individuals suggests that loss of sensitivity to β-chemokines during the course of infection occurs in parallel with a switch of virus phenotype from NSI to SI.
Recently, it has been suggested that the V3 region of gp120 is a critical determinant of virus susceptibility to β-chemokines (43, 44). Sequence analysis revealed that changes of charge of V3 amino acid residues at positions 311 and 325 correlated with a loss of virus sensitivity to inhibition by β-chemokines when we examined the sequentially obtained virus isolates. These observations of changes of V3 amino acids were analogous to findings correlating V3 substitutions with a switch from NSI to SI virus phenotype (10). Of interest, viruses with V3 sequence variants previously described to be of intermediate genotype (10) were partially inhibited by the β-chemokines. The observation of a partial sensitivity of SI virus variants could theoretically be due to the presence of both NSI and SI virus variants in these isolates. However, the results from the sequence analysis do not support this explanation; no polymorphism in the indicated amino acid residues could be detected in those SI variants that were partially inhibited by the β-chemokines. Virus of an intermediate genotype might instead use different coreceptors that can be blocked partially by one or more of the β-chemokines RANTES, MIP-1α, and MIP-1β. The usage of both fusin and the CC CKR-5 as coreceptor was described for the dual tropic isolate 89.6 (20, 21). In two of the articles (20, 21) describing CC CKR-5 as a coreceptor for NSI and macrophage tropic viruses, it also was suggested that other chemokine receptors, such as CC CKR-2b and CC CKR-3, in combination with CD4 can facilitate HIV-1 entry into human cells. In addition, it is reasonable to speculate that variability in the HIV-1 envelope proteins may alter the affinity of a possible interaction between the virus and its coreceptor. The alteration of usage of coreceptors by different variants of HIV-1 merits further investigation.
Inhibitory capacity of the CD8+ T cells in endogenous HIV-1 inhibition assays has been reported to be lost with development of disease (41, 45). This phenomenon could be explained by a loss of production of HIV-1 inhibitory factors from the CD8+ T cells but could also be due to loss of virus sensitivity to inhibition by β-chemokines with the progression of the HIV-1 infection, as described herein. It is intriguing to speculate that β-chemokines have a protective role during the early stage of HIV-1 infection. However, it is also evident from our results that infected individuals who have already developed AIDS and are close to death can harbor viruses that are predominantly sensitive to RANTES, MIP-1α, and MIP-1β. The presence of β-chemokine-sensitive viruses accordingly does not necessarily reflect an asymptomatic clinical stage of the HIV-1-infected individual. Several investigators (32, 37, 38) have reported that a proportion of patients that progresses to AIDS does not harbor virus with a typical SI phenotype. Our findings nevertheless indicate that, with time and a prolonged period of disease manifestation, virus variants that are less sensitive to β-chemokine inhibition might be selected. However, it is still an open question whether it is a selective pressure, mediated by chemokines, that induces a switch of coreceptor usage and cell tropism. The loss of HIV-1 β-chemokine sensitivity and switch of coreceptor might merely be a reflection of a collapse in the host immunosurveillance. Nevertheless, analysis of the LESTR/fusin ligand SDF-1 and its ability to inhibit replication of primary SI virus isolates will be of great interest. It is important to focus further studies on evaluation of the potential links among virus chemokine sensitivity, coreceptor usage, and HIV-1 pathogenesis.
Acknowledgments
We are indebted to K. Aperia for help with propagation of viruses, S. Gartner for selecting blood donor PBMC, R. A. Harris for linguistic advice, and S. Schwartz for discussions and critical comments on the manuscript. This work was supported by grants from the Swedish Medical Research Council, the Swedish Agency for Cooperation with Developing Countries, and Istituto Superiore di Sanitá. M.J. was supported by a fellowship from the Istituto Superiore di Sanitá.
Footnotes
Abbreviations: SI, syncytium-inducing; NSI, nonsyncytium-inducing; V3, third variable; MIP, macrophage inflammatory protein; PBMC, peripheral blood mononuclear cells; PHA, phytohemagglutinin; ID50, 50% infectious dose. RANTES, regulated upon activation, normal T-cell expressed and secreted; CKR-5, chemokine receptor 5.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base (accession nos. U76078–U76087U76078U76079U76080U76081U76082U76083U76084U76085U76086U76087).
