

Abstract
Nitric oxide (NO·) does not react significantly with thiol groups under physiological conditions, whereas a variety of endogenous NO donor molecules facilitate rapid transfer to thiol of nitrosonium ion (NO+, with one less electron than NO·). Here, nitrosonium donors are shown to decrease the efficacy of evoked neurotransmission while increasing the frequency of spontaneous miniature excitatory postsynaptic currents (mEPSCs). In contrast, pure NO· donors have little effect (displaying at most only a slight increase) on the amplitude of evoked EPSCs and frequency of spontaneous mEPSCs in our preparations. These findings may help explain heretofore paradoxical observations that the NO moiety can either increase, decrease, or have no net effect on synaptic activity in various preparations.
Keywords: excitatory postsynaptic currents, glutamate receptors, nitrosonium donors, redox, spontaneous miniature excitatory postsynaptic currents
A number of experiments, most of which have used inhibitors of endogenous nitric oxide synthase, have suggested that nitric oxide (NO·) acts presynaptically to enhance the amplitude of evoked synaptic potentials during long-term potentiation (LTP) (1–5). Yet, a variety of exogenous NO donors produce a reversible depression (and only rarely an enhancement) of synaptic transmission (1, 6–10). Apparent discrepancies between effects of various endogenous and exogenous NO donors on neurotoxicity or neuroprotection were attributed at least in part to different redox-related species of the NO group and their disparate chemical reactivities (11, 12). With this in mind, the present study was undertaken to determine if different redox-related forms of NO could contribute to differential effects on neurotransmission.
Under physiological conditions, free nitric oxide (NO·) reacts only very slowly with thiolate anions [the negatively charged form of thiol or -SH (sulfhydryl) groups]. Instead, the NO group in an alternative redox state from NO· reacts with protein thiol, producing transfer of NO+ equivalents to the thiolate anion (RS−), resulting in RS–NO formation (11, 13–16). Under physiological conditions NO+ does not exist in a free state in the nervous system, but extensive evidence exists in vivo that the NO group exists in a form that can be donated as NO+. For example, S-nitroso-glutathione, S-nitroso-hemoglobin, iron-nitrosyl complexes, nitroso-albumen, and N-nitrosamines are all present in biologic tissue, including the nervous system, and each of these chemical species can donate NO+ (15, 17, 18). We report here that donors with NO+ character, but not NO·, decrease evoked neurotransmission, apparently via reaction with thiol groups. Concomitantly, the frequency of spontaneous miniature excitatory postsynaptic currents (mEPSCs) is increased.
MATERIALS AND METHODS
NO Donors.
Preparations of nitroglycerin (NTG), S-nitrosocysteine (SNOC), diethylamine/nitric oxide complex (DEA/NO), N-ethylmaleimide (NEM), and their control solutions have been described (11, 19–21). These reagents were mixed in the extracellular solution, adjusted to pH 7.2, applied via bath superfusion for ≈2 min, and then washed out immediately before agonist-evoked recordings. The concentration of NO· generated by the various NO donors over a given unit of time (generally 2 min to coincide with the period of application to the neurons) was monitored with an NO·-specific electrode (World Precision Instruments, Sarasota, FL), as we have described previously (11). This electrode senses NO· but not the other redox-related forms of the NO group (11).
“Mass Cultures” and “Micro-Cultures” of Central Neurons.
For mass cultures, mixed neurons and glia were prepared from the cortex of fetal day 15 or 16 Sprague–Dawley rats and maintained in culture, as described (19). For micro-cultures, single, isolated hippocampal neurons were prepared and cultured as detailed previously (22–25). Before electrophysiological recording, the culture medium was replaced with an extracellular solution based upon Hanks’ balanced salt solution (19).
Patch-Clamp Electrophysiology.
In mass cultures, patch-clamp recordings were performed on cortical neurons in the whole-cell configuration at room temperature using standard procedures (11, 19). Patch pipettes contained a CsCl, tetraethylammonium-based solution (11, 19). In micro-cultures, autaptic EPSCs were evoked at 5-s intervals by 5-ms voltage jumps to 0 mV from a holding potential of −60 mV in extracellular solution that also contained 500 nM tetrodotoxin (TTX), 100 μM d-2-amino-5-phosphonovalerate (APV), 100 μM bicuculline, and 20 μM picrotoxinin. APV was added to suppress the N-methyl-d-aspartate (NMDA) receptor-mediated component of the EPSC; this was done because NMDA responses are known to be inhibited by nitroso-compounds postsynaptically, whereas the non-NMDA responses are not (refs. 19 and 26, but see ref. 27). This fact could be used to isolate a component of the EPSC (the non-NMDA component) that we knew was not blocked by nitroso-compounds at a postsynaptic locus. In all experiments, responses under the various conditions were interleaved in time to avoid time-dependent changes in the synaptic currents such as rundown.
