

Abstract
Elevation of the neuropeptide corticotropin-releasing factor (CRF) in the brain is associated with a reduction of food intake and body weight gain in normal and obese animals. A protein that binds CRF and the related peptide, urocortin, with high affinity, CRF-binding protein (CRF-BP), may play a role in energy homeostasis by inactivating members of this peptide family in ingestive and metabolic regulatory brain regions. Intracerebroventricular administration in rats of the high-affinity CRF-BP ligand inhibitor, rat/human CRF (6-33), which dissociates CRF or urocortin from CRF-BP and increases endogenous brain levels of “free” CRF or urocortin significantly blunted exaggerated weight gain in Zucker obese subjects and in animals withdrawn from chronic nicotine. Chronic administration of CRF suppressed weight gain nonselectively by 60% in both Zucker obese and lean control rats, whereas CRF-BP ligand inhibitor treatment significantly reduced weight gain in obese subjects, without altering weight gain in lean control subjects. Nicotine abstinent subjects, but not nicotine-naive controls, experienced a 35% appetite suppression and a 25% weight gain reduction following acute and chronic administration, respectively, of CRF-BP ligand inhibitor. In marked contrast to the effects of a CRF-receptor agonist, the CRF-BP ligand inhibitor did not stimulate adrenocorticotropic hormone secretion or elevate heart rate and blood pressure. These results provide support for the hypothesis that the CRF-BP may function within the brain to limit selected actions of CRF and/or urocortin. Furthermore, CRF-BP may represent a novel and functionally selective target for the symptomatic treatment of excessive weight gain associated with obesity of multiple etiology.
Considerable evidence suggests a role for endogenous brain corticotropin-releasing factor (CRF) systems in appetite regulation, energy balance, and the etiology of eating disorders (1, 2). Food intake is diminished by administration of CRF or urocortin agonists or treatments that increase endogenous hypothalamic CRF production, such as stress, tumor induction, or appetite-suppressing drugs (3–6). It is noteworthy that central CRF treatment induces, concurrently with a reduction in food intake, an increase in the activity of the sympathetic nervous system (7, 8). This finding suggests that the anorectic effect of CRF may be mediated in part, as are its thermogenic effects, by central control over the autonomic nervous system (9, 10). Central administration of the CRF receptor antagonist, α-helical CRF (9-41), potentiates the increases in appetite induced by neuropeptide Y and attenuates stress-induced appetite suppression at doses that do not alter intake in non-food-deprived or non-stressed subjects (4, 11, 12). These clues point to a physiological role for CRF or urocortin in the induction of negative energy balance, especially under conditions of exaggerated hunger/weight gain, which may be counteracted by anorexic and sympathomimetic effects of activated CRF systems. Indeed, brain CRF production is dependent on feeding/weight status in animal models of dysregulated energy balance, such as the Zucker obese rat, in tumor-bearing cachexia, in chronic exercise, and in the context of drug- or stress-induced changes in appetite (1, 3, 5, 12).
Studies of obese rats have provided data implicating the hypothalamic (CRF)–pituitary–adrenal axis in energy balance dysregulation. A decline in CRF immunoreactivity in hypothalamic brain areas has been linked with the etiology of obesity in the Zucker and Wistar fatty rats (13, 14). Zucker rats exhibit an abnormal response of the hypothalamo–pituitary–adrenal axis to stressors and a substantially increased body weight gain, which is normalized by treatments that increase hypothalamic CRF, such as adrenalectomy or chronic administration of glucocorticoid antagonists (15, 16). Furthermore, central infusion of CRF interrupts the excessive body weight gain of obese Zucker rats (15, 16). Negative energy balance resulting from central administration of CRF agonists can be attributed jointly to appetite loss as well as elevated metabolic rate encompassing increased cardiac output and energy mobilization (9). In particular, one feature of the primary etiology of obesity in rodents, insufficient heat production within sympathetically innervated brown adipose tissue, is restored by treatment with exogenous CRF to values seen in lean animals (9). These findings suggest that brain CRF dysregulation contributes to the pathogenesis of obesity in the Zucker rat.