References
- 1.Åsjö B, Morfeldt-Månson L, Albert J, Biberfeld G, Karlsson A, Lidman K, Fenyö E-M. Lancet. 1986;ii:660–662. [PubMed] [Google Scholar]
- 2.Fenyö E-M, Morfeldt-Månson L, Chiodi F, Lind B, von Gegerfelt A, Albert J, Olausson E, Åsjö B. J Virol. 1988;62:4414–4419. doi: 10.1128/jvi.62.11.4414-4419.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng-Mayer C, Seto D, Tateno M, Levy J A. Science. 1988;240:80–82. doi: 10.1126/science.2832945. [DOI] [PubMed] [Google Scholar]
- 4.Tersmette M, de Goede R E Y, Al B J M, Winkel I N, Gruters R A, Cuypers H T, Huisman H G, Miedema F. J Virol. 1988;62:2026–2032. doi: 10.1128/jvi.62.6.2026-2032.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz S, Felber B, Fenyö E M, Pavlakis G N. Proc Natl Acad Sci USA. 1989;86:7200–7203. doi: 10.1073/pnas.86.18.7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher C A, Lange J M, Miedema F F. AIDS. 1992;6:1259–1264. doi: 10.1097/00002030-199211000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Koot M, Vos A H V, Keet I P M, de Goede R E Y, Dercksen M W, Terpstra F G, Coutinho R A, Miedema F, Tersmette M. AIDS. 1992;6:49–54. doi: 10.1097/00002030-199201000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Schuitemaker H, Koot M, Koostra N A, Dercksen M W, de Goede R E Y, van Steenwijk R P, Lange J M A, Eeftink-Schattenkerk J K M, Miedema F, Tersmette M. J Virol. 1992;65:356–363. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connor R I, Mohri H, Cao Y, Ho D D. J Virol. 1993;67:1772–1777. doi: 10.1128/jvi.67.4.1772-1777.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fouchier R, Groenink M, Kootstra N A, Tersmette M, Huisman H G, Miedema F, Schuitemaker H. J Virol. 1992;66:3183–3187. doi: 10.1128/jvi.66.5.3183-3187.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng-Mayer C, Quiroga M, Tung J W, Dina D, Levy J A. J Virol. 1990;64:4390–4398. doi: 10.1128/jvi.64.9.4390-4398.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang S S, Boyle T J, Lyerly K, Cullen B R. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien W A, Koyanagi Y, Namazie A, Zhao J-Q, Diagne A, Idler K, Zack J A, Chen I S Y. Nature (London) 1990;348:69–73. doi: 10.1038/348069a0. [DOI] [PubMed] [Google Scholar]
- 14.Westervelt P, Gendelman H E, Ratner L. Proc Natl Acad Sci USA. 1991;88:3097–3101. doi: 10.1073/pnas.88.8.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesebro B, Wehrly K, Nishio J, Perryman S. J Virol. 1992;66:6547–6554. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Y, Broder C C, Kennedy P E, Berger E A. Science. 1996;272:872–877. [Google Scholar]
- 17.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, Davis C B, Peiper S C, Schall T J, Littman D R, Landau N R. Nature (London) 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 18.Dragic T, Litwin V, Allaway G P, Martin S R, Huang X, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 19.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 20.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath P D, Wu L, Mackay C R, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 21.Doranz B J, Rucker J, Yi Y, Smyth R J, Samson M, Peiper S C, Parmentier M, Collman R G, Doms R W. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 22.Loetscher M, Geiser T, O’Reilly T, Zwahlen R, Baggiolini M, Moser B. J Biol Chem. 1994;269:232–237. [PubMed] [Google Scholar]
