

Abstract
Specific antagonists of central dopaminergic receptors constitute the major class of antipsychotic drugs (APD). Two principal effects of APD are used as criteria for the pre-clinical screening of their antipsychotic action: (i) inhibition of basal and depolarization-induced activity of mesolimbic dopaminergic neurons; (ii) antagonism of the locomotor effects of dopaminergic agonists. Given that glucocorticoid hormones in animals increase dopamine release and dopamine-mediated behaviors and that high levels of glucocorticoids can induce psychotic symptoms in humans, these experiments examined whether inhibition of endogenous glucocorticoids might have APD-like effects on mesolimbic dopaminergic transmission in rats. It is shown that suppression of glucocorticoid secretion by adrenalectomy profoundly decreased (by greater than 50%): (i) basal dopaminergic release and the release of dopamine induced by a depolarizing stimulus such as morphine (2 mg/kg, s.c.), as measured in the nucleus accumbens of freely moving animals by microdialysis; (ii) the locomotor activity induced by the direct dopaminergic agonist apomorphine. The effects of adrenalectomy were glucocorticoid specific given that they were reversed by the administration of glucocorticoids at doses within the physiological range. Despite its profound diminution of dopaminergic neurotransmission, adrenalectomy neither modified the number of mesencephalic dopaminergic neurons nor induced gliosis in the mesencephalon or in the nucleus accumbens, as shown by tyrosine hydroxylase and glial fibrillary acidic protein immunostaining. In conclusion, these findings suggest that blockade of central effects of glucocorticoids might open new therapeutic strategies of behavioral disturbances.
Keywords: drug abuse, corticosterone, nucleus accumbens, morphine, apomorphine
The discovery of treatments reducing the activity of the central dopaminergic (DA) transmission is one of the main goals of neuroscience and pharmacological researches. Interest in dopamine has been sustained by the discovery that antipsychotic drugs (APD) were potent antagonists of the DA receptors (1). Decades of intensive research has considerably enlarged our knowledge about the functional role of DA neurons (2), suggesting that their hyperactivity may be involved in various behavioral pathologies, such as psychosis and addiction (3, 4). Nevertheless, antagonists of DA receptors are still the only class of drugs used in clinical practice to antagonize DA activity.
There is evidence indicating that glucocorticoid hormones may be one of the factors determining a pathological hyperactivity of DA neurons. First, an increase in glucocorticoids can induce behavioral changes that are among those attributed to the enhancement of DA activity. For example, high levels of glucocorticoids can generate mood changes ranging from euphoria to psychosis in humans (5, 6), and increase the propensity to develop self-administration of drugs of abuse in animals (7). Second, administration of glucocorticoids increases extracellular concentrations of dopamine in vivo (8) and in vitro (9) and facilitates dopamine-dependent behaviors (10). Third, blockade of stress-induced corticosterone secretion suppresses the sensitization of the DA activity induced by stress (11, 12), which is considered an experimental model of acute psychosis (13, 14).
Given that glucocorticoids stimulate DA activity (8–10) and induce dopamine-related behavioral pathologies (5–7), it is reasonable to hypothesize that an inhibition of the secretion of these hormones might decrease DA activity and have APD-like effects. This idea is also supported by the relationship between glucocorticoids and psychotic symptoms. Treatment with synthetic glucocorticoids can induce psychotic symptoms in humans (15) and a hypersensitivity to the dopaminergic effects of glucocorticoids has been reported in schizophrenic patients (16). To test this hypothesis, the effects of the suppression of endogenous glucocorticoids on some of the DA parameters believed to reflect the antipsychotic action of APD were studied. Among the several DA effects of APD few of them are believed to reflect the antipsychotic action of these drugs (for review see ref. 17): (i) inhibition of basal and depolarization-induced activity of the mesolimbo-cortical component of DA neurons, which results in a decreased cell firing and in lower extracellular concentrations of dopamine and (ii) block of the locomotor effects of dopaminergic agonists such as apomorphine.
Basal and depolarization-induced release of dopamine and apomorphine-induced locomotion were studied in animals in which endogenous glucocorticoids were suppressed by ablation of the adrenal gland and in adrenalectomized (ADX) animals receiving a glucocorticoid replacement treatment. Dopamine release was estimated by measuring changes in extracellular concentrations of dopamine in freely moving rats using the microdialysis technique. Dopamine was studied in the nucleus accumbens, a mesolimbic DA projection involved in motivational and cognitive functions (2). Depolarization-induced dopamine release was obtained by injecting the opioid morphine. This drug increases the firing rate of dopaminergic neurons and induces a marked depolarization (18). The effects of adrenalectomy on the number of mesencephalic DA cells and on the density of astrocytes in the ventral mesencephalon and in the nucleus accumbens were also evaluated.