A high-affinity, biologically inactivating CRF-binding protein (CRF-BP) is extensively but selectively distributed throughout the central nervous system and has been proposed to serve to limit the action of CRF (17) and, more recently, the new CRF family member urocortin (18). The development of ligand inhibitors of CRF-BP (19) provides a means of evaluating the physiologic roles of the binding protein (20). Furthermore, such inhibitors hold promise as a means of increasing concentrations of endogenous unbound CRF in select brain areas where the binding protein limits the action of CRF, without producing generalized activation of CRF neurons that would occur with a postsynaptic receptor agonist for CRF (20). Potential beneficial actions of central CRF receptor agonists would be abated by the adverse consequences of generalized CRF receptor activation such as a fear-like state of hyperemotionality (21), stimulation of pituitary–adrenocortical hormone secretion (22), and increased heart rate and blood pressure (23).
In the present studies, we examined the role of brain CRF and CRF-BP in excess body weight gain associated with the Zucker obese phenotype by comparing the effects of chronic administration of CRF with that of the selective, high-affinity CRF-BP ligand inhibitor, rat/human (r/h) CRF (6-33). Because hyperphagia and weight gain following smoking cessation are a reproducible component of the nicotine abstinence syndrome modeled in animals (24–28), we also evaluated the effects of acute and chronic administration of r/h CRF (6-33) on increased food intake and weight gain in nicotine-withdrawn rats. Results suggest that CRF-BP, by neutralizing CRF-related ligands, may serve physiologically to restrain these endogenous appetite and body weight suppressants.
MATERIALS AND METHODS
Subjects.
Lean (Fa/?; n = 26, 300–400 g) and obese (fa/fa; n = 19, 400–500 g) Zucker strain male rats (Harlan Breeders, Indianapolis) were age-matched (3 months old) on arrival. Male Wistar rats (Charles River Breeding Laboratories; 300–450 g) were used in the nicotine dependence/withdrawal validation (n = 23), the plasma nicotine (n = 5), and the acute (n = 34) and chronic (n = 23) CRF-BP ligand inhibitor experiments. Sprague–Dawley male rats (Harlan; n = 9, 250–280 g) were used in the blood pressure experiments. All subjects were single-housed and allowed a 1-week acclimation to a temperature, humidity, and light cycle-controlled (lights on from 0600–1800 h) vivarium with ad libitum access to normal laboratory chow (Picolab Rodent Diet 20; PMI Feeds, St. Louis) and water before experimental testing. Subjects in the nicotine experiments were tested only once, while some of the Zucker lean (n = 10) and obese (n = 9) rats were retested following a 3-week washout period and pseudorandom treatment group reassignment. Testing protocols employed herein were approved by the Institutional Animal Care and Use Committees of Neurocrine Biosciences or The Salk Institute.
Intracerebroventricular (i.c.v.) Cannulations and Injections.
Rats tested in the appetite/body weight studies were anesthetized with isoflurane and secured in a Kopf stereotaxic instrument (Kopf Instruments, Tujunga, CA). A guide cannula aimed above the lateral ventricle was then implanted and anchored to the skull with two stainless steel screws and dental cement. Stereotaxic coordinates were, with the tooth bar 5 mm above interaural zero, −0.6 mm posterior to bregma, ±2.0 mm lateral and −3.2 mm below skull surface at the point of entry. Guide cannulae were kept patent until injection by insertion of a dummy stylet. Animals were undisturbed for a 7-day postsurgical recovery period. Using a similar procedure for arterial blood pressure measurement, a 22-gauge guide cannula fitted with a dummy insert (Plastics One, Roanoke, VA) was implanted under Rompun anesthesia (ketamine/xylazine/acepromazine, 25:5:1 mg/kg; subcutaneous) into the lateral ventricle and affixed to the skull with dental acrylic 1 week before testing.
Osmotic Mini-Pump Implantation and Drug/Peptide Infusion.
Chronic administration of nicotine (nicotine tartrate salt; Sigma) was accomplished by subcutaneous implantation, under isoflurane anesthesia, of an Alzet osmotic mini-pump (Alza, model no. 2002; ref. 24), which allows for a measured and precise means of administration that circumvents the need for repeated handling for syringe injection or aversive properties of orally administered nicotine in the rat (29). The pump was located subdermally in the interscapular region of the back and infused a solution of nicotine tartrate salt dissolved in saline or saline alone (vehicle) at a rate of 0.5 μl/h for 2 weeks. To achieve a nicotine dose of 3.15 mg/kg per day (nicotine free base), a stock solution of 225 mg of nicotine tartrate per ml was used for subjects with a body weight of 200 g and adjusted accordingly (24). Pumps were activated before implantation by overnight incubation in a 37°C water bath to standardize the initiation and rate of nicotine infusion over time. Nicotine pumps were implanted at 1800 h, at the beginning of the dark portion of the light/dark cycle. The present studies examined nicotine abstinent subjects beginning 24 h following pump removal when plasma nicotine levels have been reported in the undetectable range following chronic levels of 20–60 ng/ml at a 6 mg/kg per day dose (30).