Spontaneous mEPSCs were recorded from single hippocampal neurons in micro-cultures using the permeabilized-patch technique (with nystatin in the recording pipette) (28, 29). Cultures were placed in solutions containing TTX (1 μM) for 20–50 min before recording to allow decay of the posttetanic potentiation of mEPSCs that we have observed for 10–20 min after spontaneous firing. TTX was added to suppress Na+ currents in the micro-culture experiments, eliminating any possible confounding effect due to depressed excitability by Na+ current inhibition. The illustrated current traces were obtained at a holding potential of −60 mV, although other potentials were also tested.
RESULTS
Effects of NO Donors on Synaptic Activity in Mass Cultures.
Our initial observations of the effect of NO donors were made on cultured cerebrocortical neurons, which manifest multiple synaptic contacts (termed mass cultures). Whole-cell recordings display synaptic currents that are evoked by endogenous activity in the network of neuronal contacts. For subsequent synaptic analysis, experiments were performed on hippocampal cultures consisting of a single neuron with autaptic connections (termed micro-cultures).
In mass cultures, we found that administration of NTG (an NOx+ equivalent, with x = 1 or 2) (11) inhibited synaptic activity in a dose-dependent manner (Figs. 1 A–C and 2A). There was no effect of a control solution comprised of the diluent (Fig. 1D). A second NO donor compound, SNOC (capable of transferring NO+ equivalents to thiol groups by transnitrosation) (11) also decreased synaptic activity in a dose-dependent fashion (Figs. 1 E–G and 2B). These effects of nitroso-compounds reversed within a few minutes of washout. As a control, SNOC was allowed to decay (dissipate its NO group) for 24 hr. Under these conditions, there was no effect on synaptic activity; similarly, controls of equimolar cysteine and cystine had no effect. In contrast to NO+ donors, DEA/NO, a compound that selectively generates NO·, did not inhibit synaptic activity even at high (1 mM) concentrations for several minutes duration.
Figure 1.
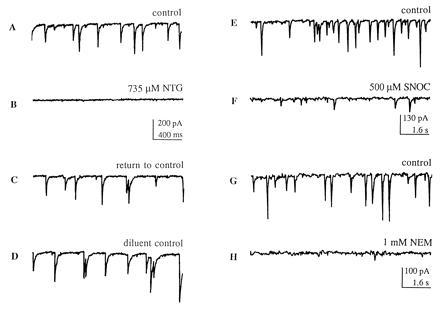
Inhibitory effect of NO donor compounds and NEM on synaptic activity in “mass cultures.” (A–C) In a representative cortical neuron, 735 μM NTG reversibly blocked synaptic transmission evoked by endogenous activity in the network of neuronal contacts (recording obtained immediately after washout of NTG); 200 μM NTG also resulted in significant suppression of synaptic activity (Fig. 2A). Holding potential, −60 mV. (D) A control solution, consisting of the diluent, propylene glycol and ethanol, plus glycerol (the 3-carbon backbone to which three -ONO2 groups are attached to form NTG) produced no effect. (E and F) A second NO group donor, SNOC, also reversibly decreased the frequency of synaptic activity in a second cortical neuron. (G and H) NEM (1 mM) mimicked the inhibitory effect of nitroso-compounds by blocking synaptic activity; this inhibitory effect was irreversible. Similar results to those illustrated in this figure were obtained in at least 8 cortical neurons from multiple platings of mass cultures.
It should be noted that in addition to transferring NO+ equivalents to thiol groups by heterolytic cleavage of the S-NO bond, SNOC can spontaneously decompose by homolytic cleavage to liberate NO· (11). Under our conditions, NTG does not directly generate NO· (11). In contrast, DEA/NO directly generates NO· but not NO+ equivalents (20, 21). To discount the possibility that the NO· generated by SNOC, rather than the transfer of NO+ equivalents, was responsible for the observed decrement in synaptic activity, the following experiment was performed. We measured the amount of NO· generated by SNOC (e.g., 250 or 500 μM) in a 2 min period with an NO·-specific electrode. We then monitored various concentrations of DEA/NO with the NO·-specific electrode to obtain a concentration that generated a similar amount of NO· as SNOC in a 2 min interval under our conditions. Next, we tested comparable NO·-generating concentrations of SNOC and DEA/NO (e.g., 250–500 μM and 30–50 μM, respectively) for effects on synaptic activity during a 2-min superfusion. We found that SNOC, but not DEA/NO (even at concentrations as high as 1 mM), led to a prolonged decrease in synaptic activity. To the contrary, occasionally with application of NO· donors, synaptic activity transiently increased in both amplitude and frequency. Thus, in these mass culture experiments the effects of donors of NO+ equivalents were clearly differentiated from those of NO·.
