

Abstract
Uncoupling protein 2 (UCP2) is upregulated in the brain after sublethal ischemia, and overexpression of UCP2 is neuroprotective in several models of neurodegenerative disease. We investigated if increased levels of UCP2 diminished neuronal damage after global brain ischemia by subjecting mice overexpressing UCP2 (UCP2/3tg) and wild-type littermates (wt) to a 12-min global ischemia. The histopathological outcome in the cortex, hippocampus, striatum, and thalamus was evaluated at 4 days of recovery, allowing maturation of the selective neuronal death. Global ischemia led to extensive cell death in the striatum, thalamus, and in the CA1 and CA2, and less-pronounced cell death in the CA3 and dentate gyrus (DG) hippocampal subfields. Histologic damage was significantly lower in the ventral posterolateral VPL and medial VPM thalamic nuclei in UCP2/3tg animals compared with wt. These thalamic regions showed a larger increase in UCP2 expression in UCP2/3tg compared with wt animals relative to the nonprotected DG. In the other regions studied, the histologic damage was lower or equal in UCP2/3tg animals compared with wt. Consequently, neuroprotection in the thalamus correlated with a high expression of UCP2, which is neuroprotective in a number of models of neurodegenerative diseases.
Keywords: cerebral ischemia, mitochondria, neurodegeneration, neuroprotection, uncoupling protein 2
Introduction
Accumulating evidence has shown that uncoupling protein 2 (UCP2) is neuroprotective (Bechmann et al, 2002; Clavel et al, 2003; Diano et al, 2003; Mattiasson et al, 2003; Sullivan et al, 2003; Vogler et al, 2005), in models of focal cerebral ischemia (Mattiasson et al, 2003), traumatic brain injury (Mattiasson et al, 2003), Parkinson's disease (Andrews et al), seizures (Diano et al, 2003; Sullivan et al, 2003), and encephalomyelitis (Vogler et al, 2006). UCP2 is part of the endogenous neuroprotective response after ischemic preconditioning (IPC) in rat (Mattiasson et al, 2003). Ischemic preconditioning is induced through a short, sublethal global cerebral ischemia that renders the brain resistant to a subsequent, longer, and normally deleterious insult (Kitagawa et al, 1990). The inducible property of UCP2 makes it suitable as a potential therapeutic target for neuroprotection (Mattiasson and Sullivan, 2006). Because of the lack of reproducible experimental models of global cerebral ischemia in mouse, the neuroprotective properties of UCP2 overexpression after global ischemia has not been investigated previously.
Uncoupling proteins (UCPs; thermogenins) are encoded by nuclear DNA and are located in the inner membrane of the mitochondria. Uncoupling protein 1 is located in brown adipose tissue, where it is clearly involved in mitochondrial uncoupling and thermogenesis. The role of the other UCPs (UCP2–4, UCP5/BMCP1 (brain mitochondrial carrier protein 1)) is less clear, but one of their functions is to translocate protons from the inter-membrane space to the matrix of the mitochondria (Mattiasson and Sullivan, 2006), although the extent of mitochondrial depolarization mediated by these proteins is presently unknown. The UCPs, UCP2, UCP4, and BMCP1, are all expressed in the central nervous system. UCP4 and BMCP1 have around 30% similarity with UCP1, whereas UCP2 shares about 56% amino-acid identity with UCP1, and has a high homology between rat, mouse, and humans (Hidaka et al, 1998). Under normal circumstances, UCP2 is expressed predominantly in neurons in several brain regions in both rodents and primates (Horvath et al, 1999; Arsenijevic et al, 2007), although microglia, residing monocytes of the brain (Bechmann et al, 2002; Clavel et al, 2003), invading monocytes and neutrophils (Arsenijevic et al, 2007), cells of the choroids plexus (Richard et al, 1999) as well as endothelial cells (Fink et al, 2005) also express this mitochondrial uncoupling protein. In mouse, it is expressed in several parts of the fore-, mid- and hindbrain, including the hypothalamus (Horvath et al, 1999; Richard et al, 1998, 2001), thalamus (Richard et al, 1998; Horvath et al, 1999), and cerebellum (Richard et al, 1998). UCP2 is expressed at low levels in the mouse hippocampus (Richard et al, 1998), whereas hippocampal expression is higher in rat (Richard et al, 2001).
Excitotoxic cell death is believed to be one of the central pathogenic mechanisms behind human neurodegenerative disorders, including cerebral ischemia. The cell death mechanisms are complex and not fully understood, but include glutamate toxicity, which leads to deranged ion homeostasis with K+ efflux and Ca2+ influx. The influx of Ca2+ is a key event in acute brain injuries, which affects signaling cascades within the cell, such as second messenger systems and protein kinases. It also has an effect on mitochondrial function, integrity and production of reactive oxygen species (ROS), activation of caspase cascades, and changes in gene expression (Wieloch, 2002). Increased levels of intracellular calcium as well as oxidative stress may induce mitochondrial permeability transition (mPT), which leads to mitochondrial swelling and release of apoptogenic factors that activate cell death cascades (mitochondria-mediated cell death). Inhibition of mPT by Cyclosporin A and some of its analogues is neuroprotective after brain trauma, as well as global and focal ischemia (Wieloch et al, 2007).
The molecular mechanisms behind the neuroprotective effect of UCP2 are not fully known. It has been suggested that UCP2 leads to a slight depolarization of the inner mitochondrial membrane, with a reduction in the rate of calcium uptake and production of ROS. This will subsequently reduce the probability of mPT and activation of cell death cascades (Richard et al, 2001; Mattiasson et al, 2003). This hypothesis is supported by the fact that a slight mitochondrial depolarization with 2,4-dinitrophenol is neuroprotective both in vitro (Mattiasson et al, 2003) and in vivo (Korde et al, 2005). Besides the effect of UCP2 on ROS production, evidence has also accumulated to implicate UCP2 in mitochondrial proliferation within neurons (Diano et al, 2003; Andrews et al, 2005b), a mechanism that may further improve cellular resistance to neurodegeneration (Andrews et al, 2005a).