MATERIALS AND METHODS
General Methods
Animals and Housing Conditions.
Male Sprague–Dawley rats (Iffa Credo) weighing 280–300 g upon arrival were used. Animals had ad libitum access to food and water and were housed individually. The light-dark cycle (lights were on from 6 a.m. to 8 p.m.), temperature (22°C), and humidity (60%) were kept constant in the animal house. Animals were allowed at least 1 week of acclimatization before the experiments were started.
Drugs and Drug Administration.
Corticosterone 21-hemisuccinate (Agrar), morphine sulfate and apomorphine (Apokinon, Aguettant, France) were used, and concentrations are expressed as base. Morphine (2 mg/kg/ml) and apomorphine (0.375–6.000 mg/kg/ml) were dissolved in sterile 0.9% NaCl saline solution and injected subcutaneously. Isotonic saline was also used as vehicle for control injections.
Constitution of Experimental Groups.
Because locomotor response to novelty is correlated to the DA activity in the nucleus accumbens (19), we ensured a homogeneous distribution of this factor throughout the different experimental groups. For this purpose, 1 week after arrival, the locomotor activity in a novel environment (a circular corridor, 10 cm wide and 70 cm in diameter) was measured for 2 hr beginning at 4 p.m. Locomotor activity scores (expressed as photocell counts) were used to evenly distribute rats among the different experimental groups.
Locomotor Response to Apomorphine.
For the dose response study metal wire mesh activity cages (20 × 25 × 36 cm) were used. A locomotor activity count was recorded each time a rat crossed the full distance (15 cm) between photocells located on each of the two long sides. For the corticosterone replacement study, the circular corridors were used. Two different apparati were used to generalize the results. In particular, the circular corridor allows a more precise measure of locomotion.
Microdialysis.
Under sodium pentobarbital anesthesia (50 mg/kg, i.p.) rats were implanted with a guide cannula (CMA/11-Sweden), which was lowered to 2 mm above the bottom of the nucleus accumbens. The coordinates relative to bregma, in millimeters, were: A = 3.6, L = 1.9, V = 6.5, at a lateral angle of 6° (20). After a post-operative period of 10–14 days, the animals were ADX or sham operated. Five days later, the microdialysis probe (CMA/11, 2 mm cuprophane membrane length) was inserted through the guide cannula. The in vitro recovery of each probe had been determined before the implantation to homogenize this factor over the groups. Two days later, each animal was transferred to the dialysis cage (32 × 32 × 22 cm), the probe was connected to a syringe pump (Harvard 22) via a two-channel swivel, and the perfusion (2 μl/min) started. The perfusion fluid was a modified artificial cerebrospinal fluid (145 mM NaCl/1.2 mM CaCl2/2.7 mM KCl/1 mM MgCl2/0.2 mM Na2HPO4/NaH2PO4 buffered at pH 7.4). Brain dialysis was performed with a fully automated on-line system (see ref. 12 for a detailed description). High performance liquid chromatography coupled to a coulometric detector (Coulochem II, ESA, Bedford, MA) was used to detect dopamine (0.5 pg detection limit). The first three consecutive dialysate samples that showed less than 10% variation in peak height were considered as the baseline. At the end of the experiments, cannula placements were verified histologically on 100-μm thionin-stained coronal sections. Only the animals with correctly placed probes were included in the statistical analyses of microdialysis data. Simultaneously to dopamine, locomotor activity was recorded by two photocell beams located on two sides of the dialysis cage.
Adrenalectomy and Corticosterone Replacement Treatment.
Adrenalectomy was performed between 8 and 10 a.m. under ether anesthesia via the dorsal approach. Surgeries were accomplished in less than 4 min and 30 sec from the time the cage was taken from the animal room. Following surgery, NaCl (0.9%) was added to the drinking water. Some of the ADX animals (ADX+CORT group) received a corticosterone replacement treatment reproducing the corticosterone circadian secretion. Animals in this group were implanted with a subcutaneous solid pellet of corticosterone and cholesterol (50 mg each), which provides a constant release of the hormone in the range of the diurnal basal levels (10). The nocturnal peak was reproduced by adding, from 7 p.m. to 8 a.m., corticosterone (50 μg/ml) to the drinking solution (10). ADX animals that did not receive any replacement treatment constituted the ADX group. Control animals (SHAM group) underwent the same surgical procedure as the ADX animals except that the adrenals were not removed. All experiments were performed 7 days after adrenalectomy. At the end of the experiments, a blood sample was withdrawn from the tail vein and corticosterone assayed by radioimmunoassay (RIA kit, ICN). Only ADX animals with corticosterone levels below the detection threshold (0.1 μg/100 ml) were included in the experiment; ADX+CORT rats had corticosterone levels in the range of controls (2.1 ± 0.56 μg/100 ml).
Immunohistochemistry.
