

Abstract
Pain differs from other sensations in many respects. Primary pain-sensitive neurons respond to a wide variety of noxious stimuli, in contrast to the relatively specific responses characteristic of other sensory systems, and the response is often observed to sensitize on repeated presentation of a painful stimulus, while adaptation is typically observed in other sensory systems. In most cases the cellular mechanisms of transduction and sensitization in response to painful stimuli are not understood. We report here that application of pulses of noxious heat to a subpopulation of isolated primary sensory neurons rapidly activates an inward current. The ion channel activated by heat discriminates poorly among alkali cations. Calcium ions both carry current and partially suppress the current carried by other ions. The current is markedly increased by bradykinin, a potent algogenic nonapeptide that is known to be released in vivo by tissue damage. Phosphatase inhibitors prolong the sensitization caused by bradykinin, and a similar sensitization is caused by activators of protein kinase C. We conclude that bradykinin sensitizes the response to heat by activating protein kinase C.
Keywords: pain, sensory transduction, protein kinase C, sensory neuron
The dorsal root ganglion (DRG) contains a mixed population of primary sensory neurons that send myelinated and unmyelinated axons to the skin surface and elsewhere within the body, and convey information about a wide variety of sensory stimuli, including touch, temperature, and noxious stimuli. Noxious stimuli are detected both by terminals of unmyelinated C fibers, which typically respond to noxious chemical, mechanical, and heat stimuli, and by terminals of small myelinated A fibers, which often exhibit more specific responses (1–5). From this mixed population of sensory neurons, a population consisting mainly of nociceptors can be selected simply by choosing small neurons, because a large fraction of the smallest neurons (<25 μm in diameter) in the DRG possess unmyelinated axons and respond to noxious stimuli (6, 7).
The cellular basis of the response to painful stimuli has, in contrast with other sensations, been little investigated. This is partly because of the difficulty of isolating the fine pain-sensitive nerve terminals, and partly because of the difficulty of defining what is a painful stimulus. Rather than attempting to study nociceptive nerve terminals themselves, we have used cultured neurons from the DRG as a model because many of the properties of nociceptive neurons in vivo are known to be replicated in small cultured neurons from the DRG (8–12). We have investigated the responses of these neurons to heat stimuli of an intensity that would produce a sensation of pain and avoidance responses in a human or animal subject. One objective was to investigate at a cellular level how noxious heat stimuli excite nociceptive neurons. To our knowledge, no studies to date have investigated the transduction pathways underlying the excitation of nociceptors by heat. A second objective was to investigate the cellular basis of the process of sensitization, which in vivo produces an increasing sensation of pain in response to repeated application of a noxious heat stimulus.
MATERIALS AND METHODS
Preparation and Recording.
Neonatal (<5 days old) Wistar rats were decapitated, and about 40 DRG were removed. Neurons were isolated and cultured in the presence of nerve growth factor (100 ng/ml, Promega) in 35-mm-diameter plastic culture dishes for 4–6 days (12). Small neurons (<25 μm diameter) were selected and whole-cell voltage-clamped at −70 mV using patch-clamp electrodes of tip diameter ≈0.7 μm (bubble number ≈5.5) and a List EPC7 amplifier. Pipette solution contained 135 mM KCl, 0.2 mM CaCl2, 1.6 mM MgCl2, 2 mM EGTA, 2.5 mM MgATP, 0.2 mM LiGTP, and 10 mM Hepes (pH 7.3). Hanks’ solution used for experiments contained 130 mM NaCl, 4 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 4 mM d-glucose, and 10 mM Hepes (pH 7.4). Solutions were changed within 20 msec (see Fig. 1B) using an automated multipipe solution changer (BioLogic RSC-100). One solution was heated using a Peltier device regulated to within ±0.1°C by a proportional gain feedback controller. Temperature was monitored by a miniature thermocouple immediately before the solution entered the bath. Linear temperature ramps changing from 28°C to 49°C in 10 sec were generated by positioning the neuron in front of the thermally controlled pipe and switching on the Peltier device; data such as that in Figs. 2A and 4C are plots of current versus temperature measured by the miniature thermocouple and corrected for the 200 msec transit time from the thermocouple to the cell. Data were acquired at (typically) 10 kHz using pclamp software (Axon Instruments, Foster City, CA).