Effect of cGMP on Synaptic Activity.
In some neuronal systems, the NO moiety exerts its effects by stimulating guanylate cyclase (30); cGMP has been shown to enhance evoked EPSC amplitude and mEPSC frequency by a presumably presynaptic mechanism in a preparation of cultured hippocampal neurons (31); in Aplysia neurons, NO donors increase excitatory but decrease inhibitory postsynaptic currents via a cGMP-dependent mechanism (32). Nonetheless, the addition of 1 mM 8-bromo-cGMP to the extracellular solution did not affect synaptic activity of cortical neurons in our preparation of mass cultures, and it also did not affect evoked EPSCs or increase the frequency of spontaneous mEPSCs at autapses of single hippocampal neurons in our micro-cultures (see below). Thus, in our preparations cGMP does not mimic the actions of NO donors on synaptic transmission. This is consistent with the notion that another mechanism of action of NO+ donors may be involved here.
Effect of Sulfhydryl Alkylating Agents on Synaptic Activity.
NEM mimicked the effects of NO+ donor compounds in decreasing synaptic activity in the mass cultures, except as expected, synaptic activity did not recover upon washout of NEM (Fig. 1H). Additionally, prior administration of NEM occluded any further effect of NO+ donor compounds. NEM produces irreversible alkylation of thiol groups; in the case of nitroso-compounds, a well-known reaction that is consistent with our results is the transfer of the NO group (in the NO+ redox form) to protein thiolate anion (RS−) with resulting RS–NO formation (S-nitrosylation), which may be reversible (11, 15, 17, 19, 33–35). Concerning the chemical mechanism with NTG, intermediate formation of a thionitrate (RS–NO2) is not excluded (11).
Although nitroso-compounds or NEM attenuate postsynaptic NMDA-evoked currents to some degree, they do not affect the postsynaptic responses elicited by non-NMDA agonists such as kainate (11, 19, 26). This fact suggests that the effects of the NO group and NEM in this paradigm might be at least in part presynaptic because the EPSCs are mediated largely by non-NMDA channels [they are blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 μM)—see also below]. However, a decrease in “synaptic activity” in these mass cultures could be due to a general decrease in excitability and firing of neurons in culture, although this seemed less likely—neither SNOC nor NEM significantly inhibited voltage-dependent Na+ current, as monitored with whole-cell recording, and NTG did not affect neuronal Ca2+ levels in synaptic terminals, as assessed with confocal digital imaging using the dye fluo-3 (11, 19, 36). To further obviate these questions and for a more thorough evaluation of the potential synaptic action of NO+ donors, we next monitored evoked and spontaneous miniature synaptic activities in the hippocampal micro-culture system.
Effect of NO Donors on Evoked EPSCs in Hippocampal Micro-Culture Autapses.
Autaptic, non-NMDA EPSCs were evoked in the presence of TTX. The NO+-like donor NTG (500–1000 μM) reversibly inhibited evoked EPSCs within 1 min of application (Fig. 3A; decrement in EPSCs = 41.8 ± 3.4%, mean ± SEM, n = 5). In contrast to the effect of NO+ donors, the NO· donor DEA/NO (1000 μM) did not decrease the magnitude of EPSCs; in fact, the amplitude and duration of the EPSC were, if anything, very slightly increased compared with control (Fig. 3B).
Figure 3.
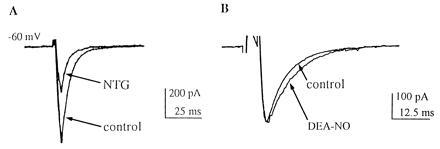
Synaptic currents elicited by depolarizing pulses at autapses in micro-cultures are decreased by NO donors of NO+ character, like NTG, but not by NO donors releasing nitric oxide (NO·), such as DEA/NO. (A) Compared with control (lower trace), NTG (500 μM) inhibited autaptic non-NMDA EPSCs on cultured, single hippocampal neurons. The EPSC amplitude recovered within 2 min of NTG washout. (B) Compared with control (upper trace), DEA/NO (1000 μM) did not inhibit the autaptic EPSC and, if anything, resulted in a minor prolongation of the synaptic current. Similar results were obtained in six autapses in micro-cultures.
Effect of NO Donors on Spontaneous mEPSCs in Hippocampal Micro-Culture Autapses.