In this study, we investigated the neuroprotective effect of UCP2 after global ischemia in mice overexpressing UCP2 (Fuller et al, 2000). Using a model of global cerebral ischemia in mouse (Olsson et al, 2003), UCP2/3tg animals and wild-type (wt) litter-mate controls were subjected to 12 mins of global ischemia. The histopathological outcome in the cortex, hippocampus, striatum, and thalamus was evaluated at 4 days after injury, a time point where the ischemic lesion matured in this model.
Materials and methods
Animals
All experiments were approved by the Malmoe/Lund Ethical Committee on Animal Experiments. The animals were housed under diurnal light conditions and provided free access to food and water before surgery. UCP2/3-overexpressing mice (UCP2/3tg) were developed using an 80 kb Bacterial Artificial Chromosome containing the human UCP2/3 genes, including native promoters and cis-acting elements (Fuller et al, 2000). Consistent with the endogenous tissue distribution, these animals overexpress UCP2, but not UCP3, in their brains (Horvath et al, 2003). Seven UCP2/3tg and 8 wt animals were included in the study.
The Global Ischemia Model
The global ischemia procedure has previously been described in detail (Olsson et al, 2003) and applied in several studies (Olsson et al, 2004a, b, c). Briefly, animals were anaesthetized with halothane (O2/N2O, 30:70), intubated, administered muscle relaxant (norcuron) to avoid spontaneous breathing movements during ischemia, and connected to a small animal respirator. The pCO2 in respiratory end-tidal volume was measured with a capnometer to enable adjustment of the respirator to ensure physiologic blood gases. The common carotid arteries were exposed via a small ventral neck incision and were encircled loosely with a silk thread so as to later enable occlusion with a nontraumatic aneurysm clip. To estimate the density of ischemia in each hemisphere, the regional cerebral blood flow (rCBF) was recorded in both hemispheres with a two-channeled laser-Doppler flowmeter. Regional CBF was measured before, during, and for 5 mins after the ischemia with flexible optical filaments attached to the skull bone, 1 mm caudal to bregma and 4 mm lateral to the midline. Only hemispheres in which rCBF decreased below 10% of baseline within 1 min after occlusion were considered as ischemic and were included in the study. Individual hemispheres were not dependent on the contralateral blood flow (Olsson et al, 2003) and could be analyzed independently. Rectal temperature was kept at 37.2°C±0.2°C before, during, and shortly after ischemia by a homeothermic blanket. The brain was kept normothermic (37.1°C±0.1°C) during the whole period of ischemia with the help of a continuous flow of humidified warm air over the head of the mouse. The duration of ischemia by bilateral common carotid artery occlusion was 12 mins. Anesthesia was discontinued 2 mins before the end of ischemia. Laser-Doppler flow confirmed restoration of rCBF after clip removal. Five minutes after ischemia the laser-Doppler probes were removed, and the skin sutured. When the animals were able to breathe normally, after 10 to 30 mins, they were disconnected from the ventilator and placed in an incubator at 33.5°C to maintain normothermia for 24 h. The mice were extubated about 90 mins after the end of ischemia. Rectal temperature was monitored within the first 2 h after ischemia, and then at least two more times during the first day after surgery.
Measurement of End-Tidal CO2, Regional Cerebral Blood Flow, and Postischemic Temperature
The end-tidal pCO2 was monitored continuously with a capnometer from 15 mins before ischemia until 5 mins after the end of ischemia. The respirator was adjusted to allow a pCO2 of 35 to 40 mm Hg in arterial blood, corresponding to a value of 1.5% CO2 measured by the capnometer in a separate group of animals. Every animal was stabilized at 1.5% CO2 before the start of ischemia. The rCBF was monitored continuously from at least 5 mins before ischemia until 5 mins after the end of ischemia by laser Doppler. Changes in rCBF during ischemia (1 and 2 mins after occlusion) and 1 and 5 mins after the start of reperfusion were compared between UCP2/3tg and wt mice. Decreased rates of rCBF during ischemia were calculated as follows: (ischemia rCBF/preischemia rCBF) × 100. Postischemic temperature was rectally measured 0 to 2, 2 to 4, and 4 to 8 h after ischemia, when the animals were housed in an incubator.
Perfusion Fixation and Histologic Preparation
After 4 days of recovery, the animals were anesthetized in 4% halothane and transcardially perfused with 4% buffered formaldehyde at a flow of 3 to 5 mL/min after a short rinse with saline. The brains were removed and stored for at least 24 h in 4% formaldehyde at 4°C before dehydration and embedding in paraffin. Ten-micrometer coronal sections were cut and stained with celestine blue/acid fuchsin.
cRNA Probe Synthesis and In Situ Hybridization Histochemistry
To analyze the expression of mouse UCP2 (mUCP2) mRNA, a riboprobe was used (Richard et al, 1999). The UCP2 cRNA probe was generated from the 945-bp EcoRI fragment of an mUCP2 cDNA subcloned into a pSELECT vector (Promega Inc., Lyon, France), which was linearized with SacI and KpnI (Pharmacia Biotech Canada Inc., Baie d'Urfé, QC, Canada) for sense and antisense probes, respectively. The radioactive riboprobes were synthesized by incubating 250 ng linearized plasmid in 10 mmol/L NaCl, 10 mmol/L dithiothreitol (DTT), 6 mmol/L MgCl, 40 mmol/L Tris (pH 7.9), 0.2 mmol/L ATP/GTP/CTP [35S]UTP, 40 U RNasin (Promega, Madison, WI, USA), and 20 U SP6 and T7 RNA polymerase for sense and antisense probes, respectively, for 60 mins at 37°C. The DNA templates were treated with 100 mL of DNase solution (1 mL DNase, 5 mL of 5 mg/mL tRNA, and 94 mL of 10 mmol/L Tris/10 mmol/L MgCl2). Preparation of the riboprobes was completed through phenol-chloroform extraction and ammonium acetate precipitation. This probe recognizes the mUCP2 mRNA but not that of rat or human.