Figure 1.
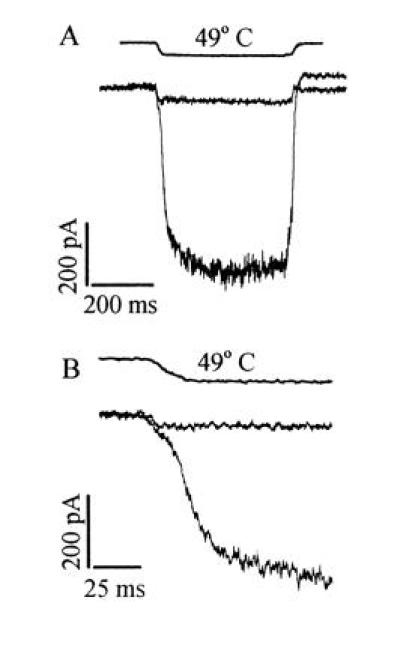
Activation of an inward current by heat in primary sensory neurons. (A) Membrane current activated in a heat-sensitive and a heat-insensitive neuron in response to a temperature change from 25°C to 49°C (timing of solution change given by top trace, obtained by measuring time course of change in solution conductance at pipette tip after detaching from the cell at the end of the experiment). In 56% (n = 155) of small neurons (<25 μm in diameter), a delayed increase in membrane current exceeding 50 pA was observed in response to a 49°C pulse; such neurons were classified as heat-sensitive. (B) Same traces as in A expanded to show delayed onset of current increase in heat-sensitive neurons.
Figure 2.
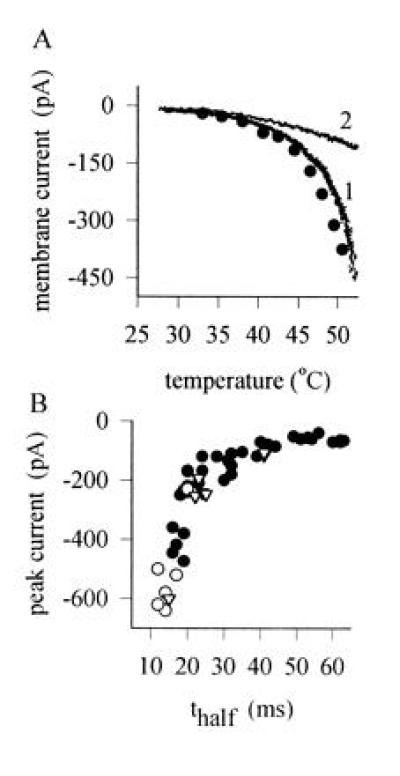
(A) Peak current as a function of temperature activated in a heat-sensitive neuron by 400-msec step temperature changes (•). A similar relation was obtained by the quicker procedure of applying a temperature ramp (trace 1). Trace 2 shows the result of a similar temperature ramp applied to a heat-insensitive neuron. (B) Relation between current activated by a 49°C temperature step and time to half-activation of delayed current in individual neurons. •, Untreated heat-sensitive neurons; ▵, neurons exposed to BK (1 μM for 20 sec; for further details, see Fig. 3). ○, Neurons exposed to the PKC activator PMA (1 μM for 20 sec; for further details, see Fig. 4).
Figure 4.