Quite surprisingly, the frequency of mEPSCs dramatically increased to 180% ± 18% (mean ± SEM, n = 10), rather than decreasing as expected, in the presence of the NO+-like donor NTG (1000 μM; P < 0.001, ANOVA; Fig. 4 A and B). This effect was still observed in the nominal absence of extracellular Ca2+ (0 Ca2+ added and 0.5 mM EGTA; n = 3). Additionally, NTG (1000 μM) did not affect intracellular Ca2+ ([Ca2+]i) in single hippocampal neurons, as monitored by confocal microscopy with the dye fluo-3 (36). In contrast to the effects of NO+ donors, the NO· donor DEA/NO (1000 μM) produced only a very slight increase in the frequency of mEPSCs (to 116 ± 8%, n = 3), and 8-bromo-cGMP (1000 μM; n = 3) did not increase the frequency of mEPSCs. The amplitude of the mEPSCs remained virtually invariant in the presence of NTG (1000 μM; Fig. 4C).
Figure 4.
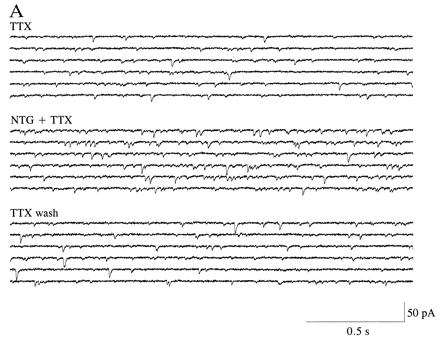
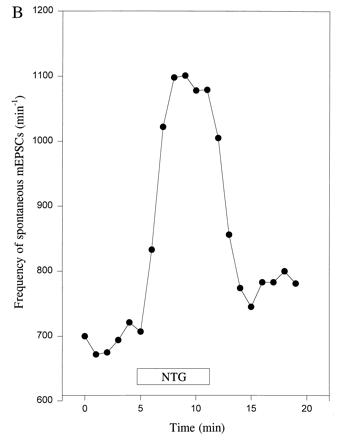
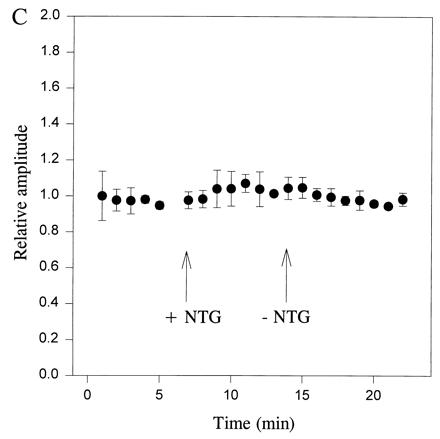
Frequency of spontaneous mEPSCs is increased at autapses in micro-cultures by an NO donor with NO+ character. (A) Spontaneous mEPSCs recorded in the presence of TTX (1 μM) before and after the addition of NTG (1000 μM). The control solution superfused prior and subsequent to NTG also contained the diluent plus glycerol, as described in Fig. 1D. The frequency of mEPSCs was noticeably enhanced in the presence of NTG. (B) Plot of frequency of spontaneous mEPSCs versus time for a hippocampal autapse. TTX (1 μM) was added to the bathing solution beginning 40 min before the epoch of recording illustrated here. NTG (1000 μM) increased the frequency of mEPSCs. Data are shown from one representative recording of 10. (C) Plot of relative amplitude of spontaneous mEPSCs versus time for hippocampal autapses. The amplitude of mEPSCs was unaffected by NTG (1000 μM), nor was there a change in the noise level that would have affected the detectability of mEPSCs. Data represent the mean ± SEM for four neurons in micro-cultures treated in an identical fashion.
DISCUSSION
Inhibitors of nitric oxide synthase prevent the development of LTP (1, 2, 4, 37) and decrease neurotransmitter release from synaptosomes and other preparations (38–41). These results have been used to argue that NO· enhances LTP by potentiating presynaptic release of glutamate neurotransmitter. Additionally, direct evidence for the enhancement of LTP in the presence of exogenous NO· has been obtained (42), although not by all laboratories. Paradoxically, direct application of exogenous NO donors can also attenuate NMDA-evoked neurotransmitter release from synaptosomes (41), decrease neurohormone release (43), and inhibit LTP (44). Moreover, direct application of exogenous NO donors has been shown to increase spontaneous (Ca2+-independent) neurotransmitter release from synaptosomes (45).