A second UCP2 riboprobe, not cross-reacting with mUCP2 mRNA, was used for the detection of human UCP2 (hUCP2) transgene in the mouse brain. Amplification of the RNA sequence for hUCP2 was performed as previously described (Diano et al, 2003) (5′-CATCTCCTGGGACGTAGC-3′ and 5′-AGAGAAGGGAAGGAGGGAAG-3′). The resulting complementary DNA (1.1 kb), purified from agarose gel using the QIAquick gel extraction kit (Qiagen Inc., Valenica, CA, USA), was digested with EcoRI and inserted in pBluescript vector and then subcloned. Linearized DNAs by HindIII and XbaI were transcribed using T7 polymerase (antisense cRNA riboprobe) and T3 polymerase (sense cRNA probe; restriction enzyme XbaI; Riboprobe Combination System T3/T7; Promega Corporation, Madison, WI, USA), respectively, and labeled with [35S]UTP (10 mCi/mL; Amersham Pharmacia Biotech, Piscataway, NJ, USA). The radiolabeled cRNA probe was then purified by passing the transcription reaction solution over a G50 column (Amersham Pharmacia Biotech) and fractions were collected and counted using a scintillation counter.
In situ hybridization (mUCP2/hUCP2) was performed on the tissue sections after first bringing them to room temperature and postfixing them in 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4; 15 mins). The sections were then rinsed in phosphate buffer and digested with Proteinase K (10 μg/mL) in Tris-EDTA buffer (100 mmol/L Tris, 50 mmol/L EDTA; pH 8.0; 30 mins, 37°C). Next, they were acetylated, dehydrated with ascending concentrations of ethanol, and dried under vacuum for 2 h. They were then hybridized for 18 h at 65°C with 100 μL of [35S]-labeled antisense riboprobe, diluted to 1 × 107 c.p.m./mL of hybridization buffer (50 mmol/L DTT, 250 μg/mL tRNA, 50% formamide, 0.3 mol/L sodium chloride, 1 × Denhardt's solution, 20 mmol/L Tris (pH 8.0), 1 mmol/L EDTA, and 10% dextran sulfate). For the hybridization procedure, glass coverslips were affixed to the slides using DPX mounting medium (BDH Laboratory Supplies, Poole, UK). They were subsequently removed after two 30-min soakings in 4 × saline-sodium citrate buffer (SSC; 0.6 mol/L NaCl, 60 mmol/L sodium citrate buffer; pH 7.0) containing 20 mmol/L DTT. The sections were then incubated in Tris-EDTA buffer (10 mmol/L Tris, 1 mmol/L EDTA, and 0.5 mol/L sodium chloride; pH 8.0) containing RNase A (10 μg/mL) for 30 mins at 37°C, followed by two 30-min washes at room temperature with 2 × SSC containing 1 mmol/L DTT. After a final 30-min wash at 70°C with 0.1 × SSC containing 1 mmol/L DTT, they were dehydrated through ascending concentrations of ethanol, containing 0.3 mol/L ammonium acetate, and air-dried for 30 mins. To visualize the hybridization pattern, the sections were apposed to Hyperfilm β-max (Amersham Pharmacia Biotech) for 6 days. As a control experiment, sections were incubated as described above with hybridization solution containing the sense-strand probe synthesized with T7 RNA polymerase (mUCP2) or T3 polymerase (hUCP2) to transcribe the coding strand of the DNA insert.
Quantification of UCP2 mRNA Expression
The density of the hybridization product for mUCP2 and hUCP2 mRNA was assessed in wt and hUCP2/3 transgenic mice. To digitally analyze, quantitate, and compare the amount of UCP2 mRNA, an Image-1/AT image processor (Universal Imaging Corp., West Chester, PA, USA) using an Olympus IMT-2 inverted microscope with dark-field optics (Olympus Corp., Lake Success, NY, USA) and a Hamamatsu CCD camera (Hamamatsu Photonics, Hamamatsu, Japan) were employed. Six sections per animal were selected from the same area to assess the intensity of the hybridization product (Arbitrary Optical Density). The total surface covered by the hybridization product was assessed within a test region measuring 2 × 105 mm2. The threshold for measurement was assessed for each slide by determining the background labeling over the third ventricle.
Assessment of Ischemic Damage
Light microscopy was used for the evaluation of the ischemic neuronal injury. The damage to the hippo-campus, cortex, and thalamus was evaluated at coronal sections 1.7 mm caudal to bregma. In the sections, hippocampal neurons in the entire CA1 (∼500 cells), CA2 (∼50 cells), CA3 (∼280 cells), and dentate gyrus (DG; ∼400 cells) were counted, as were neurons in the barrel field of the neocortex (∼120 cells). Thalamic damage was evaluated in a 500 × 500 μm2 section (∼160 cells) placed 1.7 mm lateral to bregma and 3.2 mm ventral of the dura mater. This area corresponds to the ventral posterolateral (VPL) and medial (VPM) thalamic nuclei. In the hippocampus, cortex, and thalamus, the total number of neurons showing morphologic features of ischemic cell death (shrunken cell bodies, eosinophilic cytoplasm, and triangulated nuclei (see Figure 2) as well as normal-appearing neurons was counted. The damage was calculated as percentage of the total neuronal cell numbers in the respective structures. Striatal damage was assessed by visual assessment, confirmed by area measurement (by ImageJ) of the percentage of lesioned area 0.9 mm rostral to bregma using a 0 to 5 graded scale (0 = no damage, 1 = 0% to 20%, 2 = 20% to 40%, 3 = 40% to 60%, 4 = 60% to 80%, and 5 = 80% to 100% of the total striatal area damaged).
Figure 2.