Chronic i.c.v. infusion of r/h CRF (6-33) was accomplished by subcutaneous implantation of a second osmotic pump (Alza, model no. 2001) on experimental day 14, simultaneous with nicotine pump removal. The outlet of the infusion pump was attached to a Brain Infusion Kit (Alza), which allowed peptide to be continuously infused into the lateral cerebral ventricle, which was in turn targeted using the same surgical procedure and stereotaxic coordinates described for i.c.v. cannulation.
Food Intake and Body Mass Measurement.
For studying acute effects of CRF-BP ligand inhibitor on meal size, single-housed, cannulated rats were allowed 120 min of access to a predetermined portion of laboratory chow in a home cage environment beginning 2 h into the dark/active cycle at 2000 h. Nicotine abstinent subjects and nicotine-naive controls were administered vehicle or r/h CRF (6-33) 3 days following pump removal. In the remainder of studies involving chronic CRF-BP ligand inhibitor treatment, subjects were single housed and body weight and food intake were measured daily at 1400 h during the diurnal photoperiod. Drinking water was available ad libitum in all studies.
Jugular Catheterization and Blood Sampling.
Rats were placed under general anesthesia using continuous inhalation of an isoflurane/oxygen mixture and then surgically prepared with a chronic indwelling intravenous catheter. The left jugular vein was incised, and a 3.5-cm length of polyethylene (PE)-10 tubing (Clay Adams) was inserted and secured with the tip in proximity to the heart. The enlarged (PE 50-tubing; Clay Adams) output end of the catheter was closed with a removable occluder and passed out of an incision in back of the neck so as to facilitate tethering and intravenous injection or passive blood sampling. Catheterized rats were single housed for a 2-day postsurgical recovery period during which time catheters were flushed and filled with heparinized (1:5) physiological saline. Plasma adrenocorticotropic hormone (ACTH) was assayed from blood samples drawn at five separate time points: basal (0 min)—1 h after attachment of a catheter tether and placement in opaque cylinders (15 cm in diameter, 40 cm high) situated in a quiet, dimly lit room and immediately before ovine (o)CRF (1-41) or r/h CRF (6-33) administration—and 15, 30, 60, and 120 min following administration. Using a similar procedure for arterial blood pressure measurement, an indwelling femoral arterial catheter was implanted under methohexane anesthesia.
Arterial Blood Pressure Measurement.
Experiments were carried out on individually housed, awake, freely moving subjects 24 h after implantation of a femoral arterial catheter for monitoring of blood pressure. One to two hours before the experiment, the arterial catheter [sterile, heparinized (100 units/ml), PE-50 tubing; Clay Adams] was connected to a blood pressure transducer (Statham, Hato Rey, PR). Heart rate was computed from the blood pressure pulses using a biotachometric pen recorder (Gould, Cleveland) for off-line analysis. The oCRF (1-41), oCRF (6-33), and r/h CRF (6-33) peptides (kindly provided by Jean Rivier, The Salk Institute) were dissolved in 0.9% saline that contained 0.01 M acetic acid and injected i.c.v. (10, 25, or 50 μg in 1 μl) in a randomized sequence. Similar volumes of the acidified saline vehicle were injected to controls. Depending on the dose, injections were spaced at 30- to 60-min intervals after complete recovery of arterial blood pressure to basal values. Mean arterial pressure was calculated as the sum of 2/3 diastolic pressure and 1/3 systolic pressure.
Plasma Nicotine Determinations.
Trunk blood was collected from subjects infused by osmotic pump with nicotine over 48 h at 0, 1, 3, 6, and 24 h following osmotic pump removal. Samples were centrifuged, and the supernatant plasma was extracted by a solid phase method. Blood nicotine levels were assayed by gas chromatography using a nitrogen-phosphorus detector (31). Continuous subcutaneous infusion of a 3.15 mg/kg per day dose of nicotine free base from osmotic minipumps resulted in plasma nicotine concentrations of 48 ± 7.6 ng/ml. Levels declined with time following minipump removal (data not shown) and were undetectable (<3 ng/ml) at 24 h postwithdrawal.
In Vitro CRF-BP Ligand Inhibitor Stability Assay.