Our electrophysiological results with exogenous NO donors may help explain these seemingly contradictory findings by considering the fact that the NO group can react in an alternative redox state from NO·, most likely by transfer of NO+ to thiol groups (or more properly thiolate anion, RS−) (35). This study represents a detailed synaptic electrophysiological analysis of the effects of different redox forms of the NO group. As demonstrated here, NO+ donors produced a decrease in evoked synaptic activity (paralleling the decrease in evoked neurotransmitter release observed previously in synaptosomes in response to exogenous NO donors) but an increase in spontaneous synaptic activity (paralleling the increase in spontaneous neurotransmitter release observed previously in synaptosomes). The fact that NO+ donors increased miniature frequency but not amplitude is classically considered evidence of a presynaptic effect (46). However, this tentative conclusion must be tempered by the fact that recent evidence suggests that at central synapses this could be due to a purely postsynaptic mechanism of functional activation of silent receptors (47, 48). Thus, this area of synaptic analysis remains a contentious one.
In contrast to the effect of NO+ donors, in our preparations application of exogenous NO· donors exhibited either no effect or occasionally a slight increase in both the amplitude of evoked and the frequency of spontaneous synaptic currents. The small increases in synaptic activity that we observed with NO· donors could conceivably contribute to the enhancement of EPSC amplitude and frequency of mEPSCs seen in LTP (49). However, it is possible that neurotransmission was already maximally potentiated at many synapses in our culture preparations (50, 51), precluding a more dramatic effect of NO·.
All of our findings were obtained with exogenous NO donor compounds rather than by manipulating the endogenous, physiological NO group. Part of the reason for this is that exogenous NO donors can be controlled with respect to the redox state of the NO moiety that they produce, while we do not yet completely appreciate how to accomplish this type of redox regulation of endogenous NO groups, although we know that it must exist (15, 18). Additionally, the action of redox reagents, including NO+ donors, is both time- and concentration-dependent. Therefore, relatively large concentrations of these agents (e.g., hundreds of micromolar) that are effective in 1 or 2 min may signify that much lower concentrations (e.g., nanomolar) would be effective over longer periods of time. Relatively high concentrations were used here simply to accelerate the onset of effects during our electrophysiological recordings. Nonetheless, it is well known in the redox and NO literature that concentrations of hundreds of micromolar often used for in vitro experiments may reflect physiological events influenced by much lower concentrations of NO donors (35). Moreover, the half-life of many of the NO donors used in the present study is short (11), so in many cases the effective concentration was probably far less than that applied.
A standard procedure for preventing the effects of endogenous or exogenous NO is to bind this molecule to reduced hemoglobin, as described (30, 52). However, this was a problem in our short-term physiology experiments because, as we have previously shown, reduced hemoglobin by itself increased [Ca2+]i for up to several minutes in Ca2+ imaging experiments on these neurons (19). Additionally, reduced hemoglobin (and similar NO chelators) generated an inward current in our preparations during voltage clamp at −60 mV, which obfuscated the results and precluded the use of such chelators in the present experiments (19).
Our results suggest that the redox state of the NO moiety is critical in determining its influence on neurotransmitter release and resulting synaptic activity. These synaptic effects could also contribute to the opposing roles of different redox states of the NO moiety in neuroprotection versus neurodegeneration since evoked release of glutamate could play a role there as well (11). These complex mechanisms of action may have broad implications for homeostatic function and feedback regulation by nitroso-compounds in the nervous system, for example, with the NO group enhancing LTP under one set of conditions but inhibiting LTP or even fostering long-term synaptic depression under another (53).