Ischemic cell damage in transgenic animals overexpressing UCP2 (UCP2/3tg; n = 7; 12 hemispheres) and their wild-type littermates (wt; n = 8; 15 hemispheres) exposed to 12 mins of global ischemia. Neuronal damage, expressed as percentage of viable cells, in the CA1, CA2, CA3, and dentate gyrus (DG) in hippocampus (A) and in cortex and thalamus (B) at the level of 1.7 mm caudal to bregma. (C) Neuronal injury in the striatum, expressed as area damage score, at the level of 0.9 mm rostral of bregma. See the Materials and Methods section for detailed description of the evaluation method. n = hemispheres. Mann–Whitney's U-test, *P < 0.05.
Data Analysis
Individual hemispheres showed no dependency of the contralateral blood flow (Olsson et al, 2003). Therefore, all hemispheres were counted separately and in all graphs each circle represents one hemisphere (n = 15 for 8 wt mice and n = 12 for 7 UCP2/3tg mice). Mann–Whitney's U-test was used to compare neuronal damage between the two groups. The unpaired Student's t-test was employed to compare the values for body temperature, pCO2, and rCBF between the groups. UCP2 mRNA expression was compared using a one-way ANOVA with a Student-Newman–Keuls post hoc test. In all calculations, a P-value of < 0.05 was considered statistically significant.
Results
Physiologic Parameters
There was no difference in weight between wt and UCP2/3tg animals before surgery or 4 days after ischemia when mice were killed (Table 1). Regional CBF measured by laser Doppler in the outer layer of the barrel cortex in individual hemispheres before, during, and after ischemia did not show any difference between the experimental groups at any of the time points (Table 1). The body temperature during the first 8 h of reperfusion was similar between the groups (Table 1).
Table 1.
Physiologic parameters before and after global cerebral ischemia in UCP2/3tg and wt mice
| wt | UCP2/3tg | ||
|---|---|---|---|
| Cortical cerebral blood flow | |||
| Ischemia 1 min | 4.6±2.8 | 4.1±3.4 | |
| Ischemia 2 mins | 3.3±2.4 | 3.8±2.9 | |
| Recirculation 1 min | 47.3±18.2 | 43.8±22.4 | |
| Recirculation 5 mins | 97.8±33.5 | 86.7±17.6 | |
| End-tidal pCO2 | |||
| Preischemia | 1.5 | 1.5 | |
| Ischemia 6 mins | 1.5±0.1 | 1.5±0.1 | |
| Recirculation 5 mins | 1.7±0.1 | 1.7±0.3 | |
| Body temperature (°C) at reperfusion | |||
| 0 to 2 h | 37.1±0.7 | 36.9±0.7 | |
| 2 to 4 h | 36.3±0.3 | 36.5±0.7 | |
| 4 to 8 h | 36.0±0.6 | 36.3±0.6 | |
| Weight (g) | |||
| Before ischemia | 25.3±1.8 | 26.1±1.4 | |
| After 4 days of reperfusion | 22.9±2.9 | 21.0±3.0 | |
Abbreviations: UCP2, uncoupling protein 2; wt, wild-type.
No significant difference was detected between the experimental groups. Wild-type (wt, n = 8) and UCP2-overexpressing mice (UCP2/3tg, n = 7) were subjected to global cerebral ischemia. Cortical cerebral blood flow was measured by laser Doppler in both hemispheres during ischemia and early reperfusion (expressed in percentage of baseline; mean±s.d.). End-tidal pCO2 was monitored continuously with a capnometer from 15 mins before ischemia until 5 mins after the end of ischemia (mean% CO2±s.d.). Body temperature (°C; mean±s.d.) was measured in the reperfusion phase, and body weight (gram; mean±s.d.) was registered before ischemia and at 4 days of recovery. No differences between the groups in any of these measures were detected (Student's t-test).
Ameliorated Ischemic Cell Death in the Thalamic Nuclei of UCP2/3tg Mice
Representative photomicrographs of the hippocampal subregion CA1, striatum, and thalamus in UCP2/3tg mice and in wt littermates are displayed in Figure 1. The extent of ischemic cell death in the specific brain regions is shown in Figure 2. In the thalamic VPL and VPM, cell death was significantly decreased in UCP2/3tg mice compared with wt controls (19.4%±15.5% versus 47.1%±32.1%; P = 0.0225, Mann–Whitney). In the other regions studied except the CA2, there was a trend toward neuroprotection in the UCP2/3tg animals, but the difference was not statistically significant. The fraction of degenerated cells in wt and UCP2/3tg animals in the different subregions were, respectively, as follows: CA1, 26.5%±22.6% versus 20.8%±25.8% (P = 0.39); CA2, 54.3%±37.1% versus 54.6%±35.1% (P = 0.90); CA3, 6.5%±7.4% versus 3.8%±2.5% (P = 0.69); DG, 4.7%±6.6% versus 3.8%±5.7% (P = 0.49); the striatum, 4.4±0.5 versus 4.0±1.2 (P = 0.81); and cortex, 7.7%±10.5% versus 6.8%±9.4% (P = 0.92).
Figure 1.