Persistence of bioactivity over time of r/h CRF (6-33) and oCRF (1-41) was assessed by evaluating the peptides’ ability to compete for 125I-labeled Tyr°hCRF (DuPont/NEN; specific activity, 2200 Ci/mmol; 1 Ci = 37 GBq) binding to the human recombinant CRF-BP in the previously described CRF-BP ligand immunoradiometric assay (20, 32). Osmotic mini-pumps were loaded with peptide at a concentration of 1 μg/μl in saline and incubated in a 37°C water bath to simulate the in vivo environment. Expelled solution was sampled at 1, 4, and 7 days postinitiation.
The r/h CRF (6-33) and oCRF (1-41) solutions dispensed from the pump on days 1, 4, and 7 competed for 125I-labeled hCRF binding to the recombinant hCRF-BP with potency equal to a freshly prepared r/h CRF (6-33) or oCRF (1-41) standard, respectively; IC50 values for r/h CRF (6-33) standard were 2 nM, 2.8 nM, and 2 nM on days 1, 4 and 7, respectively. Similarly, there was no significant difference found in the IC50 value for oCRF (1-41) on postincubation days 1 (116 nM), 4 (115 nM), or 7 (110 nM). These data confirm that the ability of r/h CRF (6-33) and oCRF (1-41) to bind to hCRF-BP have not degraded over a 1-week period under physiological conditions.
ACTH Assay.
Blood was collected in chilled tubes containing 2.5 μl of EDTA (50 mg/ml) and 2.5 μl of aprotinin (5000 kilounits/ml). Plasma ACTH was determined by two-site immunometric assay using commercial kits from ICN. The intra- and interassay variabilities are <10%.
Peptide Synthesis.
oCRF (1-41), r/h CRF (6-33), and oCRF (6-33) peptides were synthesized at Neurocrine Biosciences by Nick Ling or The Salk Institute by Jean Rivier using solid phase methodology on a peptide synthesizer (Beckman, model no. 990). Acutely administered peptides were solubilized in distilled water adjusted to pH 6.7, whereas chronic infusion pumps used a saline vehicle.
Analysis of Data.
Data were analyzed by mixed factor analysis of variance (ANOVA) with nicotine treatment, peptide treatment, and Zucker strain as between-subject factors and time (i.e., day) as a repeated measure. Individual mean comparisons were performed using Sheffé post-hoc or Student’s t tests following identification of significant main or interaction effects by ANOVA.
RESULTS
Chronic Administration of CRF-BP Ligand Inhibitor in Zucker Rats.
Fig. 1 exhibits the relative efficacy of the CRF agonist, oCRF (1-41), and CRF-BP ligand inhibitor, r/h CRF (6-33), in suppressing weight gain in lean and obese animals of the Zucker strain. A significant effect of peptide treatment on cumulative weight change (P < 0.005) was detected over the entire 2-week period (day 14 − day 1 difference scores). In particular, oCRF (1-41) suppressed weight gain by 60% in both lean (P < 0.02) and obese (P < 0.01) subjects, whereas both doses of CRF-BP ligand inhibitor, r/h CRF (6-33), significantly suppressed weight gain by 60% in obese Zucker rats (P < 0.04), but not in lean controls. An equivalent analysis of daily food intake over the 2-week experimental period did not reveal any significant effects involving peptide treatment (Fig. 1 legend).
Figure 1.
