

Abstract
The influence of the excitotoxic cascade on the developing brain was investigated using ibotenate, a glutamatergic agonist of both N-methyl-d-aspartate (NMDA) ionotropic receptors and metabotropic receptors. Injected in the neopallium of the golden hamster at the time of production of neurons normally destined for layers IV, III, and II, ibotenate induces arrests of migrating neurons at different distances from the germinative zone within the radial migratory corridors. The resulting cytoarchitectonic patterns include periventricular nodular heterotopias, subcortical band heterotopias, and intracortical arrests of migrating neurons. The radial glial cells and the extracellular matrix are free of detectable damage that could suggest a defect in their guiding role. The migration disorders are prevented by coinjection of dl-2-amino-7-phosphoheptanoic acid, an NMDA ionotropic antagonist, but are not prevented by coinjection of l(+)-2-amino-3-phosphonopropionic acid, a metabotropic antagonist. This implies that an excess of ionic influx through the NMDA channels of neurons alters the metabolic pathways supporting neuronal migration. Ibotenate, a unique molecular trigger of the excitotoxic cascade, produces a wide spectrum of abnormal neuronal migration patterns recognized in mammals, including the neocortical deviations encountered in the human brain.
Keywords: glutamate, heterotopia, lissencephaly, microcephaly, N-methyl-d-aspartate
In targeted postmigratory neurons, excitatory amino acids trigger a calcium-dependent death (1–4). At each developmental step after completion of neocortical neuronal migration, the excitotoxic cascade produces a timed set of laminar and multilaminar neuronal depopulations. It mimics all lesional brain patterns described after hypoxia occurring in human fetuses and neonates between 20 and 40 weeks of gestation (3–6). In contrast, no excitotoxic destructive effect has been reported before maturation of N-methyl-d-aspartate (NMDA) receptors and before acquisition of postmigratory neuronal aerobiosis. In seminal observations performed in organotypic tissue cultures, Komuro and Rakic (7) reported accelerations and decelerations of cerebellar neuronal migration under the influence of excitatory amino acids. The present paper reports neuronal migration disorders induced in vivo in hamster by ibotenate, a glutamatergic agonist, and uses this model to approach the pathophysiology of these frequent neurodevelopmental disturbances.
MATERIALS AND METHODS
Animal Handling and Injections of Ibotenate and of Other Glutamatergic Agents.
Golden hamsters were chosen for this study because of the timing of brain development in this species. As regards neuronal migration in the neopallium, the newborn hamster is at a stage similar to that of the human fetus at 15 weeks of gestation and at a stage comparable to that of the mouse at the 17th embryonic day. Pregnant female golden hamsters were allowed to deliver on day 16 of gestation. Several successive litters of newborn hamsters of both sexes were used for the experiments. Ibotenate, a glutamatergic agonist, activates both NMDA ionotropic receptors and metabotropic receptors. Under ether anesthesia, intracerebral injections of ibotenate (Sigma) diluted in 0.02% acetic acid 0.1 M PBS were performed with a 26-gauge needle on a 50-μl Hamilton syringe mounted on an Oxford micromanipulator (3). Two 1-μl injections of ibotenate were made at an interval of 60 sec. The injections were made 2 mm beneath the skull surface in the frontoparietal area of the right hemisphere with coordinates of 2 mm from the midline and 3 mm in front of the hinder suture. After ibotenate injections, the pups were returned to their dams. Doses of 0.001, 0.1, 10, or 20 μg of ibotenate per hamster (i.e., 0.006, 0.6, 60, and 120 nM) were used. Controls received 2 μl of 0.02% acetic acid PBS. The mortality rate was 100% in the hamsters injected with 20 μg of ibotenate, and 10% in the hamsters injected with 10 μg of ibotenate. Hamsters injected with 20 μg of ibotenate were seizure free but were areactive, and all died within a few hours during apneic episodes. There was no mortality in the pups injected with the lower doses of ibotenate (i.e., 0.1 and 0.001 μg) or with PBS. The NMDA antagonist, dl-2-amino-7-phosphoheptanoic acid (AP7; Sigma) was coinjected intracortically with ibotenate in an equimolar dose. The metabotropic receptor l-(+)-2-amino-3-phosphonopropionic acid (L-AP3) (Tocris Neuramin, Bristol, U.K.) was coinjected with ibotenate at a 50-time molar dose as done by Schoepp et al. (8). The brain of other animals was injected with 0.1, 1, 10, and 50 nM (i.e., 18.3 ng, 183 ng, 1.8 μg, and 9.15 μg) of (RS)-3,5-dihydroxyphenylglycine (Tocris Neuramin), a metabotropic agonist (9).