Quite unexpectedly, a clue to the mechanism of NO donors of NO+ character was obtained when we found that, unlike evoked synaptic activity, the frequency of spontaneous mEPSCs was dramatically enhanced. A decrease in the amplitude of evoked EPSCs with a concomitant increase in the frequency of spontaneous mEPSCs has been observed previously. Repetitive stimulation under high quantal conditions resulted in this phenomenon at the neuromuscular junction (54). Large, possibly transient, increases in intraterminal Ca2+ levels have been shown to be associated with a block in evoked release or an increase in mEPSC frequency (55, 56), but there was no evidence for Ca2+ accumulation in our experiments in confocal microscopy with fluo-3, and lowering the extracellular Ca2+ concentration had no effect. Other mechanisms remain possible, however. Chief among them is the observation that effects on synaptotagmin I can produce a similar result. For example, targeted disruption in Drosophila of synaptotagmin, a synaptic vesicle-associated protein that interacts with Ca2+ and affects exocytosis and recycling (57), results in decreased evoked transmitter release but an increase in spontaneous miniatures (refs. 58–61, but see refs. 62 and 63). Synaptotagmin binds to a prefusion complex comprised of syntaxin, a vesicle membrane receptor (VAMP or synaptobrevin) for NEM-sensitive factor attachment protein (SNAP), and the plasma membrane protein SNAP-25 (61, 64–66). It is possible, therefore, that effects on any of these proteins in the core complex of synaptic proteins involved in docking and fusion would contribute to a phenotype of decreased evoked and increased spontaneous release. Along these lines, it has been shown that transfer of the NO moiety (in the form of NO+) to the neuronal presynaptic protein SNAP-25 results in a covalent bond between the NO moiety and a thiol group on a cysteine residue. This reaction, termed S-nitrosylation (15), prevents subsequent fatty acid acylation of the affected cysteine residue (67). Such reactions of the NO group can exert physiological effects (11), in this case possibly contributing to a decrease in evoked neurotransmitter release and an increase in spontaneous release since acylation is required for activity of SNAP-25 (68). In fact, NO donors (likely in the NO+ form) were very recently shown to increase formation of VAMP/SNAP-25/syntaxin 1a core complex while inhibiting the binding of n-sec1 to syntaxin 1a (69). It is also possible that other synaptic vesicle-associated proteins are S-nitrosylated at regulatory cysteine sulfhydryl groups, resulting in a similar phenotype. Whatever the exact mechanism of action, our findings are consistent with a possible presynaptic effect of the NO group, mediated by transfer of NO+ to critical protein thiol(s), which can modulate neurotransmission. This alternative redox form of NO can thus act as a molecular switch to exert allosteric control over exocytosis and other cellular functions regulated by interactions with protein thiols (11, 18).
Figure 2.
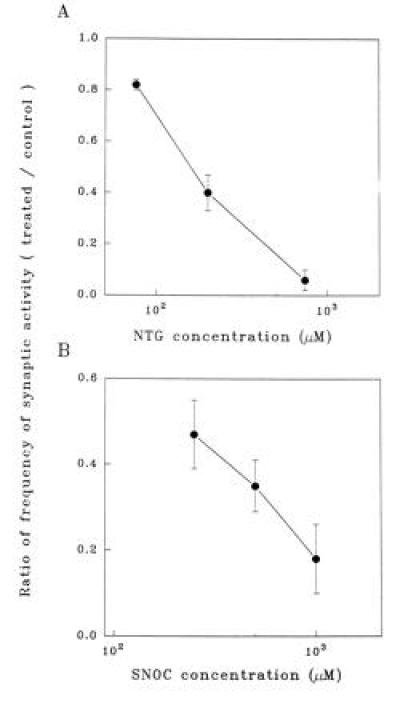
NO+ donor compounds inhibit synaptic activity of mass cultures in a dose-dependent manner. (A) Dose-dependent inhibitory effect of NTG on the frequency of postsynaptic currents, normalized to control conditions. Data points represent means ± SEM (for n = 15 cortical neurons in mass culture). (B) Dose-dependent inhibitory effect of SNOC on the frequency of postsynaptic currents (for n = 11 neurons).
Acknowledgments
We wish to thank J. S. Stamler, N. J. Sucher, and H.-S. V. Chen for helpful discussions, and D. D’Emilia for the cortical mass cultures. This work was supported in part by an American Foundation for AIDS Research Frank G. Wells Scholar Award (to Z.-H.P.), by the Charles H. Hood Foundation (to M.M.S.), and by National Institutes of Health Grants R01 EY05477 and P01 HD29587 (to S.A.L.).
Footnotes
Abbreviations: DEA/NO, diethylamine/nitric oxide complex; EPSCs, excitatory postsynaptic currents; mEPSCs, miniature EPSCs; NEM, N-ethylmaleimide; NTG, nitroglycerin; NMDA, N-methyl-d-aspartate; SNOC, S-nitrosocysteine; TTX, tetrodotoxin; LTP, long-term potentiation.