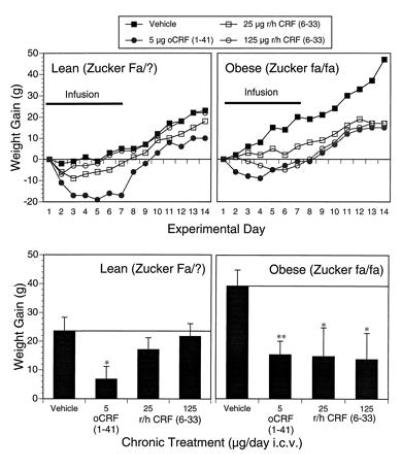
Comparison of CRF receptor agonist and CRF-BP ligand inhibitor actions on body weight in the Zucker model of obesity. Mean ± SEM body weight gain during and following 7-day, chronic, i.c.v. administration of vehicle, oCRF (1-41) (5 μg/day), or r/h CRF(6-33) (25 and 125 μg/day) is exhibited for Zucker lean [vehicle, n = 10; oCRF (1-41), n = 10; 25 μg of r/h CRF (6-33), n = 7; 125 μg of r/h CRF (6-33), n = 9] and obese [vehicle, n = 9; oCRF (1-41), n = 7; 25 μg of r/h CRF (6-33), n = 6; 125 μg of r/h CRF (6-33), n = 6] rats. (Upper) Daily weight change relative to pretreatment baseline weight and (Lower) cumulative weight gain over the 14 experimental days. Peptide treatment significantly reduced body weight gain [F(3,56) = 5.28, P < 0.005]. Mean ± SEM cumulative intake of vehicle-treated subjects was greater than that of peptide treated animals in both obese [vehicle, 325 ± 14 g; oCRF (1-41), 290 ± 15 g; 25 μg of r/h CRF (6-33), 301 ± 34 g; 125 μg of r/h CRF (6-33), 303 ± 26 g] and lean [vehicle, 236 ± 25 g; oCRF (1-41), 204 ± 22 g; 25 μg of r/h CRF (6-33), 182 ± 21 g; 125 μg of r/h CRF (6-33), 218 ± 21 g] cohorts, but these differences were not statistically significant. ∗, P < 0.05; ∗∗, P < 0.01, relative to respective vehicle control groups.
Validation of Chronic Nicotine Infusion/Withdrawal.
Using cumulative measures of body weight change and food intake over the nicotine dependence phase of the study (day 14 − day 1 difference scores), body weight gain (P < 0.04) and food intake (P < 0.05) were significantly reduced by 90% and 5%, respectively, following chronic 14-day nicotine treatment relative to saline-treated controls. The ensuing 14-day period of nicotine abstinence induced a significantly elevated rate of weight gain in nicotine withdrawn subjects (35% increase; P < 0.05) relative to controls treated previously with saline (data not shown).
Acute Administration of CRF-BP Ligand Inhibitor During Nicotine Withdrawal.
In a separate group of subjects made nicotine dependent over 14 days, withdrawal from nicotine produced results consistent with those of the validation study by significantly increasing body weight gain (30% increase; P < 0.04) without a concurrent effect on cumulative food intake. However, during a 2-h nocturnal meal conducted on the third day of the 14-day withdrawal period, r/h CRF (6-33) administered acutely (25 μg i.c.v.) significantly reduced food intake (30% decrease) in nicotine-abstinent subjects (P < 0.05) without altering intake of vehicle-treated controls (Fig. 2).
Figure 2.
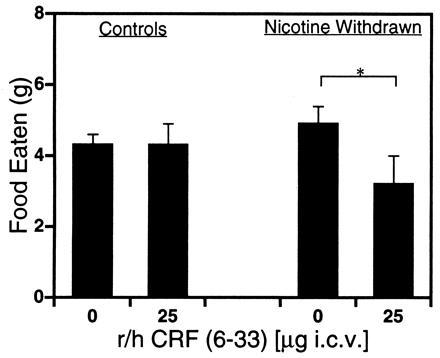
Food intake in nondeprived, nicotine-abstinent rats treated acutely with CRF-BP ligand inhibitor. Mean ± SEM intake of laboratory chow in nicotine-naive (Control) or nicotine-abstinent (Nicotine Withdrawal) subjects is exhibited during a 2-h nocturnal meal following a single administration of 0- (Control, n = 10; Nicotine Withdrawn, n = 9) or 25-μg (Control, n = 8; Nicotine Withdrawn, n = 7) i.c.v. doses of CRF-BP ligand inhibitor. ∗, P < 0.05.
Chronic Administration of CRF-BP Ligand Inhibitor During Nicotine Withdrawal.
In a separate group of subjects made nicotine-dependent over 14 days and then withdrawn concurrent with initiation of chronic administration of r/h CRF (6-33) over the first 7 days of withdrawal, a significant reduction (15% decrease) in weight gain was detected among CRF-BP ligand inhibitor-treated subjects relative to vehicle-treated controls (Fig. 3; P < 0.05). With regard to food intake, there was a significant increase (P < 0.05) in cumulative food intake over the 2-week withdrawal phase in the vehicle-treated, nicotine-abstinent group (341 ± 13 g), but not the CRF-BP ligand inhibitor-treated, nicotine-abstinent group (332 ± 9 g), relative to the vehicle-treated control group (315 ± 8 g).
Figure 3.