Animals were perfused transcardiacally with 100 ml of 0.1 M phosphate buffered saline (pH 7.3), followed by 350 ml of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.3). Following a 24 hr post-fixation period, 50-μm sections were cut using a vibratome. For each antibody, all free-floating sections were processed at the same time through a standard immunohistochemical procedure to visualize either tyrosine hydroxylase (TH) with a rabbit anti-TH antibody (dilution of 1/10,000, Boy, Paris) or glial fibrillary acidic protein (GFAP) with a rabbit anti-GFAP antibody (dilution of 1/400, Dako). Following 48 hr of incubation, sections were incubated with a biotin-labeled goat anti-rabbit antibody (1/200, Dako). TH- and GFAP-immunoreactivity was visualized by the biotin-streptavidin technique (ABC kit, Dako) using 3,3′ diaminobenzidine as the chromogen. These staining conditions allowed us to evaluate possible differences in the number of TH-positive cells, but, given the saturation of the staining, they do not allow us to evaluate quantitative differences in protein content.
Statistics.
Analysis of variance for repeated measures was used. Changes in extracellular concentrations of dopamine and locomotor activity during microdialysis were analyzed using the treatment (three levels: SHAM, ADX, or ADX+CORT) as between factor and the time of sampling (18 levels) as within factor. The locomotor response to apomorphine in the dose-response experiment was analyzed using two between factors: treatment (two levels, SHAM or ADX) and dose (six levels). For the corticosterone-replacement experiment, one between factor (treatment, three levels: SHAM, ADX, or ADX+CORT) was used. In the case of the immunohistochemistry, data were analyzed with treatment (SHAM or ADX) as between factor and section as within factor. Newman-Keuls test was used for all post hoc analyses.
Procedures
Effects of the Suppression of Glucocorticoids on Extracellular Concentration of Dopamine in the Nucleus Accumbens.
Rats in the SHAM (n = 9), ADX (n = 8), ADX+CORT (n = 7) groups were placed in the dialysis cages at 9 a.m. Extracellular concentrations of dopamine and locomotor activity were recorded over 20-min intervals. After 2 hr of habituation to this apparatus, the animals received an injection of vehicle and 2 hr later an injection of morphine (2 mg/kg, s.c.). The effects of the latter injection were studied for 3 hr.
Effects of the Suppression of Glucocorticoids on Apomorphine-Induced Locomotion.
In the first experiment, independent experimental groups of SHAM and ADX animals received one of the following doses of apomorphine: 0, 0.375, 0.750, 1.500, 3.000, and 6.000 mg/kg (n = 7–10 per group). Animals were placed in the activity cages at 11 a.m., and after 2 hr of habituation they received the assigned injection. In a second experiment, animals in the ADX, SHAM, and ADX+CORT groups were placed in the circular corridors at 9 a.m. After 2 hr of habituation they received an injection of vehicle and 1 hr later an injection of apomorphine (1.5 mg/kg). This dose of apomorphine was chosen on the basis of the results of the first experiment. In both experiments locomotor response to apomorphine was recorded for 2 hr over 10-min intervals.
Effects of Adrenalectomy on TH- and GFAP-Positive Cells.
TH- and GFAP-positive cells were counted in brain sections of animals in the ADX and SHAM groups (n = 5–7). The TH-positive cells were counted in the ventral tegmental area (VTA) and in the substantia nigra (SN) on three sections for each animal, with an interval of 150 μm between the sections. The density of GFAP-positive cells was determined on two sections per animal for the nucleus accumbens and the SN, with an interval of 250 μm between the sections, and on one section per animal for the VTA. The VTA and SN subregions were chosen on the basis of the medial lemniscus, which made a clear separation between the two regions. The VTA was studied between antero-posteriority (A/P) −5.0 and −5.5 from the bregma, and the SN between A/P −5.0 and −6.0 (21). The region of the nucleus accumbens studied was medial to the anterior commissure and between A/P +1.5 and +2.0 (21).
RESULTS
Effects of the Suppression of Glucocorticoids on Extracellular Concentrations of Dopamine in the Nucleus Accumbens.