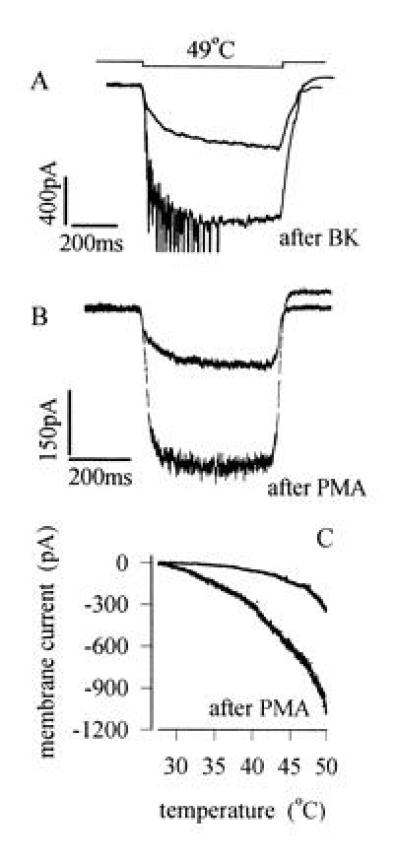
(A) Sensitization of heat response by BK. Traces show membrane current responses to steps to 49°C before and after application of BK (1 μM for 20 sec, which generated a transient inward current of maximum amplitude 203 pA in this neuron). Brief transient inward currents observed on application of heat after BK are action potentials elicited in unclamped neuronal processes. (B) Sensitization caused by application of the PKC activator PMA (1 μM for 20 sec). Mean increase in heat-sensitive current elicited by a 49°C temperature step was 532 ± 123% of control (n = 9), and significant increases were observed in all heat-sensitive neurons tested. PMA had no detectable effect on membrane current at room temperature in the majority of neurons. Experimental details as in A. (C) Current versus temperature relations, obtained using temperature ramps as in Fig. 2A, before and after application of PMA (1 μM).
For experiments on ionic selectivity, the internal solution was modified by substituting Cs+ for K+ to suppress voltage-activated K+ currents and by reducing divalent ion concentrations to avoid possible block of the heat-sensitive current from the inner membrane surface. Pipette solution composition was 135 mM CsCl, 2 mM EGTA, 2.5 mM MgATP, 0.2 mM LiGTP, and 10 mM Hepes (pH 7.3). External solutions contained 100 μM Cd2+ to suppress calcium currents and 10 mM Hepes and 4 mM d-glucose, in addition to the stated concentrations of NaCl and CaCl2. Nominal zero-Ca solutions do not contain EGTA, which would chelate Cd2+, and [Ca2+] was found to be 10 μM by atomic absorption spectrophotometry. N-Methyl-d-glucamine was used a Na+ substitute.
RESULTS
Basic Characteristics of the Response to Heat.
Recordings of membrane current obtained from small DRG neurons using the whole-cell patch-clamp technique are shown in Fig. 1. In all neurons, a rapid increase in inward current was observed in response to a noxious thermal stimulus, but two different types of response were clearly distinguishable. In neurons that we classified as heat-insensitive, the only current change was a small increase in inward current, which was activated as rapidly as the solution could be changed. The instantaneous current change was also observed in neurons classified as heat-sensitive, but in these latter neurons a much larger increase in inward current was activated with a delay following the instantaneous current change (see Fig. 1B). Neurons were considered to be heat-sensitive if the delayed membrane current exceeded 50 pA in response to a temperature step to 49°C. Approximately 56% of small neurons (<25 μm in diameter) were heat-sensitive.
The form of the dependence of the delayed heat-sensitive current on temperature is shown in Fig. 2A (trace 1). The activation of current in heat-sensitive neurons was strikingly nonlinear, with the major change in inward current occurring above 42°C. A similar dependence on temperature of both the subjective sensation of pain and of the frequency of action potentials in nociceptive nerve fibers is observed in vivo (1–4). The responses of receptors sensitive to non-noxious warmth, in contrast, increases over the range 30–40°C and declines above ≈44°C (13). No responses to temperature with properties similar to those of warm-sensitive neurons were observed in the present study. The current versus temperature relation of temperature-insensitive neurons was much more linear than in heat-sensitive neurons (trace 2 in Fig. 2A), as expected if the current in these neurons resulted from a physical process such as a nonspecific change in membrane resistance or in the pipette seal resistance.