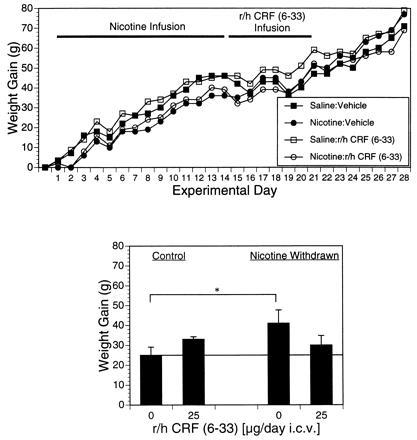
Body weight gain in nicotine-abstinent rats treated chronically with CRF-BP ligand inhibitor. Mean ± SEM body weight gain is exhibited over two 14-day periods of exposure to saline or nicotine (Nicotine Infusion) and following exposure to saline or nicotine (Nicotine Withdrawal). CRF-BP ligand inhibitor (0 or 25 μg/day i.c.v.) was administered chronically over the first 7 days of abstinence [r/h CRF (6-33) Infusion] to both saline-control and nicotine withdrawn groups (n = 6 per group). (Upper) Daily changes in body weight relative to pretreatment baseline weight and (Lower) cumulative weight gain over experimental days 14–28. ∗, P < 0.05
Plasma ACTH Determinations.
Intravenous or i.c.v. treatment with CRF significantly (P < 0.001) elevated plasma ACTH levels relative to vehicle-treated controls (Fig. 4). Treatment with substantially larger doses of CRF-BP ligand inhibitor (25- to 10,000-fold higher) did not significantly alter plasma ACTH levels by either route of administration.
Figure 4.
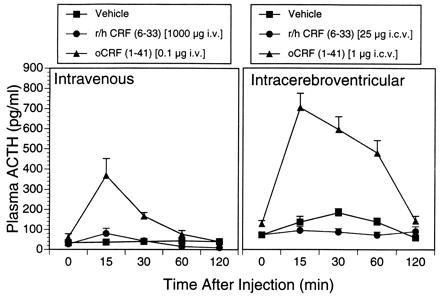
Comparison of CRF receptor agonist and CRF-BP ligand inhibitor actions on ACTH secretion. Mean ± SEM plasma ACTH levels are exhibited in subjects treated intravenously with vehicle (control; n = 10), a 1000-μg dose of r/h CRF (6-33) (n = 6), or a 0.1-μg dose of oCRF (1-41) (n = 6), or i.c.v. with vehicle (control; n = 7), a 25-μg dose of r/h CRF (6-33) (n = 4), or a 1-μg dose of oCRF (1-41) (n = 5). Blood samples were drawn passively over the 2 h subsequent to peptide injection and assayed subsequently for ACTH levels. CRF(1-41) administered both i.v. and i.c.v. significantly elevated plasma ACTH levels [F(2,76) = 18.96, P < 0.001, and F(2,63) = 126.9, P < .001, respectively].
Comparison of Centrally Administered CRF Receptor Agonist Versus CRF-BP Ligand Inhibitor Actions on Cardiovascular Tone.
While i.c.v. administration of the CRF receptor agonist, oCRF (1-41), induced striking hypertension and tachycardia, no cardiovascular stimulatory effects of r/h CRF (6-33) on either mean arterial blood pressure or heart rate were detected (Table 1). Indeed, bradycardia and hypotension were exhibited by subjects treated with r/h CRF (6-33) relative to controls treated either with vehicle or an inert but appropriate control peptide, oCRF (6-33), which binds neither to CRF-BP nor CRF receptors (20). In vehicle-treated subjects, baseline mean ± SEM arterial pressure was 93.3 ± 1.4 mmHg (1 mmHg = 133 Pa) and heart rate was 488.0 ± 7.2 beats per min.
Table 1.
Central cardiovascular effects of CRF receptor agonist and CRF-BP ligand inhibitor peptides
| Treatment | Δ MAP, mmHg | ΔHR, beats per min |
|---|---|---|
| Vehicle | −1.2 ± 0.4 | 0 |
| oCRF (1-41) | ||
| 10 μg | +17.2 ± 3.9 | +20.6 ± 2.8 |
| 25 μg | +26.9 ± 1.4 | +42.0 ± 3.4 |
| oCRF (6-33) | ||
| 25 μg | +0.3 ± 0.1 | 0 |
| 50 μg | +1.7 ± 0.6 | 0 |
| r/h CRF (6-33) | ||
| 25 μg | −4.9 ± 0.9 | −17.9 ± 2.1 |
| 50 μg | −7.2 ± 0.8 | −34.4 ± 4.0 |
Effect of i.c.v. injection of CRF agonist [oCRF (1-41)], a control peptide [oCRF (6-33)], CRF-BP ligand inhibitor [r/h CRF (6-33)], or vehicle on mean arterial pressure (MAP) and heart rate (HR) in conscious, freely moving rats.