Immunohistochemical Studies of Cell Differentiation.
The pups were killed 2, 5, or 40 days after ibotenate injection. They were either killed by decapitation or by intracardiac perfusion with 4% paraformaldehyde in 0.1 M PBS (pH 7.4) fixative under ether anesthesia. After decapitation, the brains were frozen and 20-μm coronal sections were cut on a cryostat for the immunohistochemical procedure. After intracardia perfusion with fixative, the hemispheres were removed and embedded in paraffin. Coronal sections (10 μm) were cut and processed for cresyl-violet staining and immunohistochemical studies. Several specific cell markers were immunohistochemically identified on tissue sections: (i) microtubule-associated proteins (MAPs) types 2 and 5, which are dendritic markers present in immature brain of the hamster, and MAP1, which is an axonal and dendritic marker mainly present in adult brain (10); (ii) glial fibrillary acidic protein (GFAP), a marker of astrocytes; and (iii) vimentin, which labels radial glial cells. For the MAP and GFAP studies, paraffin-embedded sections adjacent to cresyl-violet stained sections displaying excitotoxic damage were incubated with specific primary antibodies [mouse monoclonal anti-MAPs diluted 1/250-1/500 in PBS-Triton from Sigma, and rabbit polyclonal anti-GFAP diluted 1/100 in PBS-Triton from Dakopatts (Glostrup, Denmark)], as described elsewhere (3). Primary antibodies were detected and revealed using the corresponding Vectastain ABC kit (Vector Laboratories) and 3,3′-diaminobenzidine chromogen. Vimentin detection was performed on cryostat sections as described (11). Monoclonal antivimentin antibody from Amersham was diluted 1/50 in PBS. Detection of the primary antibody was performed as above. Control sections were immunostained with omission of the primary antibody. In no case was labeling observed.
Study of the Extracellular Matrix.
P5 hamsters injected with ibotenate at birth were fixed by intracardiac perfusions of 4% paraformaldehyde in 0.1 M PBS (pH 7.4) containing 0.5% cetylpyridinium, embedded in paraffin. Sections (10 μm) were stained with Alcian blue 8G-X (Sigma) at pH 2.5 to detect all the extracellular matrix polyanions, including hyaluronan, sulfated glycosaminoglycans, and other glycoproteins. Adjacent sections were incubated with a biotinylated sheep-brain hyaluronectin probe to detect hyaluronan as described in a previous study (12).
Study of Cell Proliferation.
5-Bromodeoxyuridine (BrdUrd) (Sigma), an S-phase marker, was injected into pregnant hamster females (50 μg/g). To have BrdUrd stained cells in S-phase in the periventricular zone at the time of ibotenate injection, we had to inject BrdUrd into pregnant mothers 24 hr before term. Detection was made on 10-μm embedded-paraffin sections as described by Takahashi et al. (13). The sections were incubated with monoclonal anti-BrdUrd antibodies (Becton Dickinson) diluted 1/50 in PBS-Triton. Detection of the primary antibody was performed as above. Both weak and strong BrdUrd-positive cells were counted in the injected and in the contralateral hemispheres. The numbers of cells are expressed as means ± SEM.
In other ibotenate-injected hamsters, the DNA and protein contents of each hemisphere were measured, with the method described by Burton (14) and modified by Munro (15) and with Bio-Rad protein assay (16).