Fig. 2B (solid circles) shows that a monotonic relationship between the magnitude of the current increase and the half-time of activation was observed in different neurons, with the largest current associated with the shortest activation time. The mean time to half-activation was 34.8 ± 2.6 msec (mean ± SEM, n = 36), comparable to the observed activation time in heat-sensitive C-fiber afferents in vivo of <100 msec (32).
Ionic Selectivity of the Heat-Sensitive Channel.
The ionic selectivity of the current activated by heat is examined in Fig. 3. The reversal potential of the current with Na+ as the principal extracellular cation and Cs+ as the principal internal cation was close to 0 mV, showing that the ion channel activated by heat discriminates poorly between Na+ and Cs+. When the larger cation N-methyl-d-glucamine replaced Na+ in the external solution, however, the reversal potential was more negative (see Fig. 3 legend), showing that NMG is much less permeable than Na+ or Cs+. Calcium passes through the channel, as shown by its effect on the reversal potential in the absence of Na+. From the values of reversal potential given in the legend to Fig. 3 and using the constant-field equation (see ref. 14, equations 1-13 and 13-11), we calculate PNa:PCs:PNMG:PCa = 1:1.24:0.08:1.28, respectively.
Figure 3.
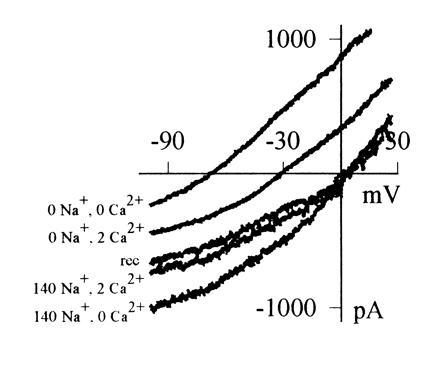
Current–voltage relations of heat-sensitive current, obtained by applying a voltage ramp passing from +30 mV to −100 mV in 455 msec during a brief (600 msec) temperature step to 49°C, and subtracting current observed in response to the ramp at room temperature. Heat-sensitive current was maximized for the purpose of these experiments by prior activation of PKC using PMA (1 μM) and inhibition of phosphatases using calyculin A (20 nM; see Figs. 4 and 5). Relations shown were obtained in the following order: in 140 mM [Na]o, 2 mM [Ca]o (mean Erev = −2 ± 1 mV, n = 9 cells); in 140 mM [Na]o, 0 mM [Ca]o (Erev = −4 ± 3 mV); in 0 mM [Na]o, 0 mM [Ca]o, using Na+ substitute N-methyl-d-glucamine (Erev = −69 ± 2 mV); in 0 mM [Na]o, 2 mM [Ca]o (Erev = −36 ± 1 mV); and again in 140 mM [Na]o, 2 mM [Ca]o (trace labeled “rec”).
Calcium ions also have a blocking effect on the current carried by other cations, as can be seen from the observation that addition of 2 mM Ca2+ reduces the heat-sensitive current when compared with that observed in nominal zero-Ca2+ solution (Fig. 3). Calcium, therefore, competes with Na+ for a binding site in or near the channel. In both the ionic selectivity and the block by calcium, the ion current activated by heat resembles most closely the current gated by cyclic nucleotides in a wide variety of sensory and other neurons (15, 16), although it also has some similarity to the voltage-activated calcium channel, where calcium both carries current and blocks current carried by alkali cations.
Sensitization of the Heat Response.
No sensitization of the response of the isolated neurons used in the present study was observed on repeated application of heat, in contrast to the behavior of heat-sensitive neurons in vivo in which strong sensitization is observed (1–4). However, application of bradykinin (BK), a potent pain-inducing nonapeptide generated by the release of proteolytic enzymes from damaged cells in injured or inflamed tissues (17–21), caused a striking sensitization of the response to noxious temperatures (Fig. 4A).