Manipulations of the glucocorticoid status modified extracellular concentrations of dopamine in the nucleus accumbens [treatment effect, F(2,21) = 5.11, P < 0.015] (Fig. 1). This effect was present in basal conditions [treatment effect, F(2,21) = 4.96, P < 0.02], after the injection of vehicle [treatment effect, F(2,21) = 5.50, P < 0.02], and after the injection of morphine [treatment effect, F(2,21) = 4.74, P < 0.02]. In all the cases, ADX animals had lower levels of dopamine than SHAM rats (Newman-Keuls: basal = P < 0.02; vehicle = P < 0.015; morphine = P < 0.03) and than ADX animals receiving corticosterone replacement (Newman-Keuls: basal = P < 0.05; vehicle = P < 0.035; morphine = P < 0.03). The latter two groups did not differ. Manipulations of the glucocorticoid status also modified the percentage increase in the concentrations of dopamine induced by morphine over time [treatment × time interaction, F(16,168) = 1.95, P < 0.02]. A significant treatment effect was found only over the first hour after morphine injection [F(2,21) = 4.68, P < 0.02]. Within this time window, the percentage increase in dopamine concentrations was lower in ADX animals than in either SHAM rats (Newman-Keuls: P < 0.03) or in ADX rats receiving corticosterone (Newman-Keuls: P < 0.03). Again, the latter two groups did not differ. In contrast, no significant effect was found over the second [F(2,21) = 2.31, P > 0.12] and third [F(2,21) = 0.81, P > 0.45] hour after the injection, and point by point comparisons did not reveal any significant differences between ADX and SHAM animals over this period of time. This indicates that ADX rats have a lower, but not longer, response to morphine. The effect of adrenalectomy on basal concentrations of dopamine were replicated in a supplementary experiment bringing the number of animals tested to n = 15 per group. Pooling the results of the two experiments increased the statistical significance of the effects described above [F(2,42) = 6.07, P < 0.005, Newman-Keuls: ADX versus SHAM or versus ADX+CORT, P < 0.009].
Figure 1.
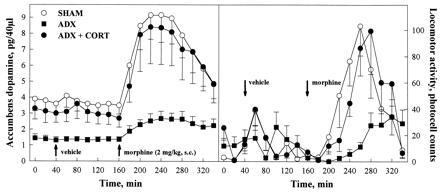
Effects of the suppression of glucocorticoids on extracellular concentrations of dopamine in the nucleus accumbens and on locomotor activity. Compared with animals with an intact glucocorticoid secretion (SHAM), animals in which endogenous glucocorticoids have been suppressed by adrenalectomy (ADX) showed lower levels of dopamine in basal conditions and after the injection (arrows) of either vehicle or morphine, morphine-induced locomotor activity was also reduced in ADX rats. Administration of corticosterone to ADX animals (ADX+CORT) reversed these effects.
Morphine-induced locomotion was similarly modified by manipulations of the glucocorticoid status [treatment effect F(2,24) = 4.99, P < 0.02] (Fig. 1). ADX animals had lower morphine-induced locomotion than SHAM rats and than ADX animals receiving corticosterone replacement (Newman-Keuls: P < 0.025 in both cases); in contrast, the latter two groups did not differ. The effect of adrenalectomy was significant only during the first 2 hr following the injection [F(2,24) = 7.125, P < 0.004], but no significant difference was found during the last hour [F(2,24) = 0.59, P > 0.55]. Locomotor activity in basal conditions [treatment effect F(2,24) = 1.44, P > 0.25] or after the injection of vehicle [treatment effect F(2,24) = 0.05, P > 0.94] was not modified. These findings on locomotor activity confirm previous published results (10).
Effects of the Suppression of Glucocorticoids on Apomorphine-Induced Locomotion.
ADX and control rats did not differ in response to the injection of vehicle [treatment effect, F(1,25) = 0.89, P > 0.36]; for this reason these values were cumulated in Fig. 2 (shaded area). In contrast, adrenalectomy modified the locomotor response to apomorphine [treatment effect, F(1,61) = 4.69, P < 0.035]. ADX animals showed a lower sensitivity to the locomotor effect of apomorphine as shown by the shift toward the right of the dose-response curve of these rats (Fig. 2). A significant reduction in apomorphine-induced locomotion in ADX animals was observed at the 0.750 mg/kg [F(1,17) = 5.83, P < 0.03] and 1.500 mg/kg [F(1,17) = 6.99, P < 0.025] doses. This effect of adrenalectomy was glucocorticoid specific. Thus, corticosterone administration reversed the effect of adrenalectomy [treatment effect F(2,15) = 4.43, P < 0.03] on the locomotor response to 1.5 mg/kg of apomorphine (Fig. 2). ADX animals receiving exogenous administration of corticosterone did not differ from SHAM rats and had a higher locomotor activity than ADX rats (Newman-Keuls: P < 0.04).
Figure 2.
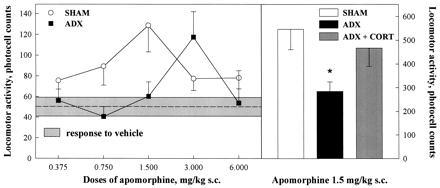
Effects of the suppression of glucocorticoids on apomorphine-induced locomotion. Compared with animals with an intact glucocorticoid secretion (SHAM), animals in which endogenous glucocorticoids have been suppressed by adrenalectomy (ADX) showed a lower sensitivity to the locomotor effects of apomorphine, as indicated by the shift toward the right in the dose-response curve. Administration of corticosterone to ADX animals (ADX+CORT) reversed the effect of adrenalectomy on the locomotor response to 1.5 mg/kg of apomorphine. Apomorphine-induced locomotion has been cumulated over the 2 hr of testing. ∗, P < 0.05 versus SHAM or versus ADX+CORT.