References
- 1.Böhme G A, Bon C, Stutzmann J M, Doble A, Blanchard J C. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- 2.Schuman E M, Madison D V. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 3.O’Dell T J, Hawkins R D, Kandel E R, Arancio O. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haley J E, Wilcox G L, Chapman P F. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 5.Gribkoff V K, Lum-Ragan J T. J Neurophysiol. 1992;68:639–642. doi: 10.1152/jn.1992.68.2.639. [DOI] [PubMed] [Google Scholar]
- 6.Bon C, Böhme G A, Doble A, Stutzmann J M, Blanchard J C. Eur J Neurosci. 1992;4:420–424. doi: 10.1111/j.1460-9568.1992.tb00891.x. [DOI] [PubMed] [Google Scholar]
- 7.Boulton C L, Irving A J, Southam E, Potier B, Garthwaite J, Collingridge G L. Eur J Neurosci. 1994;6:1528–1535. doi: 10.1111/j.1460-9568.1994.tb00543.x. [DOI] [PubMed] [Google Scholar]
- 8.Murphy K P S J, Williams J H, Bettache N, Bliss T V P. Neuropharmacology. 1994;33:1375–1385. doi: 10.1016/0028-3908(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 9.Lingren C A, Laird M V. NeuroReport. 1994;5:2205–2208. doi: 10.1097/00001756-199410270-00054. [DOI] [PubMed] [Google Scholar]
- 10.Wang T, Xie Z, Lu B. Nature (London) 1995;374:262–266. doi: 10.1038/374262a0. [DOI] [PubMed] [Google Scholar]
- 11.Lipton S A, Choi Y-B, Pan Z-H, Lei S Z, Chen H-S V, Sucher N J, Loscalzo J, Singel D J, Stamler J S. Nature (London) 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 12.Lipton S A, Stamler J S. Neuropharmacology. 1994;33:1229–1233. doi: 10.1016/0028-3908(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 13.Pryor W A, Lightsey J W. Science. 1981;214:435–437. doi: 10.1126/science.214.4519.435. [DOI] [PubMed] [Google Scholar]
- 14.Pryor W A, Church D F, Govinden C K, Crank G. J Org Chem. 1982;47:156–159. [Google Scholar]
- 15.Stamler J S, Singel D J, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 16.Lipton S A, Stamler J S, Singel D J. Nature (London) 1994;367:28. [Google Scholar]
- 17.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 18.Jia L, Bonaventura C, Bonaventura J, Stamler J S. Nature (London) 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 19.Lei S Z, Pan Z-H, Aggarwal S K, Chen H-S V, Hartman J, Sucher N J, Lipton S A. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 20.Maragos C M, Morley D, Wink D A, Dunams T M, Saavedra J E, Hoffman A, Bove A A, Isaac L, Hrabie J A, Keefer L K. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 21.Wink D A, Hanbauer I, Drishna M C, DeGraff W, Gamson J, Mitchell J B. Proc Natl Acad Sci USA. 1993;90:9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segal M M. J Neurophysiol. 1991;65:761–70. doi: 10.1152/jn.1991.65.4.761. [DOI] [PubMed] [Google Scholar]
- 23.Bekkers J M, Stevens C F. Proc Natl Acad Sci USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan Z Z, Tong G, Jahr C E. Neuron. 1993;11:85–91. doi: 10.1016/0896-6273(93)90273-t. [DOI] [PubMed] [Google Scholar]
- 25.Tong G, Jahr C E. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 26.Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-t. [DOI] [PubMed] [Google Scholar]
- 27.Hoyt K R, Tang L-H, Aizenman E, Reynolds I J. Brain Res. 1992;592:310–316. doi: 10.1016/0006-8993(92)91690-g. [DOI] [PubMed] [Google Scholar]
- 28.Horn R, Marty A. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal M M. J Neurophysiol. 1994;72:1874–1884. doi: 10.1152/jn.1994.72.4.1874. [DOI] [PubMed] [Google Scholar]
- 30.Garthwaite J, Charles S L, Chess W R. Nature (London) 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 31.Arancio O, Kandel E R, Hawkins R D. Nature (London) 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 32.Mothet J-P, Fossier P, Tauc L, Baux G. Proc Natl Acad Sci USA. 1996;93:8721–8726. doi: 10.1073/pnas.93.16.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bates, J. N., Baker, M. T., Guerra Jr., R. & Harrison, D. G. (1991) Biochem. Pharmacol. 42, Suppl., S157–S165. [DOI] [PubMed]
- 34.Noack, E. & Feelisch, M. (1991) Basic Res. Cardiol. 86, Suppl. 2, 37–50. [DOI] [PubMed]
- 35.Feelisch M, Stamler J S, editors. Methods in Nitric Oxide Research. Chichester, U.K.: Wiley; 1996. [Google Scholar]