As described by other authors (22, 23), BK was observed to activate an inward current in some, but not all, small DRG neurons (e.g., Fig. 5). About half (27 of 65) of heat-sensitive neurons responded to BK with a current exceeding 10 pA in magnitude. The heat-sensitive current elicited by a 49°C temperature step in these BK-responsive neurons increased to 209 ± 19% of control (mean ± SEM; n = 12) at the peak of the response to BK. In the same cells, BK speeded the response to a heat pulse, with the time to half-activation of current decreasing to 65 ± 2.9% of its value before BK application (see inverted triangles in Fig. 2B). To rule out the possibility that the sensitization of the heat response was directly related to the activation of membrane current by BK, we also measured sensitization 2 min after BK application, when the BK-induced current had declined to zero (see Fig. 5A). Sensitization at this time was still significant, at 155 ± 15% of control (n = 15). The sensitization caused by BK was limited to those neurons in which BK caused a significant membrane current response, because no enhancement of the heat response was observed in 38 of 38 heat-sensitive neurons in which BK application did not elicit a significant inward current.
Figure 5.
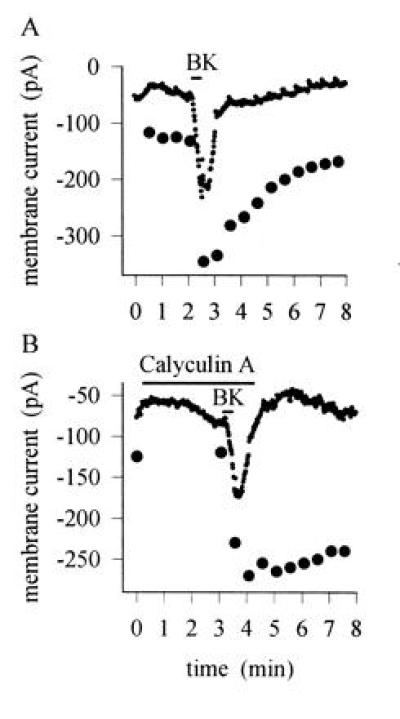
(A) Time course of sensitization induced by BK (1 μM). Simultaneous recording of inward current induced by BK (points) and current increase elicited by a 400 msec step to 49°C, repeated every 30 sec (•). Responses to temperature steps removed from current trace. Similar results obtained in 12 neurons. (B) Similar experiment in which the phosphatase inhibitor calyculin A (20 nM), which inhibits type 1 and 2A serine/threonine phosphatases, was applied before the BK pulse. Inward current induced by BK and sensitization of the heat response were unaffected, but recovery from the sensitized state was prevented by phosphatase inhibition. Similar results obtained in six neurons. The heat response was tested again 12 min after BK application and remained elevated at a level of 185 pA.
Sensitization Caused by Activation of Protein Kinase C (PKC).
A possible mediator of the sensitizing effect of BK is PKC, which is known to be activated by BK via a G protein-coupled pathway (22, 24–26). Like BK, the specific PKC activator phorbol 12-myristate 13-acetate (PMA) both increased the membrane current activated by noxious heat and speeded the rate of activation (Fig. 4B). The mean increase in current in nine neurons was to a value 532 ± 123% of control, and the half-time of activation was reduced to 39 ± 7% of control (see also Fig. 2B, open circles). Both effects are similar to those of BK, although the magnitude is larger, presumably because a more complete activation of PKC is obtained with PMA than with BK. Fig. 4C demonstrates that the threshold for activation of membrane current by temperature is shifted to lower temperatures by PKC activation, an effect that is similar to that observed in vivo after sensitization of the skin by noxious heat or injection of BK (1–4, 27).