Effects of Adrenalectomy on TH- and GFAP-Positive Cells.
The large reduction in the activity of the DA transmission induced by the suppression of glucocorticoids was not related to changes in the number of DA neurons, as identified by a TH immunostaining, in the VTA [F(1,10) = 0.00, P > 0.95] or in the SN [F(1,11) = 0.14, P > 0.7] (Fig. 3). Similarly, no changes were observed in the density of GFAP-positive cells in the regions of the VTA [F(1,12) = 0.57, P > 0.46], SN [F(1,9) = 0.01, P > 0.92], or nucleus accumbens [F(1,8) = 0.05, P > 0.81] (Fig. 3). ADX and SHAM animals also did not differ when the level of the sections was taken into account [section × treatment interaction for TH-positive cells: SN F(2,22) = 0.369, P > 0.69; VTA F(2,20) = 0.439, P > 0.65; section × treatment interaction for GFAP density: N.Acc. F(1,8) = 0.199, P > 0.66; SN F(1,9) = 0.852, P > 0.38].
Figure 3.
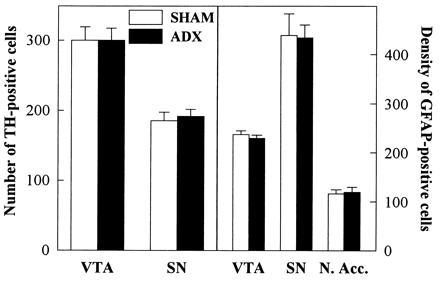
Effects of adrenalectomy on TH- and GFAP-positive cells. Compared with animals with an intact glucocorticoid secretion (SHAM) animals in which endogenous glucocorticoids have been suppressed by adrenalectomy (ADX) did not differ for the number of dopaminergic neurons (TH-positive cells) in the VTA and in the SN. Similarly, the two groups did not differ for the density of astrocytes (GFAP-positive cells, nb/mm2) in the VTA, in the SN, and in the nucleus accumbens (N.Acc.).
DISCUSSION
These results show that suppression of endogenous glucocorticoids drastically reduces the activity of the meso-accumbens DA transmission. Thus, 1 week after adrenalectomy, decreases in the following were observed: (i) the basal extracellular concentrations of dopamine in the nucleus accumbens, (ii) the release of dopamine in response to a depolarizing stimulus such as morphine, (iii) the locomotor response to morphine and to the direct DA agonist apomorphine. These observations suggest that glucocorticoids modify both the presynaptic and postsynaptic side of the DA transmission. Indeed, extracellular concentrations of dopamine, as measured by microdialysis, reflect the release of this neurotransmitter from the presynaptic side. Apomorphine, at the doses used in the present experiments, increases locomotion by stimulating DA postsynaptic receptors.
The large reduction in the activity of the DA transmission induced by the suppression of glucocorticoids is probably the result of functional changes in the activity of this neural system. Thus, adrenalectomy did not reduce the number of DA cells in the VTA or in the SN. In parallel, in these regions and in the nucleus accumbens, there were no changes in the density of GFAP-positive cells, which are an index of the gliosis that usually accompanies degenerative process in the central nervous system.
These and previous results (8) suggest that in physiological conditions glucocorticoids exercise a facilitatory input on the functional activity of mesencephalic DA neurons. Thus, suppression of endogenous glucocorticoids decreases extracellular concentrations of dopamine. In parallel, an increase in glucocorticoid levels within the stress range enhances extracellular concentrations of dopamine in the nucleus accumbens and induces dopamine-dependent locomotor activity (8). Stimulating effects of corticosterone on depolarization-induced dopamine release have also been found in cultures of mesencephalic DA neurons (9).
Glucocorticoids may modulate extracellular concentrations of dopamine by acting directly on DA neurons, which express corticosteroid receptors (22). Three principal mechanisms of action may be foreseen. First, glucocorticoids may control dopamine synthesis by increasing the levels of TH, the limiting enzyme of dopamine synthesis (23). Effects of corticosterone on TH have been demonstrated in the locus coeruleus (24), hypothalamus (25), and more recently in the VTA (26), though important individual differences in the latter effects have been reported (26). Second, glucocorticoids may decrease dopamine catabolism acting as reversible monoamine oxidase inhibitors (27–29). This action of glucocorticoids is consistent with the fact that dexamethasone decreases deaminated products of dopamine such as HVA and DOPAC (29, 30), which depend on monoamine oxidase activity, whereas it increases 3MT levels that depend on COMT (catechol-O-methyltransferase) (29). Third, glucocorticoids may decrease catecholamine reuptake (31, 32). Inhibition of dopamine reuptake by glucocorticoids has been demonstrated in synaptosome preparations obtained from projection areas of mesencephalic DA neurons, and using concentrations of the hormone in the physiological range (32). Glucocorticoids may also control extracellular concentrations of dopamine through neural mechanisms extrinsic to the DA neurons. For example, opioid, γ-aminobutyric acid, excitatory amino acid, and serotoninergic transmissions are influenced by glucocorticoids (33) and can modulate the activity of DA neurons (34).