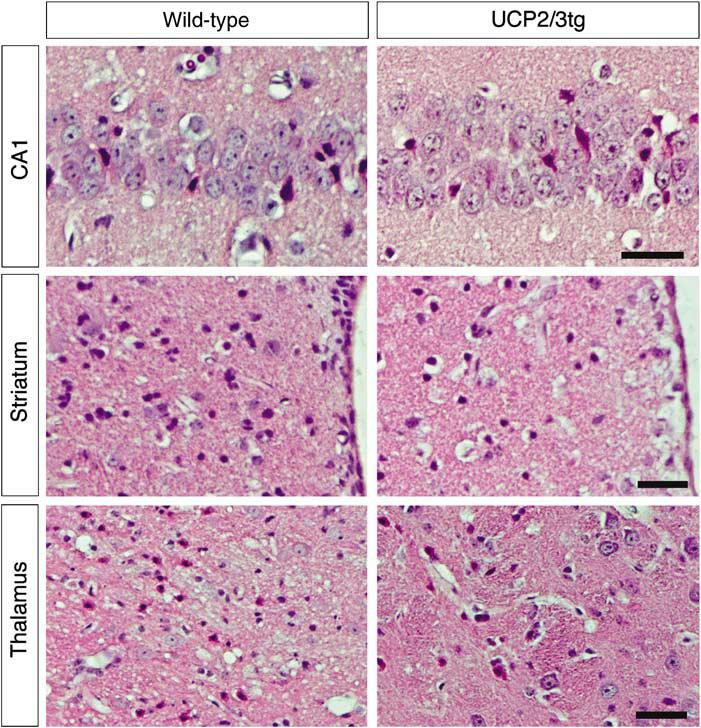
Representative pictures showing the hippocampal subregion CA1, the medial part of striatum next to the ventricle, and the ventral posterolateral and medial thalamic nuclei in wild-type and UCP2-overexpressing animals (UCP2/3tg). Normal-appearing neurons and dead neurons showing typical morphologic features of ischemic cell death with shrunken eosinophilic cytoplasms and pyknotic nuclei are seen. (Scale bar = 35 μm).
Specifically High UCP2 mRNA Expression Levels in the Thalamic Nuclei of UCP2/3tg Mice
Similar to previous observations (Andrews et al, 2005b), no difference was observed regarding mUCP2 mRNA expression between wt and transgenic animals. Expression of mUCP2 was detected in the cortex, the CA1–3 and DG hippocampal fields, and the thalamus both in wt and UCP2/3tg mice (in situ hybridization; data not shown). In the UCP2/3tg, but not the wt mice, hUCP2 mRNA was expressed in the same regions. In situ hybridizations of the CA1–3 and DG hippocampal fields and the thalamus are shown in Figures 3A to 3C. UCP2 expression level (mUCP2 and hUCP2) was evaluated in the DG and VPL thalamic nucleus in in situ hybridizations of brain sections at −1.6 mm to bregma (Figures 3D and 3E). The total UCP2 mRNA expression (mUCP2 + hUCP2) in the VPL of UCP2/3tg animals was approximately 160% higher compared with the endogenous mUCP2 mRNA expression in wt animals. In the DG, the total UCP2 mRNA of UCP2/3tg was approximately 50% higher than that of mUCP2 mRNA in wt mice (Figure 3F). The wt DG had a higher expression level of mUCP2 compared with the VPL, and also displayed less cell death compared with the VPL thalamic nucleus (Figure 2). Interestingly, the high relative increase in UCP2 in UCP2/3tg versus wt correlated with a significant neuroprotection in the VPL thalamic nucleus (Figures 1 and 2).
Figure 3.
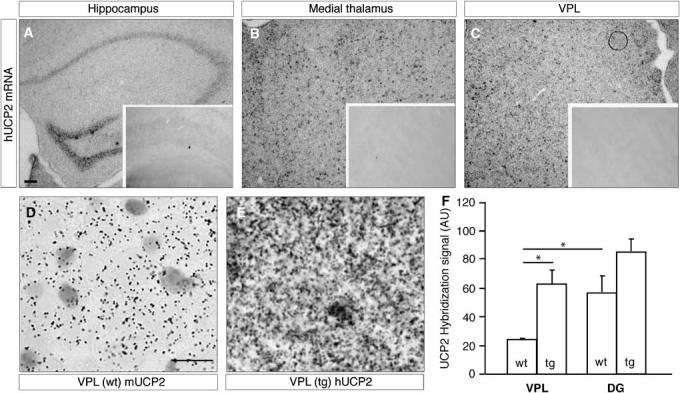
in situ hybridization of human UCP2 (hUCP2) mRNA in UCP2/3tg animals, which labels cells in the hippocampus (A), medial (B), and ventral posterolateral nuclei (VPL) of the thalamus (C). Insets show the corresponding regions in wt mice after hybridization with the hUCP2 probe, showing that no expression was detected. in situ hybridization for mouse UCP2 mRNA in wild-type mouse VPL (D) shows decreased expression level (silver grain intensity) than human UCP2 mRNA (E) in transgenic mice (UCP2/3tg). The expression of mUCP2 in UCP2/3tg animals was similar to that in wt animals (not shown). The bar diagram shows the combined levels of mouse and human UCP2 (mUCP2 and hUCP2, respectively) in the VPL and dentate gyrus (DG) in wt and UCP2/3tg animals (F). The relative increase in total UCP2 in UCP2/3tg versus wt animals was significantly higher in the VPL (2.6-fold), but not in the DG (1.5-fold). Scale bar on panel A represents 100 μm for panels A to C. Scale bar on left panel represents 10 mm for panels D and E. Values are presented as means of UCP2 hybridization signal (AU; arbitrary units); error bars show s.e.m., *P < 0.05.
Discussion
Ischemic tolerance or IPC is time- and protein-synthesis-dependent (Barone et al, 1998), represents the mobilization of endogenous neuroprotective pathways, and includes changes in the expression of a large number of genes (Kawahara et al, 2004). After IPC, UCP2 (but not UCP1, 3, 4, or BMCP1/UCP5) is upregulated in vitro (Mattiasson et al, 2003), and in vivo in the rat brain (Horvath et al, 2003; Mattiasson et al, 2003). UCP2 is also upregulated in the human brain after ischemia (Nakase et al, 2007). Overexpression of UCP2 in UCP2/3tg mice (Fuller et al, 2000) at levels similar to those observed after IPC in the rat led to a corresponding change in UCP2 protein levels in the brain, and neuroprotection after traumatic brain injury and focal ischemia in the mouse (Mattiasson et al, 2003). To evaluate if an increased expression of UCP2 is also neuroprotective after global ischemia in mouse, we subjected UCP2/3tg animals to transient global ischemia (Olsson et al, 2003). The results of this study show that at 4 days after ischemia, over-expression of UCP2 led to a significant neuroprotection in the VPL and VPM of the thalamus (Figures 1 and 2) but not in the other regions studied. Neuroprotection in the VPL nucleus of the thalamus correlated with a robust increase in UCP2 expression in UCP2/3tg compared with wt animals.