Terminology.
The terms “band heterotopias,” “diffuse heterotopias,” “(poly)microgyrias,” “pachygyrias,” and others are used as defined by Friede (17), Rorke (18), and Marret et al. (3).
RESULTS
Four main types of disorders of neuronal migration were observed after ibotenate injections (Table 1 and Fig. 1): (i) spherical nodular heterotopias in the periventricular and intermediate zones (Fig. 1 A and B); (ii) band and diffuse heterotopias in the white matter (Fig. 1C) [in these vast heterotopic fields, many neurons are arrested in all migratory corridors of the entire hemisphere, between the external sagittal stratum and the plexiform zone, producing a 1000-μm-thick heterotopic field; the resulting lamination of the cortex is made of three or four layers, mimicking the pachygyric pattern and the double cortex (17, 18)]; (iii) sub- and intracortical arrests of migration (Fig. 1E); and (iv) ectopias in the molecular layer (Fig. 1E).
Table 1.
Neopallial dysgeneses induced by ibotenate injected in hamster brain
| Doses of ibotenate, μg | No. of treated animals | No. of brain lesion, % | Mean rostro-caudal diameter of lesions,* μm | Types of
lesions (%)
|
|
|---|---|---|---|---|---|
| Neuronal heterotopias | Rostral microgyric-like lesion | ||||
| 10 | 49 | 100 | 1200 | Diffuse(12) | |
| Periventricular or subcortical (84) | (83)† | ||||
| Intracortical(4) | |||||
| Ectopias(0) | |||||
| 0.1 | 6 | 100 | 600 | Diffuse(0) | |
| Periventricular(0) | (100) | ||||
| Intracortical(66) | |||||
| Ectopias(100)‡ | |||||
| 0.001 | 6 | 50 | 200 | Diffuse(0) | |
| Periventricular(0) | (50) | ||||
| Intracortical(50) | |||||
| Ectopias(50)‡ | |||||
Mean rostro-caudal diameter was estimated by counting the number of 10-μm sections displaying the lesion.
Microgyric-like lesions were not observed in hamster brain with diffuse heterotopias.
Ectopias could be associated with intracortical heterotopias.
Figure 1.

Neuronal migration disorders induced in the neopallium by ibotenate. Injection at birth in hamster; light microscopic study at P5. (A) Cresyl-violet stained section of a brain injected with 10 μg of ibotenate showing periventricular heterotopia (arrowhead) and hypoplasia of the injected hemisphere. (B) Cresyl-violet stained section at a higher magnification than in A. (C) Cresyl-violet stained section of a “hemipachygyric” brain injected with 10 μg of ibotenate showing a severe architectural disorder without any recognizable normal neuronal layer. (D) BrdUrd-stained section adjacent to the cresyl-violet stained section shown in B. (E) Cresyl-violet stained section of a brain injected with a low dose (0. 1 μg) of ibotenate showing ectopias in the molecular layer of the cortex. (F) Cresyl-violet stained section showing the depopulation observed in the rostral part of the lesions (between arrowheads). (G) MAP2 stained section adjacent to the cresyl-violet stained section (B) showing no dendritic labeling in the heterotopia. (A and C, ×37; B and D, ×260; E and F, ×250; and G, ×520.)
Periventricular and band heterotopias are produced only with high ibotenate doses; low doses produce only intracortical and molecular layer heterotopias (Table 1). In all four types of heterotopic neuronal arrangements, most of the heterotopic cells are small, with no sign of pyknosis in the nucleus or vacuolization in the cytoplasm.
The only type of lesion of postmigratory neurons observed in this material is the depopulation of layers V–VIa; this layered neuronal depopulation is observed in the rostral, more mature, part of the brain, where completion of neuronal migration occurred before the ibotenate injection (Table 1 and Fig. 1F).
Abnormal gyri appeared after ibotenate injection in two circumstances corresponding to distinct developmental mechanisms: (i) when the cortical plate is almost missing because most migrating neurons are locally arrested in rounded heterotopias (Fig. 1A); and (ii) when postmigratory neurons of layers V–VIa are destroyed in the rostral regions (Fig. 1F).