- 36.Pan Z-H, Lipton S A. J Neurosci. 1995;15:2668–2679. doi: 10.1523/JNEUROSCI.15-04-02668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Dell T J, Kandel E R, Grant S G. Nature (London) 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- 38.Hanbauer I, Wink D, Osawa Y, Edelman G M, Gally J A. NeuroReport. 1992;3:409–412. doi: 10.1097/00001756-199205000-00008. [DOI] [PubMed] [Google Scholar]
- 39.Prast H, Philippu A. Eur J Pharmacol. 1992;216:139–140. doi: 10.1016/0014-2999(92)90223-q. [DOI] [PubMed] [Google Scholar]
- 40.Dickie B G M, Lewis M J, Davies J A. Neurosci Lett. 1992;138:145–148. doi: 10.1016/0304-3940(92)90492-p. [DOI] [PubMed] [Google Scholar]
- 41.Montague P R, Gancayco C D, Winn M J, Marchase R B, Friedlander M J. Science. 1994;263:973–977. doi: 10.1126/science.7508638. [DOI] [PubMed] [Google Scholar]
- 42.Zhuo M, Small S A, Kandel E R, Hawkins R D. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
- 43.Ceccatelli S, Hulting A L, Zhang X, Gustafsson L, Villar M, Hokfelt T. Proc Natl Acad Sci USA. 1993;90:11292–11296. doi: 10.1073/pnas.90.23.11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izumi Y, Clifford D B, Zorumski C F. Science. 1992;257:1273–1276. doi: 10.1126/science.1519065. [DOI] [PubMed] [Google Scholar]
- 45.Meffert M K, Premack B A, Schulman H. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 46.Bolshakov V Y, Siegelbaum S A. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 47.Liao D, Hessler N A, Malinow R. Nature (London) 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 48.Isaac J T, Nicoll R A, Malenka R C. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 49.Malgaroli A, Tsien R W. Nature (London) 1992;357:134–139. doi: 10.1038/357134a0. [DOI] [PubMed] [Google Scholar]
- 50.Bekkers J M, Stevens C F. Nature (London) 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- 51.Goda Y, Stevens C F. Neuron. 1996;16:103–111. doi: 10.1016/s0896-6273(00)80027-6. [DOI] [PubMed] [Google Scholar]
- 52.Shibuki K. Neurosci Res. 1990;9:69–76. doi: 10.1016/0168-0102(90)90048-j. [DOI] [PubMed] [Google Scholar]
- 53.Shibuki K, Okada D. Nature (London) 1991;349:326–328. doi: 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- 54.Zengel J E, Sosa M A. J Physiol (London) 1994;477:267–277. doi: 10.1113/jphysiol.1994.sp020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams D J, Takeda K, Umbach J A. J Physiol (London) 1985;369:145–159. doi: 10.1113/jphysiol.1985.sp015893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cummings D D, Wilcox K S, Dichter M A. J Neurosci. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jorgensen E M, Hartwieg E, Schuske K, Nonet M L, Jin Y, Horvitz H R. Nature (London) 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- 58.Littleton J T, Stern M, Schultze K, Perin M, Bellen H J. Cell. 1993;74:1125–1134. doi: 10.1016/0092-8674(93)90733-7. [DOI] [PubMed] [Google Scholar]
- 59.DiAntonio A, Schwarz T L. Neuron. 1994;12:909–920. doi: 10.1016/0896-6273(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 60.Popov S V, Poo M-m. Cell. 1993;73:1247–1249. doi: 10.1016/0092-8674(93)90352-q. [DOI] [PubMed] [Google Scholar]
- 61.Morimoto T, Popov S, Buckley K M, Poo M-m. Neuron. 1995;15:689–696. doi: 10.1016/0896-6273(95)90156-6. [DOI] [PubMed] [Google Scholar]
- 62.Geppert M, Goda Y, Hammer R E, Li C, Rosahl T W, Stevens C F, Südof T C. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 63.Ullrich B, Li C, Zhang J Z, McMahon H, Anderson R G W, Geppert M, Südof T C. Neuron. 1994;13:1281–1291. doi: 10.1016/0896-6273(94)90415-4. [DOI] [PubMed] [Google Scholar]
- 64.Scheller R H. Neuron. 1995;14:893–897. doi: 10.1016/0896-6273(95)90328-3. [DOI] [PubMed] [Google Scholar]
- 65.Schiavo G, Gmachi M J S, Stenbeck G, Söllner T H, Rothman J E. Nature (London) 1995;378:733–736. doi: 10.1038/378733a0. [DOI] [PubMed] [Google Scholar]
- 66.Littleton J T, Bellen H J. Trends Neurosci. 1995;18:177–183. doi: 10.1016/0166-2236(95)93898-8. [DOI] [PubMed] [Google Scholar]
- 67.Hess D T, Patterson S I, Smith D S, Skene J H P. Nature (London) 1993;366:562–565. doi: 10.1038/366562a0. [DOI] [PubMed] [Google Scholar]
- 68.Niemann H, Blasi J, Jahn R. Trends Cell Biol. 1994;4:179–185. doi: 10.1016/0962-8924(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 69.Meffert M K, Calakos N C, Scheller R H, Schulman H. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]