The molecular mechanisms behind UCP2-mediated neuroprotection are not fully understood. Previous experimental data indicate that the neuro-protective effect of UCP2 after an excitotoxic insult may be related to multiple effects of UCP2 on mitochondrial, and consequently cellular, metabolism. For example, UCP2 may induce a slight depolarization of the inner mitochondrial membrane, with a decreased electrophoretic uptake of calcium and reduced induction of mitochondrial mPT (Bechmann et al, 2002; Mattiasson et al, 2003; Sullivan et al, 2003). However, the magnitude of mitochondrial depolarization mediated by UCP2 is still unclear, and evidence for its uncoupling activity is obtained through measurements of mitochondrial respiration.
In the context of excitotoxic/ischemic brain injury, a mitochondrial depolarization could lead to a decrease in the electrophoretic movement of calcium ions into the mitochondria, preventing mitochondrial calcium overload and cytotoxicity (Castilho et al, 1998). Further, a reduction in the mitochondrial membrane potential, by increasing proton transport, may decrease the generation of ROS, presumably by decreasing the time of interaction between electrons and molecular oxygen, decreasing the formation of ROS (Skulachev, 1996). Even a slight depolarization (∼10 mV) of mitochondrial potential is sufficient to significantly decrease the production of ROS (Liu, 1997), suggesting that even small mitochondrial depolarizations, mediated by UCP2, may have important physiologic effects. Under intact conditions, UCP2/3tg mice were shown to have decreased levels of lipid peroxidation (Diano et al, 2003) and decreased amounts of free radicals (Andrews et al, 2005b) in neurons. This, together with a positive correlation between UCP2 levels and neuronal mitochondria number (Diano et al, 2003; Andrews et al, 2005b; Coppola et al, 2007) and brain ATP and ADP levels and ratios (Diano et al, 2003), suggests that UCP2/tg animals may have a higher ability to maintain mitochondrial function. A decrease in intramitochondrial calcium and ROS release as a consequence of UCP2 activation would decrease the probability of mPT, collapse of respiration, the release of apoptogenic factors such as cytochrome c and apoptosis-inducing factor, and the subsequent activation of caspase-3 (Wieloch, 2001). This hypothesis is supported by experimental data showing preserved membrane potential in cortical neurons overexpressing UCP2 as well as prevention of caspase-3 activation after oxygen–glucose deprivation (Mattiasson et al, 2003), possibly because of the inhibition of mPT. An inverse correlation between UCP2 expression and caspase-3 activation after acute brain injury in rat has been suggested (Bechmann et al, 2002) previously. The neuroprotective effect of UCP2-mediated mitochondrial depolarization is also supported by the fact that slight mitochondrial depolarization with 2,4-dinitrophenol is neuroprotective both in vitro (Mattiasson et al, 2003) and in vivo (Korde et al, 2005). Alternatively, UCP-2 has been shown to increase mitochondrial calcium uptake and retention capacity in noncerebral cell cultures and liver mitochondria (Trenker et al, 2007). If present in the CNS as well, this mechanism may reduce the induction of mPT and promote cell survival.
The neuroprotective effects of UCP2 outlined above assume that there is some mitochondrial membrane potential left in the area-at-risk. This would be the case, for example, after focal ischemia or trauma, where the area surrounding the ischemic core or traumatic lesion (the penumbra) is still sufficiently perfused to maintain metabolism, although at a reduced rate. In the present model of global ischemia, blood flow in the brain is completely stopped for a period of 12 mins, leading to the consumption of all available oxygen and oxidizable substrates. ATP in the brain drops to < 10% of normal levels within 2 mins of cardiac arrest (Folbergrova et al, 1990), and ATP levels are further reduced as ATP synthase is reversed in an effort to maintain mitochondrial membrane potential. In a matter of minutes, all available ATP and ADP are consumed, and the mitochondria depolarizes, suggesting that neuroprotection through a slight depolarization, mediated by UCP2, is no longer feasible. Possibly, increased levels of UCP2 may delay mitochondrial mPT by preventing mitochondrial calcium overload. Thereby, the collapse of oxidative phosphorylation is delayed, postponing the final depolarization of mitochondria and cellular energy crisis. UCP2 is neuroprotective in a number of models and species (Mattiasson and Sullivan, 2006), and it is therefore possible that UCP2 has other neuroprotective effects besides those outlined above, probably involving several cell types in the brain besides neurons. UCP2 has been suggested to affect neuronal plasticity (Yamada et al, 2006), which could be neuroprotective after cerebral ischemia by decreasing detrimental excitotoxic synaptic signaling or by stimulating functional recovery. UCP2 has also been implicated in substrate transport across the mitochondrial membrane (Criscuolo et al, 2006), which may be important for cell energy homeostasis under conditions of limited substrate and oxygen availability, that is, ischemia. UCP2 is expressed in endothelial cells (Cui et al, 2006), and may influence adhesion and migration of inflammatory cells across the endothelium. Our results show that there were no significant differences in CBF between the genotypes before, during, or after ischemia, but it is possible that UCP2 influences microvascular function or architecture in ways that were not detected. Further, UCP2 may reduce detrimental inflammatory response after ischemia by limiting acute microglial response (Bai et al, 2005), or stimulating the expression of neuroprotective genes (Mattiasson et al, 2003).