- 23.Bleul C C, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer T A. Nature (London) 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 24.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier J-L, Arenzana-Seisdedos F, Schwartz O, Heard J-M, Clark-Lewis I, Legler D F, Loetscher M, Baggiolini M, Moser B. Nature (London) 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 25.Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M. Biochemistry. 1996;35:3362–3367. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- 26.Murphy P M. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 27.Baggiolini M, Dewald B, Moser B. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 28.Schall T J, Bacon K B. Curr Opin Immunol. 1994;6:865–873. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 29.Cocchi F, DeVico A L, Garzino-Demo A, Arya S K, Gallo R C, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 30.Albert J, Fenyö E-M. J Clin Microbiol. 1990;28:1560–1564. doi: 10.1128/jcm.28.7.1560-1564.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sundquist V A, Albert J, Ohlsson E, Hinkula J, Fenyö E M, Wahren B. J Med Virol. 1989;29:170–175. doi: 10.1002/jmv.1890290305. [DOI] [PubMed] [Google Scholar]
- 32.Karlsson A, Parsmyr K, Sandström E, Fenyö E M, Albert J. J Clin Microbiol. 1994;32:364–370. doi: 10.1128/jcm.32.2.364-370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahlberg J, Albert J, Lundeberg J, von Gegerfelt A, Broliden K, Utter G, Fenyö E M, Uhlén M. AIDS Res Hum Retroviruses. 1991;7:983–990. doi: 10.1089/aid.1991.7.983. [DOI] [PubMed] [Google Scholar]
- 34.Scarlatti G, Leitner T, Halapi E, Wahlberg J, Marchisio P, Clerici-Schoeller M A, Wigzell H, Fenyö E-M, Albert J, Uhlén M, Rossi P. Proc Natl Acad Sci USA. 1993;90:1721–1725. doi: 10.1073/pnas.90.5.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hultman T, Ståhl S, Hornes E, Uhlén M. Nucleic Acids Res. 1989;17:4937–4945. doi: 10.1093/nar/17.13.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myers G, Korber B, Jeang K-T, Mellors J W, McCutchan F E, Hendersson L E, Pavlakis G N. Theoretical Biology and Biophysics. Los Alamos, NM: Los Alamos Natl. Lab.; 1995. [Google Scholar]
- 37.Koot M, Keet I P M, Vos A H V, de Goede R E Y, Roos M T L, Coutinho R A, Miedema F, Schellekens P T A, Tersmette M. Ann Intern Med. 1993;118:681–688. doi: 10.7326/0003-4819-118-9-199305010-00004. [DOI] [PubMed] [Google Scholar]
- 38.Tersmette M, Bruters B A, de Wolf F, de Goede R E Y, Lange J M A, Schellekens P T A, Goudsmit J, Huisman H G, Miedema F. J Virol. 1989;63:2118–2125. doi: 10.1128/jvi.63.5.2118-2125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker C M, Moody D, Stites D P, Levy A J. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 40.Brinchmann J E, Gaudernack G, Vartdal F. J Immunol. 1990;144:2961–2966. [PubMed] [Google Scholar]
- 41.Mackewicz C, Levy J A. AIDS Res Hum Retroviruses. 1992;8:1039–1050. doi: 10.1089/aid.1992.8.1039. [DOI] [PubMed] [Google Scholar]
- 42.Paxton W A, Martin S R, Tse D, O≪Brien T R, Skurnick J, VanDevanter N L, Padian N, Braun J F, Kotler D P, Wolinsky S M, Koup R A. Nat Med. 1996;2:412–417. doi: 10.1038/nm0496-412. [DOI] [PubMed] [Google Scholar]
- 43.Oravecs T, Pall M, Norcross M A. J Immunol. 1996;157:1329–1332. [PubMed] [Google Scholar]
- 44.Cocchi F, De Vico A L, Garzino-Demo A, Cara A, Gallo R C, Lusso P. Nat Med. 1996;2:1244–1247. doi: 10.1038/nm1196-1244. [DOI] [PubMed] [Google Scholar]
- 45.Mackewicz C E, Ortega H, Levy J A. J Clin Invest. 1991;87:1462–1466. doi: 10.1172/JCI115153. [DOI] [PMC free article] [PubMed] [Google Scholar]