The time course of sensitization of the response to heat by BK is shown in Fig. 5A. The time course of onset of the sensitization was rapid, but the effect considerably outlasted the transient inward current elicited by BK. The sensitization therefore is not simply connected to the opening of ionic channels by BK, as might occur if the current activated by BK was heat-sensitive. In Fig. 5B, application of the phosphatase inhibitor calyculin A greatly slowed the rate of recovery of sensitization after BK application, as expected if the sensitization caused by BK is due to phosphorylation of the heat-sensitive channel or of a component in the pathway modulating the channel. Note also that calyculin A did not affect the time course of current elicited by BK, showing that the deactivation of the membrane current activated by BK does not depend on a process involving dephosphorylation.
Further evidence in support of a role of phosphorylation by PKC in sensitization of the heat response is shown in Fig. 6A. Application of PMA caused a long-lasting sensitization, and further applications of PMA were without effect, as expected from the known long-term activation of PKC by PMA (28). Staurosporine, which inhibits PKC by competing for the ATP binding site (29), rapidly reverses the sensitization caused by PMA (Fig. 6B).
Figure 6.
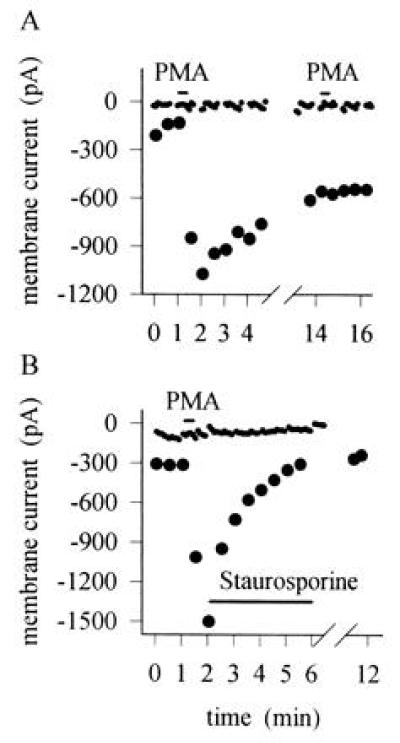
(A) Time course of sensitization induced by PMA. Time of application of PMA (1 μM) shown by bar. Similar results obtained in nine neurons. Interruptions in membrane current trace occur where test pulses of heat were applied. (B) Reversal of sensitization by the PKC inhibitor staurosporine. Similar experiment to that in A, but subsequent application of staurosporine (0.25 μM) as shown abolished the sensitization. Similar results obtained in five neurons. In separate experiments, application of staurosporine 2 min before PMA abolished the sensitization caused by PMA (n = 10).
DISCUSSION
The results reported here show that primary nociceptive neurons respond to noxious heat with a rapid, but not instantaneous, activation of an inward membrane current. The ion channel activated by heat distinguishes poorly between monovalent cations. Calcium ions can also carry current through the channel, but calcium has in addition a blocking action, because the current carried by monovalent ions is seen to be partially suppressed by the presence of external calcium ions. These observations suggest that the heat-activated channel belongs to the same family as the channel activated in visual and olfactory receptors by cyclic nucleotides, both of which exhibit qualitatively similar properties. It remains an open question whether the heat-sensitive channel is gated directly by heat or whether an intracellular messenger is involved, as in the case of the cyclic nucleotide-gated channels.
Unlike nociceptive neurons in vivo, isolated neurons do not exhibit sensitization in response to repeated application of heat, suggesting that the process of sensitization is not intrinsic to the neuron but is instead caused by factors released by damage to adjacent cells. One such factor known to be released by tissue damage is BK, a potent algogenic nonapeptide that acts at B2 receptors to release intracellular calcium and activate PKC. We find that BK sensitizes the heat response and that the mechanism involves activation of PKC. PKC activation produces a shift in the threshold of the heat-sensitive current to lower temperatures, in agreement with in vivo observations in which damaged tissue becomes sensitive to normally non-noxious temperatures (1–4).