Reduction of apomorphine-induced locomotion by suppression of endogenous glucocorticoids suggests that these hormones modify the functional activity of DA receptors. Two studies support this idea. First, the decrease in dopamine release induced by the direct dopamine agonist apomorphine is reduced by suppression of endogenous glucocorticoids (35). Second, in cultures of vascular smooth muscle, the synthetic glucocorticoid dexamethasone enhances the increase in cAMP induced by dopamine (36).
Glucocorticoids could modulate the DA postsynaptic transmission by two principal mechanisms. First, these hormones could increase the expression of DA receptors. Suppression of endogenous glucocorticoids reduces D2 and D1 receptors binding in the striatum (37), whereas an increase in corticosterone levels increases it (37, 38). Second, glucocorticoids may act downstream by increasing the activity of adenylate cyclase (39, 40), a main component of the second messenger systems coupled to DA receptors (41). However, at this stage of our knowledge, other more indirect mechanisms cannot be excluded. For example, glucocorticoids could act by modifying the activity of other neurotransmitter systems that modulate nucleus accumbens functions, such as the excitatory amino acid glutamate (42). Furthermore, a possible action on membrane potential of nucleus accumbens neurons should be taken into account (43).
Our results show that the suppression of corticosterone secretion can be compared with APD in reducing the activity of the mesolimbic DA transmission. After chronic APD treatments, reduction between 36 and 50% in basal and depolarization-induced extracellular concentrations of dopamine have been observed in the terminal field of DA mesolimbic neurons (44–48).
APD-like effect of adrenalectomy suggests that anti-glucocorticoid drugs may open new therapeutic strategies of behavioral pathologies. This idea is supported by the fact that specific antagonists of corticosteroid receptors mimic the effects of adrenalectomy on dopamine release (M.M., B. Aouizerate, M.B., M.L.M., and P.V.P., unpublished results). Existing antagonists of glucocorticoid receptors are currently used for the treatment of various non-behavioral pathologies. These drugs have a poor bioavailability and scarcely reach the brain (49, 50). Furthermore, the available type II antagonist RU 38486 is poorly specific, since this drug has been designed and is used as an antagonist of progesterone receptors (50). Inhibitors of glucocorticoid synthesis, such as metyrapone, might be an alternative to glucocorticoid antagonists. However, synthesis inhibitors also are poorly specific drugs that present two major problems. First, they have direct non-specific effects on brain functions (51). Second, they increase glucocorticoid precursors (52) that, even if with lower affinity, can still bind to corticosteroid receptors. These observations could explain why the potential APD features of anti-glucocorticoid drugs have not yet been revealed, and suggest that research efforts should be put in developing new, more specific, pharmacological tools that could allow the blocking of the central effects of glucocorticoids.
In conclusion, our findings enlarge current knowledge on the physiological regulation of DA neurons by glucocorticoids and suggest that the blockade of certain central effects of these hormones may open new therapeutic strategies for the treatment of behavioral disturbances.
Acknowledgments
This work was supported by Institut National de la Santé et de la Recherche Médicale (INSERM), Université de Bordeaux II, Conseil Régional d’Aquitaine, Pôle Médicament d’Aquitaine, Ministère de la Recherche et de l’Enseignement Supérieur.
Footnotes
Abbreviations: DA, dopaminergic; APD, antipsychotic drugs; ADX, adrenalectomized; TH, tyrosine hydroxylase; VTA, ventral tegmental area; SN, substantia nigra; GFAP, glial fibrillary acidic protein.