In 75% of the brains injected with 10 μg of ibotenate, hypoplasia of the injected hemisphere was observed and associated with heterotopias. No hemispheric hypoplasia was noted with lower doses (i.e., 0.1 and 0. 001 μg of ibotenate), even when the cortical abnormalities were conspicuous. In one animal injected with 10 μg of ibotenate the corpus callosum was severely hypoplastic with generalized migration defect.
Effects of Ibotenate Antagonists.
Six pups were coinjected with both ibotenate and AP7, an antagonist of the NMDA receptor. Three coinjected animals displayed no damage at all. In two coinjected hamsters, layers II and III were slightly depopulated without any detectable disturbance of migration. A similar minor depopulation of layers II and III was observed in two-thirds of the six control hamsters injected with AP7 alone. A small subcortical heterotopia was observed in one out of six coinjected animals.
Seven pups were coinjected with both ibotenate and with AP3, an antagonist of the metabotropic receptor. This antagonist was devoid of any protective effect.
Effects of Metabotropic Agonists.
Six pups were injected only with (RS)-3,5-dihydroxyphenylglycine, a metabotropic agonist. These injections did not produce any migration disorders.
Immunohistochemical Studies.
No gliosis was detected in the injected brains, either when examined 5 days after the ibotenate injection or when examined at the adult age. The absence of gliosis was confirmed by anti-GFAP staining. No dendritic MAP2 (Fig. 1G), MAP5, or MAP1 stainings were detectable in the heterotopic areas or in subjacent cortex. Staining of neurofilaments was weak in the heterotopic regions and showed an abrupt interruption of callosal connections. The radial glial cells, studied with the antivimentin antibodies 2 days after ibotenate injection, appeared entirely normal throughout the whole brains, including the dysgenetic areas (Fig. 2). Extracellular matrix, analyzed with Alcian blue 8G-X at pH 2.5 and with a hyaluronectin probe, also appeared normal.
Figure 2.
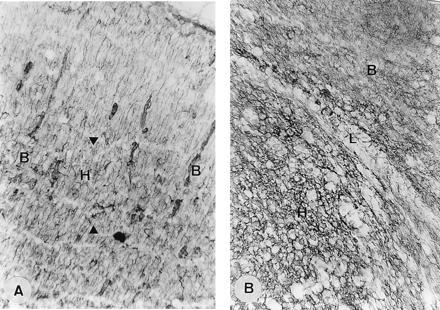
Radial glial fibers and extracellular matrix in neuronal heterotopias induced by ibotenate. (A) Injection of 10 μg in hamster neopallium at birth; killed at P2. Vimentin stained section. In the nodular periventricular heterotopia, radial glial cells are morphogically normal. Arrowheads indicate the limits of heterotopia (H); B, normal brain. (B) Injection of 10 μg in hamster neopallium at birth; killed at P5. Hyaluronan immunostained section of a nodular heterotopia reveals a morphogically normal extracellular matrix. H, heterotopia; L, limit of heterotopia; B, normal brain. (A, ×220; B, ×840.)
Incorporation of BrdUrd and Estimate of DNA Content.
The total number of BrdUrd-positive neurons (Fig. 1D) counted in 10 injected hemispheres (i.e., BrdUrd-positive neurons of heterotopias plus BrdUrd-positive normally located neurons) on adjacent sections to cresyl-violet stained sections showing heterotopias was similar to the total number of BrdUrd positive neurons counted in the 10 contralateral intact hemispheres (128 ± 43 neurons per vertical neopallial column of 400 μm in diameter in the heterotopic hemispheres, versus 121 ± 38 neurons per similar column in the normal hemispheres, a statistically nonsignificant difference). The number of BrdUrd-positive neural cells found in heterotopias reached <15% of the number of neural cells counted in the same heterotopias on cresyl-violet sections.