DISCUSSION
The major finding of this study is that a CRF-BP ligand inhibitor is capable of blunting excessive weight gain in two animal models of obesity. Interestingly, this is not associated with a generalized suppression of weight gain in nonobese rats. These results may be explained by the differential sensitivity to CRF/urocortin of ingestive and autonomic regulatory centers during circumstances when feeding and body weight are below the brain set point for those parameters (5, 33, 34). Additionally, components of the CRF system may be selectively modulated under conditions of excessive appetite or weight gain. Further evidence of selectivity was provided by separate experiments in which the antiobesity doses of r/h CRF (6-33) did not induce fear-like behaviors, which would be expected following central administration of CRF itself (20). The present and previously reported (20) results suggest that pharmacological treatment with r/h CRF (6-33) may exert selective and moderate appetite and weight gain suppression by eliciting a physiologically relevant increase in levels of endogenous CRF or urocortin. Most importantly, these results support the hypothesis that CRF-BP plays a physiologic role in limiting the action of CRF related peptides in regions where there is a coincidence of CRF-BP, CRF receptors, and CRF or urocortin.
The weight control profile produced by CRF-BP ligand inhibitor in the present studies is quite distinct from that exerted by comparable antiobesity treatments. Relative to treatment with CRF itself, r/h CRF (6-33) produced changes in body weight over the actual infusion interval that were more gradual and less likely to precipitate rebound weight gain during the recovery interval (Fig. 1). Similarly, in contrast to the leptin protein reported recently to reduce the weights of obese mice (35), a high, chronically administered dose of ligand inhibitor did not induce significant food intake suppression and did not exert continual daily weight loss over a 1-week treatment period (Figs. 1 and 3). In our other model, the hyperphagia occurring during nicotine withdrawal is attenuated by treatment with the psychostimulant, phenylpropanolamine, and the serotonin reuptake inhibitor, sertraline, but only at doses that decrease intake in free-feeding, nicotine-naive controls groups (26, 28). The acute anorexic effect of CRF-BP ligand inhibitor in nicotine-abstinent subjects (Fig. 2) suggests the use of this approach as a countermeasure for food cravings that accompany smoking cessation (36), while lack of significant appetitive effects of chronic ligand inhibitor treatment suggests that reduced weight gain is achieved by an increase in energy expenditure via lipolysis or thermogenesis (1, 37). Indeed, metabolic abnormalities such as hyperinsulinemia and autonomic arrest, which accompany overeating and obesity in Zucker obese rats, are effectively normalized by central administration of CRF (15, 38), supporting the broad physiologic significance of this system and raising the possibility that improved CRF-BP ligand inhibitors may be of therapeutic use in the management of human obesity.
A recently characterized CRF-like peptide, urocortin (18), binds with high affinity to CRF2 receptors, which are present in high concentration within the autonomic regulatory nucleus of the ventromedial hypothalamus (18, 39, 40). Urocortin binds with high affinity to CRF-BP (18); furthermore, ligand inhibitors can prevent CRF-BP inhibition of a biological activity of urocortin in vitro (41). The present results on energy balance may reflect the ability of the ligand inhibitors to increase free urocortin levels in selective brain regions, including the ventromedial hypothalamus in addition to augmenting unbound CRF (42). It may be relevant that urocortin exerts anorexia in vivo without concomitant anxiogenic effects (6), similar to the responses we observe here to the CRF-BP ligand inhibitors.
The lack of endocrine and cardiovascular activation by the selective CRF-BP ligand inhibitor in the present studies stands in contrast to the in vivo actions of CRF itself (43, 44). For example, there is a pronounced elevation of plasma ACTH levels following central or peripheral administration of oCRF (1-41), which is not mimicked by behaviorally active doses of CRF-BP ligand inhibitor (Fig. 4). The observation that tachycardia and hypertension produced by centrally administered oCRF (1-41) (43) are not reproduced by higher, behaviorally active doses of r/h CRF (6-33) suggests a further functional dissociation of the two treatments (Table 1). In fact, the ligand inhibitor reduces blood pressure and produces bradycardia perhaps because CRF or urocortin may be liberated in areas such as the nucleus of the solitary tract, which express CRF-BP, CRF receptors, and urocortin/urotensin-like immunoactivity, and which is known to respond to microinjection of CRF with a decrease in sympathetic outflow (45). The differential anatomic distribution of CRF-BP and CRF receptors and ligands throughout the brain may underlie the functional dissociation of ligands acting exclusively at CRF-BP compared with agents which directly activate postsynaptic CRF/urocortin receptors. The effects of the CRF-BP ligand inhibitors in the present study may be mediated through the liberation of CRF and/or urocortin within homeostatic regions in the brain stem and hypothalamus where CRF-BP is found (17).