In rat, IPC leads to an increased expression of UCP-2, and the brain is protected against a subsequent longer global ischemia. Induction of UCP-2 by rosiglitazone also protected the rat brain against global ischemia (Chen et al, 2006). In the present study, the lack of significant protection in the cortex, hippocampus, and striatum after global ischemia in mice overexpressing UCP2 could be explained by the fact that UCP2 is maximally induced in some of these regions (cortex and hippocampus) in the wt animals. Neuroprotection in the thalamus correlated with a higher relative increase in UCP2 expression in UCP2/3tg compared with wt animals. Alternatively, the thalamus may suffer a less ischemic insult, sufficient to maintain some mitochondrial electron transport, thereby promoting a neuro-protective UCP-2 function as discussed above. In this case the less ischemic insult could be comparable to the hypoperfused area of the peri-infarct zone during focal occlusion of the middle cerebral artery, where overexpression of UCP-2 has been shown to be effectively protective (Mattiasson et al, 2003). Another possible explanation along this line is that the insult in the regions that showed no protection was too severe, with a rapid onset of energy depletion and mitochondrial membrane depolarization.
Based on the data from the present study, we conclude that after global ischemia in mouse, over-expression of UCP2 leads to significant neuro-protection in the thalamus. In the other brain regions studied, there was a trend toward neuro-protection, but the effect was not significant. The molecular basis for this protection is presently unclear. UCP2 can be induced by PPARγ-agonists, which are currently used as antidiabetic drugs. Treatments with such compounds have been shown to increase the expression of UCP2 and be neuro-protective in an experimental rat model of global ischemia (Chen et al, 2006). The clinical usefulness of a similar approach, if any, remains to be investigated.
Acknowledgments
This work was supported by the Swedish Research Council, Tore Nilsson, Lars Hierta, The Wiberg, G&J Kock, Thuring, Bergvall and Swedish Brain Foundations, The Royal Physiographic Society, The Swedish Society for Medicine, PROMEMORIA (EU 6th FP), and NIH Grant nos. NS-41725, AG-22880.
References
- Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci. 2005a;6:829–40. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elseworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci. 2005b;25:184–91. doi: 10.1523/JNEUROSCI.4269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D, Clavel S, Sanchis D, Plamondon J, Huang Q, Ricquier D, Rouger L, Richard D. Induction of UCP2 expression in brain phagocytes and neurons following murine toxoplasmosis: an essential role of IFN-gamma and an association with negative energy balance. J Neuroimmunol. 2007;186:121–32. doi: 10.1016/j.jneuroim.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Bai Y, Onuma H, Bai X, Medvedev AV, Misukonis M, Weinberg JB, Cao W, Robidoux J, Floering LM, Daniel KW, Collins S. Persistent nuclear factor-kappa B activation in Ucp2−/− mice leads to enhanced nitric oxide and inflammatory cytokine production. J Biol Chem. 2005;280:19062–9. doi: 10.1074/jbc.M500566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, Feuerstein GZ. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke. 1998;29:1937–50. doi: 10.1161/01.str.29.9.1937. discussion 1950–1951. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Diano S, Warden CH, Bartfai T, Nitsch R, Horvath TL. Brain mitochondrial uncoupling protein 2 (UCP2): a protective stress signal in neuronal injury. Biochem Pharmacol. 2002;64:363–7. doi: 10.1016/s0006-2952(02)01166-8. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18:10277–86. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SD, Wu HY, Yang DI, Lee SY, Shaw FZ, Lin TK, Liou CW, Chuang YC. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem Biophys Res Commun. 2006;351:198–203. doi: 10.1016/j.bbrc.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Clavel S, Paradis E, Ricquier D, Richard D. Kainic acid upregulates uncoupling protein-2 mRNA expression in the mouse brain. Neuroreport. 2003;14:2015–7. doi: 10.1097/00001756-200311140-00002. [DOI] [PubMed] [Google Scholar]
- Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, Diano S. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33. doi: 10.1016/j.cmet.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo F, Mozo J, Hurtaud C, Nubel T, Bouillaud F. UCP2, UCP3, avUCP, what do they do when proton transport is not stimulated? Possible relevance to pyruvate and glutamine metabolism. Biochim Biophys Acta. 2006;1757:1284–91. doi: 10.1016/j.bbabio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Cui Y, Xu X, Bi H, Zhu Q, Wu J, Xia X, Qiushi R, Ho PC. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: the role of reactive oxygen species in diabetic retinopathy. Exp Eye Res. 2006;83:807–16. doi: 10.1016/j.exer.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Diano S, Matthews RT, Patrylo P, Yang L, Beal MF, Barnstable CJ, Horvath TL. Uncoupling protein 2 prevents neuronal death including that occurring during seizures: a mechanism for preconditioning. Endocrinology. 2003;144:5014–21. doi: 10.1210/en.2003-0667. [DOI] [PubMed] [Google Scholar]
- Fink BD, Reszka KJ, Herlein JA, Mathahs MM, Sivitz WI. Respiratory uncoupling by UCP1 and UCP2 and superoxide generation in endothelial cell mitochondria. Am J Physiol Endocrinol Metab. 2005;288:E71–9. doi: 10.1152/ajpendo.00332.2004. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Minamisawa H, Ekholm A, Siesjo BK. Phosphorylase alpha and labile metabolites during anoxia: correlation to membrane fluxes of K+ and Ca2+ J Neurochem. 1990;55:1690–6. doi: 10.1111/j.1471-4159.1990.tb04957.x. [DOI] [PubMed] [Google Scholar]