References
- 1.Seeman P. In: Psychopharmacology: The Fourth Generation of Progress. Bloom F E, Kupfer D J, editors. New York: Raven; 1995. pp. 295–302. [Google Scholar]
- 2.Le Moal M, Simon H. Physiol Rev. 1991;71:155–234. doi: 10.1152/physrev.1991.71.1.155. [DOI] [PubMed] [Google Scholar]
- 3.Swerdlow N R, Koob G F. Behav Brain Sci. 1987;10:197–245. [Google Scholar]
- 4.Piazza P V, Le Moal M. Annu Rev Pharmacol Toxicol. 1996;36:359–378. doi: 10.1146/annurev.pa.36.040196.002043. [DOI] [PubMed] [Google Scholar]
- 5.Hall R C W, Popkin M K, Stickney S K, Gardner E R. J Nerv Ment Dis. 1979;167:229–236. doi: 10.1097/00005053-197904000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Ling M H M, Perry P J, Tsuang M T. Arch Gen Psychiatry. 1981;38:471–477. doi: 10.1001/archpsyc.1981.01780290105011. [DOI] [PubMed] [Google Scholar]
- 7.Piazza P V, Maccari S, Deminière J M, Le Moal M, Mormède P, Simon H. Proc Natl Acad Sci USA. 1991;88:2088–2092. doi: 10.1073/pnas.88.6.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piazza P V, Rougé-Pont F, Deroche V, Maccari S, Simon H, Le Moal M. Proc Natl Acad Sci USA. 1996;93:8716–8720. doi: 10.1073/pnas.93.16.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronken E, Mulder A H, Schoffelmeer A N M. Eur J Pharmacol. 1994;263:149–156. doi: 10.1016/0014-2999(94)90535-5. [DOI] [PubMed] [Google Scholar]
- 10.Marinelli M, Piazza P V, Deroche V, Maccari S, Le Moal M, Simon S. J Neurosci. 1994;14:2724–2731. doi: 10.1523/JNEUROSCI.14-05-02724.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deroche V, Marinelli M, Maccari S, Le Moal M, Simon H, Piazza P V. J Neurosci. 1995;15:7181–7188. doi: 10.1523/JNEUROSCI.15-11-07181.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rougé-Pont F, Marinelli M, Le Moal M, Simon H, Piazza P V. J Neurosci. 1995;15:7189–7195. doi: 10.1523/JNEUROSCI.15-11-07189.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson T E, Becker J B. Brain Res Rev. 1986;11:157–198. doi: 10.1016/s0006-8993(86)80193-7. [DOI] [PubMed] [Google Scholar]
- 14.Kalivas P W, Stewart J. Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- 15.Lewis D A, Smith R E. J Affective Disord. 1983;5:319–332. doi: 10.1016/0165-0327(83)90022-8. [DOI] [PubMed] [Google Scholar]
- 16.Halbach M, Henning U. Pharmacopsychiatry. 1989;22:169–173. doi: 10.1055/s-2007-1014601. [DOI] [PubMed] [Google Scholar]
- 17.Kinon B J, Lieberman J A. Psychopharmacology. 1996;124:2–34. doi: 10.1007/BF02245602. [DOI] [PubMed] [Google Scholar]
- 18.Gysling K, Wang R Y. Brain Res. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- 19.Rougé-Pont F, Piazza P V, Kharouby M, Le Moal M, Simon H. Brain Res. 1993;602:169–174. doi: 10.1016/0006-8993(93)90260-t. [DOI] [PubMed] [Google Scholar]
- 20.Pellegrino L J, Pellegrino A S, Cushman A J. A Stereotaxic Atlas of the Brain. New York: Plenum; 1979. [Google Scholar]
- 21.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2nd Ed. London: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- 22.Härfstrand A, Fuxe K, Cintra A, Agnati L F, Zini I, Wilkström A C, Okret S, Zhao-Ying Y, Goldstein M, Steinbusch H, Verhofstad A, Gustafsson J A. Proc Natl Acad Sci USA. 1986;83:9779–9783. doi: 10.1073/pnas.83.24.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iuvone P M, Morasco J, Dunn A. Brain Res. 1977;120:571–576. doi: 10.1016/0006-8993(77)90412-7. [DOI] [PubMed] [Google Scholar]
- 24.Markey K A, Towle A C, Sze P Y. Endocrinology. 1982;111:1519–1523. doi: 10.1210/endo-111-5-1519. [DOI] [PubMed] [Google Scholar]
- 25.Dunn A J, Gildersleeve B, Gray H E. J Neurochem. 1978;31:977–982. doi: 10.1111/j.1471-4159.1978.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 26.Ortiz J, Decaprio J L, Kosten T A, Nestler E J. Neuroscience. 1995;67:383–397. doi: 10.1016/0306-4522(95)00018-e. [DOI] [PubMed] [Google Scholar]