The experiments reported here show that BK has two distinct actions of excitation and sensitization on nociceptive neurons. (i) BK excites neurons by eliciting an inward current, which depolarizes the neuron and generates action potentials (9, 10, 22, 23). (ii) Sensitization of the heat response is caused by PKC activation, and recovery from sensitization depends on dephosphorylation because it is prevented by phosphatase inhibitors (Fig. 5B). Excitation and sensitization follow different time courses (Fig. 5). Although the internal transmitter mediating the activation of inward current by BK remains to be established, it is clear from the results reported here that sensitization of the heat response is mediated by PKC.
The sensitizing effect of BK on the heat response is distinct from the sensitization caused by prostaglandins, in which the excitability of the nociceptive neuron is increased by lowering the threshold of activation of a tetrodotoxin-insensitive Na+ current (30). The current modified by prostaglandins is strongly voltage-sensitive, is not activated at the resting potential and is inactivated on maintained depolarization. The heat-sensitive current modified by BK, on the other hand, is neither activated nor inactivated by changes in membrane potential. It is perhaps not surprising, in view of the range of algogenic and sensitizing stimuli to which nociceptive neurons must respond, that different sensitizing agents act by distinct pathways.
The importance for inflammation of the heat response of nociceptors is underlined by the recent observation that neurogenic inflammation (the flare response) depends on activation of a specific class of heat-sensitive nociceptor and is not elicited by stimulation of polymodal nociceptors (5). A further link between the heat response and neurogenic inflammation is suggested by the finding that activation of PKC in isolated neurons, which from the results in the present paper will sensitize the heat response, enhances the release of the neuropeptides thought to be responsible for eliciting neurogenic inflammation (31).
The results reported here raise the possibility of biophysical study of the processes underlying activation and sensitization of pain-sensitive neurons by heat. They also suggest possible therapeutic approaches to the relief of pain by the use of PKC inhibitors, perhaps by targeting particular isoforms of PKC involved in the sensitization process.
Acknowledgments
We thank Prof. Carlos Belmonte for helpful discussions and Roger Stoughton for participation in early experiments. This work was supported by the European Commission.
Footnotes
Abbreviations: DRG, dorsal root ganglion; PKC, protein kinase C; PMA, phorbol 12-myristate 13-acetate; BK, bradykinin.
References
- 1.Besson J M, Chaouch A. Physiol Rev. 1987;67:67–155. doi: 10.1152/physrev.1987.67.1.67. [DOI] [PubMed] [Google Scholar]
- 2.Treede R D, Meyer R A, Raja S N, Campbell J N. Prog Neurobiol. 1992;38:397–421. doi: 10.1016/0301-0082(92)90027-c. [DOI] [PubMed] [Google Scholar]
- 3.Levine J D, Fields H L, Basbaum A I. J Neurosci. 1993;13:2273–2286. doi: 10.1523/JNEUROSCI.13-06-02273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer R A, Campbell J N, Srinivasa N R. In: Textbook of Pain. 3rd Ed. Melzack R, Wall P, editors. Edinburgh: Churchill Livingstone; 1994. pp. 13–44. [Google Scholar]
- 5.Lynn B, Schutterle S, Pierau F K. J Physiol (London) 1996;494:587–593. doi: 10.1113/jphysiol.1996.sp021516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawson S N, Perry M J, Prabhakar E, McCarthy P W. Brain Res Bull. 1993;30:239–243. doi: 10.1016/0361-9230(93)90250-f. [DOI] [PubMed] [Google Scholar]