- Fuller PM, Warden CH, Barry SJ, Fuller CA. Effects of 2-G exposure on temperature regulation, circadian rhythms, and adiposity in UCP2/3 transgenic mice. J Appl Physiol. 2000;89:1491–8. doi: 10.1152/jappl.2000.89.4.1491. [DOI] [PubMed] [Google Scholar]
- Hidaka S, Kakuma T, Yoshimatsu H, Yasunaga S, Kurokawa M, Sakata T. Molecular cloning of rat uncoupling protein 2 cDNA and its expression in genetically obese Zucker fatty (fa/fa) rats. Biochim Biophys Acta. 1998;1389:178–86. doi: 10.1016/s0005-2760(97)00188-4. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Miyamoto S, Barry S, Gatti S, Alberati D, Livak F, Lombardi A, Moreno M, Goglia F, Mor G, Hamilton J, Kachinskas D, Horwitz B, Warden CH. Uncoupling proteins-2 and 3 influence obesity and inflammation in transgenic mice. Int J Obes Relat Metab Disord. 2003;27:433–42. doi: 10.1038/sj.ijo.0802257. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Warden CH, Hajos M, Lombardi A, Goglia F, Diano S. Brain uncoupling protein 2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci. 1999;19:10417–27. doi: 10.1523/JNEUROSCI.19-23-10417.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara N, Wang Y, Mukasa A, Furuya K, Shimizu T, Hamakubo T, Aburatani H, Kodama T, Kirino T. Genome-wide gene expression analysis for induced ischemic tolerance and delayed neuronal death following transient global ischemia in rats. J Cereb Blood Flow Metab. 2004;24:212–23. doi: 10.1097/01.WCB.0000106012.33322.A2. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, Kamada T. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–4. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Maragos WF. The mitochondrial uncoupler 2,4-dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem. 2005;94:1676–84. doi: 10.1111/j.1471-4159.2005.03328.x. [DOI] [PubMed] [Google Scholar]
- Liu SS. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci Rep. 1997;17:259–72. doi: 10.1023/a:1027328510931. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–8. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid Redox Signal. 2006;8:1–38. doi: 10.1089/ars.2006.8.1. [DOI] [PubMed] [Google Scholar]
- Nakase T, Yoshida Y, Nagata K. Amplified expression of uncoupling proteins in human brain ischemic lesions. Neuropathology. 2007;27:442–7. doi: 10.1111/j.1440-1789.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- Olsson T, Cronberg T, Rytter A, Asztely F, Fredholm BB, Smith ML, Wieloch T. Deletion of adenosine A1 receptor gene does not alter neuronal damage following ischaemia in vivo or in vitro. Eur J Neurosci. 2004a;20:1197–204. doi: 10.1111/j.1460-9568.2004.03564.x. [DOI] [PubMed] [Google Scholar]
- Olsson T, Hansson O, Nylandsted J, Jaattela M, Smith ML, Wieloch T. Lack of neuroprotection by heat shock protein 70 overexpression in a mouse model of global cerebral ischemia. Exp Brain Res. 2004b;154:442–9. doi: 10.1007/s00221-003-1683-2. [DOI] [PubMed] [Google Scholar]
- Olsson T, Nygren J, Hakansson K, Lundblad C, Grubb A, Smith ML, Wieloch T. Gene deletion of cystain C aggravates brain damage following focal ischemia but mitigates the neuronal injury after global ischemia in the mouse. Neuroscience. 2004c;128:65–71. doi: 10.1016/j.neuroscience.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Olsson T, Wieloch T, Smith ML. Brain damage in a mouse model of global cerebral ischemia. Effect of NMDA receptor blockade. Brain Res. 2003;982:260–9. doi: 10.1016/s0006-8993(03)03014-2. [DOI] [PubMed] [Google Scholar]
- Richard D, Clavel S, Huang Q, Sanchis D, Ricquier D. Uncoupling protein 2 in the brain: distribution and function. Biochem Soc Trans. 2001;29:812–7. doi: 10.1042/0300-5127:0290812. [DOI] [PubMed] [Google Scholar]
- Richard D, Huang Q, Sanchis D, Ricquier D. Brain distribution of UCP2 mRNA: in situ hybridization histochemistry studies. Int J Obes Relat Metab Disord. 1999;23(Suppl 6):S53–5. doi: 10.1038/sj.ijo.0800947. [DOI] [PubMed] [Google Scholar]
- Richard D, Rivest R, Huang Q, Bouillaud F, Sanchis D, Champigny O, Ricquier D. Distribution of the uncoupling protein 2 mRNA in the mouse brain. J Comp Neurol. 1998;397:549–60. [PubMed] [Google Scholar]
- Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–7. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol. 2007;9:445–52. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler S, Goedde R, Miterski B, Gold R, Kroner A, Koczan D, Zettl UK, Rieckmann P, Epplen JT, Ibrahim SM. Association of a common polymorphism in the promoter of UCP2 with susceptibility to multiple sclerosis. J Mol Med. 2005;83:806–11. doi: 10.1007/s00109-005-0661-5. [DOI] [PubMed] [Google Scholar]
- Vogler S, Pahnke J, Rousset S, Ricquier D, Moch H, Miroux B, Ibrahim SM. Uncoupling protein 2 has protective function during experimental autoimmune encephalomyelitis. Am J Pathol. 2006;168:1570–5. doi: 10.2353/ajpath.2006.051069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieloch T, Mattiasson G, Hansson M, Elmer E. Mitochondrial permeability transition in the CNS—composition, regulation, and pathophysiological relevance. In: Gibson G, editor. Handbook of neurochemistry. Springer-Verlag; Berlin, Heidelberg: 2007. pp. 667–702. [Google Scholar]
- Wieloch T. Mitochondrial involvement in acute neurodegeneration. IUBMB Life. 2001;52:247–54. doi: 10.1080/15216540152846064. [DOI] [PubMed] [Google Scholar]
- Wieloch T. Molecular mechanisms of ischemic brain damage. In: Edvinsson LKDN, editor. Cerebral blood flow and metabolism. Lipincott Williams & Wilkins; London: 2002. pp. 423–51. [Google Scholar]
- Yamada S, Isojima Y, Kanamori S, Waguri S, Uchiyama Y, Nagai K. Uncoupling protein 2 negatively regulates neurite extensions in PC12h cells. Neurosci Lett. 2006;410:110–4. doi: 10.1016/j.neulet.2006.09.083. [DOI] [PubMed] [Google Scholar]