- 27.Ho-Van Hap A, Babineau L M, Berlinguet L. Can J Biochem. 1967;45:355–361. doi: 10.1139/o67-042. [DOI] [PubMed] [Google Scholar]
- 28.Caesar P M, Collins G G S, Sandler M. Biochem Pharmacol. 1970;19:921–926. doi: 10.1016/0006-2952(70)90255-8. [DOI] [PubMed] [Google Scholar]
- 29.Veals J W, Korduba C A, Symchowicz S. Eur J Pharmacol. 1977;41:291–299. doi: 10.1016/0014-2999(77)90322-3. [DOI] [PubMed] [Google Scholar]
- 30.Rothschild A J, Langlais P J, Schatzeberg A F, Miller M M, Saloman M S, Lerbinger J E, Cole J O, Bird E D. Life Sci. 1985;36:2491–2505. doi: 10.1016/0024-3205(85)90145-6. [DOI] [PubMed] [Google Scholar]
- 31.Iversen L L, Salt P J. Br J Pharmacol. 1970;40:528–530. doi: 10.1111/j.1476-5381.1970.tb10637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilad G M, Rabey J M, Gilad V H. Life Sci. 1987;40:2401–2408. doi: 10.1016/0024-3205(87)90754-5. [DOI] [PubMed] [Google Scholar]
- 33.Joëls M, De Kloet E R. Prog Neurobiol. 1994;43:1–36. doi: 10.1016/0301-0082(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 34.Kalivas P W. Brain Res Rev. 1993;18:75–113. doi: 10.1016/0165-0173(93)90008-n. [DOI] [PubMed] [Google Scholar]
- 35.Tanganelli S, Fuxe K, Von Euler G, Eneroth P, Agnati L F, Ungerstedt U. J Neural Transm. 1990;81:183–194. doi: 10.1007/BF01245041. [DOI] [PubMed] [Google Scholar]
- 36.Yasunari K, Kohno M, Balmforth A, Murakawa K, Yokokawa K, Kurihara N, Takeda T. Hypertension. 1989;13:575–581. doi: 10.1161/01.hyp.13.6.575. [DOI] [PubMed] [Google Scholar]
- 37.Biron D, Dauphin C, Di Paolo T. Neuroendocrinology. 1992;55:468–476. doi: 10.1159/000126158. [DOI] [PubMed] [Google Scholar]
- 38.Chiriboga C A, Pranzatelli M R, De Vivo D C. Brain Dev. 1989;11:197–200. doi: 10.1016/s0387-7604(89)80099-3. [DOI] [PubMed] [Google Scholar]
- 39.Mobley P L, Sulser F. Nature (London) 1980;286:608–609. doi: 10.1038/286608a0. [DOI] [PubMed] [Google Scholar]
- 40.Saito N, Guitart X, Hayward M, Tallman J F, Duman R S, Nestler E J. Proc Natl Acad Sci USA. 1989;86:3906–3910. doi: 10.1073/pnas.86.10.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz J C, Giros B, Martres M P, Sokoloff P. Semin Neurosci. 1992;4:99–108. [Google Scholar]
- 42.Youngren K D, Daly D A, Moghaddam B. J Pharmacol Exp Ther. 1993;264:289–293. [PubMed] [Google Scholar]
- 43.Joëls M, de Kloet E R. Trends Neurosci. 1992;15:25–30. doi: 10.1016/0166-2236(92)90345-9. [DOI] [PubMed] [Google Scholar]
- 44.Blaha C D, Lane R F. Neurosci Lett. 1987;78:199–204. doi: 10.1016/0304-3940(87)90633-1. [DOI] [PubMed] [Google Scholar]
- 45.Chen J P, Paredes W, Gardner E L. Neurosci Lett. 1991;122:127–131. doi: 10.1016/0304-3940(91)90209-c. [DOI] [PubMed] [Google Scholar]
- 46.Hernandez L, Hoebel B G. Brain Res Bull. 1989;22:763–769. doi: 10.1016/0361-9230(89)90097-x. [DOI] [PubMed] [Google Scholar]
- 47.Ichikawa J, Meltzer H Y. Brain Res. 1990;507:138–142. doi: 10.1016/0006-8993(90)90532-g. [DOI] [PubMed] [Google Scholar]
- 48.Moghaddam B, Bunney B S. Synapse. 1993;14:195–200. doi: 10.1002/syn.890140302. [DOI] [PubMed] [Google Scholar]
- 49.Karim A. Drug Metab Rev. 1978;8:151–188. doi: 10.3109/03602537808993782. [DOI] [PubMed] [Google Scholar]
- 50.Deraedt R, Bonnat C, Busigny M, Chatelet P, Cousty C, Mouren M, Philibert D, Pottier J, Salmon J. In: The Antiprogestin Steroid RU 486 and Human Fertility Control. Beaulieu E E, Segal S J, editors. New York: Plenum; 1985. pp. 103–122. [Google Scholar]
- 51.Jain M R, Patil P P, Subhedar N. Neuroreport. 1993;5:69–71. doi: 10.1097/00001756-199310000-00017. [DOI] [PubMed] [Google Scholar]
- 52.Schöneshöfer M, Schefzig B, Arabin S. J Endocrinol Invest. 1980;3:229–236. doi: 10.1007/BF03348268. [DOI] [PubMed] [Google Scholar]