- 7.Hoheisel U, Mense S, Scherotzke R. Anat Embryol. 1994;189:41–49. doi: 10.1007/BF00193128. [DOI] [PubMed] [Google Scholar]
- 8.Baccaglini P I, Hogan P G. Proc Natl Acad Sci USA. 1983;80:594–598. doi: 10.1073/pnas.80.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rang H P, Bevan S, Dray A. Br Med Bull. 1991;47:534–548. doi: 10.1093/oxfordjournals.bmb.a072491. [DOI] [PubMed] [Google Scholar]
- 10.Rang H P, Bean S, Dray A. In: Textbook of Pain. 3rd Ed. Melzack R, Wall P, editors. Edinburgh: Churchill Livingstone; 1994. pp. 57–78. [Google Scholar]
- 11.Gold M S, Dastmalchi S, Levine J D. Neuroscience. 1996;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- 12.Gilabert, R. & McNaughton, P. A. (1996) J. Neurosci. Methods, in press. [DOI] [PubMed]
- 13.Darian-Smith I. Handbook of Physiology. Vol. 3. Bethesda, MD: Am. Physiol. Soc.; 1984. pp. 879–913. [Google Scholar]
- 14.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 15.Hodgkin A L, McNaughton P A, Nunn B J. J Physiol (London) 1985;358:447–468. doi: 10.1113/jphysiol.1985.sp015561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finn J T, Grunwald M E, Yau K W. Annu Rev Physiol. 1996;58:395–426. doi: 10.1146/annurev.ph.58.030196.002143. [DOI] [PubMed] [Google Scholar]
- 17.Steranka L R, Manning D C, DeHaas C J, Ferkany J W, Borosky S A, Connor J R, Vavrek R J, Stewart J M, Snyder S H. Proc Natl Acad Sci USA. 1988;85:3245–3249. doi: 10.1073/pnas.85.9.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dray A, Perkins M. Trends Neurosci. 1993;16:99–104. doi: 10.1016/0166-2236(93)90133-7. [DOI] [PubMed] [Google Scholar]
- 19.Walker K, Perkins M, Dray A. Neurochem Int. 1995;26:1–16. doi: 10.1016/0197-0186(94)00114-a. [DOI] [PubMed] [Google Scholar]
- 20.Hall J M, Geppetti P. Neurochem Int. 1995;26:17–26. [Google Scholar]
- 21.Dray A. Br J Anaesth. 1995;75:125–131. doi: 10.1093/bja/75.2.125. [DOI] [PubMed] [Google Scholar]
- 22.Burgess G M, Mullaney I, McNeill M, Dunn P M, Rang H P. J Neurosci. 1989;9:3314–3325. doi: 10.1523/JNEUROSCI.09-09-03314.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicol G D, Cui M. J Physiol (London) 1994;480:485–492. doi: 10.1113/jphysiol.1994.sp020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thayer S A, Perney T M, Miller R J. J Neurosci. 1988;8:4089–4097. doi: 10.1523/JNEUROSCI.08-11-04089.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGuirk S M, Dolphin A C. Neuroscience. 1992;49:117–128. doi: 10.1016/0306-4522(92)90079-h. [DOI] [PubMed] [Google Scholar]
- 26.Jones S, Brown D A, Milligan G, Wilier E, Buckley N J, Caulfield M P. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- 27.Kumazawa T, Mizumura K, Minagawa M, Tsujii Y. J Neurophysiol. 1991;66:1819–1824. doi: 10.1152/jn.1991.66.6.1819. [DOI] [PubMed] [Google Scholar]
- 28.Nishizuka Y. Nature (London) 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson S E, Hallam T J. Trends Pharmacol Sci. 1994;15:53–57. doi: 10.1016/0165-6147(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 30.Gold M S, Reichling D B, Shuster M J, Levine J D. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barber L A, Vasko M R. J Neurochem. 1996;67:72–80. doi: 10.1046/j.1471-4159.1996.67010072.x. [DOI] [PubMed] [Google Scholar]
- 32.Treede R D, Meyer R A, Raja S N, Campbell T N. J Physiol (London) 1995;483:747–758. doi: 10.1113/jphysiol.1995.sp020619. [DOI] [PMC free article] [PubMed] [Google Scholar]