Dosages of DNA and protein contents were also similar between 10 injected hemispheres versus 10 control hemispheres (DNA content, 1 ± 0.167 μg per ml versus 0.966 ± 0.15 μg per ml, a statistically nonsignificant difference; protein content, 15.4 ± 2.97 mg per ml versus 14.5 ± 2.24 mg per ml, a statistically nonsignificant difference).
DISCUSSION
The salient feature of this study is the induction of disorders of neuronal migration in hamsters injected with an NMDA glutamatergic agonist before the end of neuronal migration. This animal model permits an analysis of a whole spectrum of topographic and topological types of disorders of neuronal migration and provides data concerning the complement of neurons reaching the neocortex along radial migratory corridors (19). In all neocortical fields with migration disorders, the lateral scattering of neurons above the heterotopic area is very modest, an argument against a conspicuous tangential migration in the mammalian brain. In the most affected specimens (Fig. 1B), where the radial migration arrest seems to be complete, the number of neurons reaching the very thin “cortical plate” does not exceed a fifth of the neuronal complement destined to the neocortex, combining in this estimate of the residual neocortex, neurons of the very first radial wave that reached the plate and tangentially migrating neurons. In this study, we did not obtain data concerning a possible interference of ibotenate with tangential migration. Migration defects are often focal (88%) and of the nodular heterotopic type. In 12% of the animals, the migration deficits cover the entire hemisphere and result in a band or diffuse heterotopic neocortical pattern exactly mimicking hemipachygyric brains. The injection procedure is fairly well reproducible and has no influence on the type and topography of migration disorders. As shown in Table 1, the type of deviant architectural pattern is dose-dependent. The focal/nodular versus hemispheric/band migration disturbances produced by high ibotenate doses seem to be dependent upon the developmental age. Indeed, the postconceptional age at delivery cannot be estimated with a better than 12-hr accuracy; at this developmental epoch, such a time interval can reduce the water content of extracellular matrix and influence the migration mechanisms and the level of migratory completion (19–21). The nodular heterotopias coexist in the anterior neocortical regions with areas of laminar depopulation of installed layers V–VIa, a postmigratory event extensively described in perfusion failures/hypoxias in the human (22, 23), and in excitotoxic hypoxic-like lesions described in the mouse (3). This topographic distribution can be attributed to an earlier completion of neuronal migration and to an earlier acquired postmigratory maturation (i.e., aerobic metabolism and NMDA receptor stabilization) in the rostral rather than in the caudal regions in the hamster at the moment of injection (3, 21, 23, 24). Seventy-five percent of the injected animals are “hemimicrocephalic;” the brain asymmetry being more pronounced in the “pachygyric” type but often present also in the “nodular” type. The complement of neural cells, estimated by the DNA content, the protein content, and with the BrdUrd method, is similar in both hemispheres in all specimens, even in the most asymmetric brains. Defective cell proliferation is, therefore, unlikely. The reduction of size of the injected hemisphere is probably due to defective development of the neuropile following the migration defect. The absence of MAP2, MAP5, and MAP1 stainings in all heterotopic neurons reflects disturbances of dendritic outgrowth, which is consistent with this hypothesis. As an alternative explanation of the “hemimicrocephaly,” however, we cannot exclude the role of an increased proliferation followed by an excessive apoptosis (25). Nor can we completely exclude the possibility of increased cell death in installed postmigratory neuronal layers V–VIa, which are associated with migrational disorders in this model. But in a previous excitotoxic model developed in the mouse at birth after the end of neuronal migration, we observed isolated depopulation of layers V–VIa without any migrational disorder and without any hemimicrocephaly (3).
The NMDA glutamatergic agonist arrests neurons at all levels of the radial migratory corridors (germinative zone, white matter, cortical plate, molecular layer) resulting in a spectrum of architectonic patterns: nodular heterotopias in the periventricular and intermediate zones, band and diffuse heterotopias in the white matter, sub- and intracortical arrests of migration, and ectopias in the molecular layer. The periventricular heterotopias induced by ibotenate injection at P0 in hamster are similar to the neuronal ectopic nodules encountered in Aicardi and other human syndromes (for a review, see refs. 17, 18, 23, and 26). The subcortical band heterotopias and the three- to four-layered cortex mimick the spectrum of neocortical patterns encountered in the subcortical, corticosubcortical, and intracortical migration blockades of the human double cortex and pachygyrias/lissencephalies (27, 28) and of the rat after prenatal irradiation (29). Ectopias and protrusions of neurons noted in layer I (obtained with ibotenate at low doses) display some similarities with the disorders of neuronal migration produced by cryolesions in newborn rat by Dvorak et al. (30, 31) and with status verrucosus deforman observed in the human (17).
This study demonstrates that excessive stimulation of NMDA receptors by the excitotoxin ibotenate disturbs neuronal migration. An antagonist of the NMDA receptor robustly prevents neopallial dysplasias while an antagonist of the metabotropic receptor is devoid of any protective effect and a selective metabotropic agonist does not interfere with neuronal migration. This implies that an excess of ionic influx through the NMDA channels of neurons alters the metabolic pathways supporting neuronal movement. The calcium overflux could act directly or through genetic alterations on the control of neuronal excursion (32). The NMDA-mediated action of ibotenate could be due to an effect on some phases of the cell cycle, on pre- or permigratory phases after the end of the cell cycle, or on the environment of the cell. An ibotenate effect on the environment of migrating cell could not be totally excluded, even if extracellular matrix and the radial glial guiding system are everywhere normal, including in the areas where neuronal migration is the most defective. In their in vitro study, Komuro and Rakic (7) demonstrated that the NMDA receptor is implicated in migration of granule cells in slices of rat cerebellum. Alternatively, McConnell and Kaznowski (33) demonstrated that environmental factors during the late S-phase are determinants of the ultimate position in the neocortex. In the severely affected areas, most migrating neurons are arrested by ibotenate injection. This could be due to the presence of NMDA receptors on all the migrating neurons. It could also be due to the activation of a selected population of neurons with NMDA receptors on their membrane; a secondary alteration of calcium concentration in the environment could therefore modify the migration of other non-NMDA neurons through the activation of other receptor/calcium channels (34).
In addition to the major neuronal migration disturbances, destructions of postmigratory neurons mimicking the microgyric brains are also observed in the rostral part of the brain, a phenomenon we have previously described in detail at later developmental stages (3, 4). These staged, different, and consecutive effects of the ibotenate excitotoxin depend on neuronal maturation and can be explained by a developmental change of the NMDA receptor (Table 2) (35–40).
Table 2.
Deviant neopallial patterns induced by ibotenate in rodent brains
| Developmental steps of CNS | NMDA* receptor | Ibotenate-induced lesions | Disorders of neuronal migration | Neuronal excitotoxicity | White matter sensitivity |
|---|---|---|---|---|---|
| Preparation of the neural epithelium | ND | No lesion | − | − | − |
| Migration of infragranular layers | + | ND | ND | ND | ND |
| Migration of supragranular layers | + | Heterotopia | + | + | − |
| (layers V–VI) | |||||
| Maturation of infragranular layers | + | Microgyria | + | + | − |
| (layers V–VI) | |||||
| Maturation of supragranular layers plus full activity of white matter | + | Cortico/subcortical damage | − | + | + |
| PVL | (layers II–VI) |
This table is the synthesis of all the data we obtained in different animal models of excitotoxicity using ibotenate as glutamate agonist. The step of the preparation of the neuroepithelium was investigated in vitro by whole-embryo cultures (41, 42); the step of migration supragranular layers was investigated in vivo by intracerebral injection in hamster (present study); the steps of maturation of neuronal layers were investigated in vivo by intracerebral injection in mice at P0 and P5 (3). ND, not done; +, present; −, absent; PVL, periventricular leukomalacia; CNS, central nervous system.
Data shown are derived from Watanabe et al. (38).
In the present study, we used an excitotoxin to disturb neuronal migration and produce neuronal arrests at all levels of the migratory corridors. At later developmental stages, this excitotoxin has quite different dysmorphogenetic properties, producing hypoxic-like neuronal death. This study also stresses the interactions between environmental/epigenetic and genetic factors in the pathogenesis of cytoarchitectonic disorders.
Acknowledgments
We thank Annie Delpech from the University of Rouen for her support, Bertrand Delpech from the University of Rouen for his advice and for the gift of sheep brain hyaluronectin, and Anne-Marie Rona for her excellent technical assistance. This work was partly supported by the Fonds National de la Recherche Scientifique of Belgium, by the Fondation de France, by the Fondation de la Recherche Médicale (France), and by the Centre Hospitalier Universitaire of Rouen, France.
Footnotes
Abbreviations: NMDA, N-methyl-d-aspartate; AP7, dl-2-amino-7-phosphoheptanoic acid; l-AP3, l-(+)-2-amino-3-phosphonopropionic acid; MAP, microtubule-associated protein; GFAP, glial fibrillary acidic protein; BrdUrd, 5-bromodeoxyuridine.
References
- 1.McDonald J W, Johnston M V. Brain Res Rev. 1990;15:41–70. doi: 10.1016/0165-0173(90)90011-c. [DOI] [PubMed] [Google Scholar]
- 2.Johnston M V. Clin Invest Med. 1993;62:122–132. [PubMed] [Google Scholar]
- 3.Marret S, Mukendi R, Gadisseux J F, Gressens P, Evrard P. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Marret S, Gressens P, Gadisseux J F, Evrard P. Dev Med Child Neurol. 1995;37:473–484. doi: 10.1111/j.1469-8749.1995.tb12035.x. [DOI] [PubMed] [Google Scholar]
- 5.Lipton S A, Rosenberg P A. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 6.Evrard P, de Saint-Georges P, Kadhim H J, Gadisseux J F. In: Child Neurology and Developmental Disabilities. French J, editor. Baltimore: Brookes; 1989. pp. 153–176. [Google Scholar]
- 7.Komuro H, Rakic P. Science. 1993;260:95–97. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- 8.Schoepp D D, Johnson B G, Smith E C R, McQuaid L A. Mol Pharamacol. 1990;38:222–228. [PubMed] [Google Scholar]
- 9.Birse E F, Eaton S A, Jane D E, Jones P L S, Portes R H P, Pook P C-K, Sunter D C, Udvarhelyi P M, Wharton B, Roberts P J, Salt T E, Watkins J C. Neuroscience. 1993;52:481–488. doi: 10.1016/0306-4522(93)90400-a. [DOI] [PubMed] [Google Scholar]
- 10.Oblinger M M, Kost S A. Dev Brain Res. 1994;77:45–54. doi: 10.1016/0165-3806(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 11.Gressens P, Gofflot F, Van-Maele-Fabry G, Misson J P, Gadisseux J F, Evrard P, Picard J J. J Neuropathol Exp Neurol. 1992;51:206–219. doi: 10.1097/00005072-199203000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Marret S, Delpech B, Delpech A, Asou H, Girard N, Maingonnat C, Fessard C. J Neurochem. 1994;62:1285–1295. doi: 10.1046/j.1471-4159.1994.62041285.x. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi T, Nowakowski R S, Caviness V S. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 14.Burton K. Biochem J. 1956;62:315–323. doi: 10.1042/bj0620315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munro H N. Methods Biochem Anal. 1968;14:113–176. doi: 10.1002/9780470110324.ch5. [DOI] [PubMed] [Google Scholar]
- 16.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 17.Friede R L. Developmental Neuropathology. 2nd Ed. Berlin: Springer; 1989. [Google Scholar]
- 18.Rorke L B. J Neuropathol Exp Neurol. 1994;53:105–117. doi: 10.1097/00005072-199403000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Rakic P. Proc Natl Acad Sci USA. 1995;92:11323–11327. doi: 10.1073/pnas.92.25.11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehmenküller A, Sykova E, Svoboda J, Zilles K, Nicholson C. Neuroscience. 1993;55:339–351. doi: 10.1016/0306-4522(93)90503-8. [DOI] [PubMed] [Google Scholar]
- 21.Shimada M, Langman J. J Comp Neurol. 1972;139:227–244. doi: 10.1002/cne.901390206. [DOI] [PubMed] [Google Scholar]
- 22.Caviness V S, Misson J P, Gadisseux J F. In: From Reading to Neuron. Galaburda A M, editor. Cambridge, MA: MIT Press; 1989. pp. 405–442. [Google Scholar]
- 23.Evrard P, Miladi N, Bonnier C, Gressens P. In: Handbook of Neuropsychology. Rapin I, Segalowitz S J, editors. Vol. 6. London: Elsevier; 1992. pp. 11–44. [Google Scholar]
- 24.Angevine J B, Sidman R L. Nature (London) 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 25.Gould E, Cameron H A, McEwen B S. J Comp Neurol. 1994;340:551–565. doi: 10.1002/cne.903400408. [DOI] [PubMed] [Google Scholar]
- 26.Palmini A, Andermann F, Aicardi J, Dulac O, Chaves F, Ponsot G, Pinard J M, Goutières F, Livingstone D, Tampieri D, Andermann E, Robitaille Y. Neurology. 1991;41:1656–1662. doi: 10.1212/wnl.41.10.1656. [DOI] [PubMed] [Google Scholar]
- 27.Dobyns W B, Elias E R, Newlin A C, Pagon M D, Ledbetter D H. Neurology. 1992;42:1375–1388. doi: 10.1212/wnl.42.7.1375. [DOI] [PubMed] [Google Scholar]
- 28.Palmini A, Andermann F, de Grissac H, Tampieri D, Robitaille Y, Langevin P, Desbiens R, Andermann E. Dev Med Child Neurol. 1993;35:331–339. doi: 10.1111/j.1469-8749.1993.tb11645.x. [DOI] [PubMed] [Google Scholar]
- 29.Ferrer I, Alcantara S, Marti E. Neuropathol Appl Neurobiol. 1993;19:74–81. doi: 10.1111/j.1365-2990.1993.tb00407.x. [DOI] [PubMed] [Google Scholar]
- 30.Dvorak K, Feit J. Acta Neuropathol. 1977;38:203–212. doi: 10.1007/BF00688066. [DOI] [PubMed] [Google Scholar]
- 31.Dvorak K, Feit J, Jurankova Z. Acta Neuropathol. 1978;44:121–129. doi: 10.1007/BF00691477. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh A, Greenberg M E. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 33.McConnell S K, Kaznowski C E. Science. 1991;254:282–285. doi: 10.1126/science.254.5029.282. [DOI] [PubMed] [Google Scholar]
- 34.Rakic P, Komuro H. J Neurobiol. 1995;26:299–315. doi: 10.1002/neu.480260303. [DOI] [PubMed] [Google Scholar]
- 35.Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishina M. Nature (London) 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 36.Mishra O P, Delivoria-Papadopoulos M. Neurochem Res. 1992;17:1223–1228. doi: 10.1007/BF00968404. [DOI] [PubMed] [Google Scholar]
- 37.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakman B, Seeburg P H. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 38.Watanabe M, Inoue Y, Sakimura K, Mishina M. NeuroReport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- 39.Farrant M, Feldmeyer D, Takahashi T, Cull-Candy S G. Nature (London) 1994;368:335–339. doi: 10.1038/368335a0. [DOI] [PubMed] [Google Scholar]
- 40.Sheng M, Cummings J, Roldan L A, Jan Y N, Jan L Y. Nature (London) 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 41.Marret, S., Gressens, P., Evrard, P. (1995) Dev. Med. Child Neurol. 37 (Suppl.), 81 (abstr.). [DOI] [PubMed]
- 42.Evrard, P., Marret, S., Gressens, P. (1996) Acta Paediatr. Scand., in press. [DOI] [PubMed]