Acknowledgments
We are indebted to George F. Koob for helpful discussion and suggestions and to Helen Min and Karen Jeske for expert technical assistance. W.W.V. has equity in and is a Member of the Board of Directors and Chairman of the Scientific Advisory Board of Neurocrine Biosciences, Inc. This research was supported by National Institute of Neurological Disorders and Stroke Grant NS33426 to E.B.D.S., D.P.B., and S.C.H., National Institute of Diabetes and Digestive and Kidney Diseases Grant DK 26741 to P.E.S. and W.W.V., and the Foundation for Research. W.W.V. is a Foundation for Research Senior Investigator.
Footnotes
Abbreviations: CRF, corticotropin-releasing factor; CRF-BP, CRF-binding protein; r/h, rat/human; o, ovine; ACTH, adrenocorticotropic hormone; i.c.v., intracerebroventricular.
References
- 1.Richard D. Ann NY Acad Sci. 1993;697:155–172. doi: 10.1111/j.1749-6632.1993.tb49930.x. [DOI] [PubMed] [Google Scholar]
- 2.Chrousos G P, Gold P W. J Am Med Assoc. 1992;267:1244–1252. [PubMed] [Google Scholar]
- 3.Appel N M, Owens M J, Culp S, Zazcek R, Contrera J E, Bissette G, Nemeroff C B, De Souza E B. Endocrinology. 1991;128:3237–3246. doi: 10.1210/endo-128-6-3237. [DOI] [PubMed] [Google Scholar]
- 4.Krahn D D, Gosnell B A, Grace M, Levine A S. Brain Res Bull. 1986;17:285–289. doi: 10.1016/0361-9230(86)90233-9. [DOI] [PubMed] [Google Scholar]
- 5.McCarthy H D, McKibbin P E, Perkins A V, Linton E A, Williams G. Am J Physiol. 1993;264:E638–E643. doi: 10.1152/ajpendo.1993.264.4.E638. [DOI] [PubMed] [Google Scholar]
- 6.Spina M, Merlo-Pich E, Chan R, Basso A M, Rivier J, Vale W, Koob G F. Science. 1996;273:1561–1564. doi: 10.1126/science.273.5281.1561. [DOI] [PubMed] [Google Scholar]
- 7.Brown M R, Fisher L A. In: Corticotropin-Releasing Factor: Basic and Clinical Studies of a Neuropeptide. De Souza E B, Nemeroff C B, editors. Boca Raton, FL: CRC; 1990. pp. 291–298. [Google Scholar]
- 8.Egawa M, Yoshimatsu H, Bray G A. Am J Physiol. 1990;259:R799–R806. doi: 10.1152/ajpregu.1990.259.4.R799. [DOI] [PubMed] [Google Scholar]
- 9.Rothwell N J. Neurosci Biobehav Rev. 1990;14:263–271. doi: 10.1016/s0149-7634(05)80037-5. [DOI] [PubMed] [Google Scholar]
- 10.Brown M R, Fisher L A, Spiess J, Rivier C, Rivier J, Vale W. Endocrinology. 1982;111:928–931. doi: 10.1210/endo-111-3-928. [DOI] [PubMed] [Google Scholar]
- 11.Heinrichs S C, Cole B J, Merlo Pich E, Menzaghi F, Koob G F, Hauger R L. Peptides (Tarrytown, NY) 1992;13:879–884. doi: 10.1016/0196-9781(92)90044-4. [DOI] [PubMed] [Google Scholar]
- 12.Heinrichs S C, Menzaghi F, Merlo Pich E, Koob G F. Brain Res. 1993;611:18–24. doi: 10.1016/0006-8993(93)91771-j. [DOI] [PubMed] [Google Scholar]
- 13.Fukushima M, Nakai Y, Tsukada T, Naito Y, Nakaishi S, Tominage T, Murakami N, Kawamura H, Fukata J, Ikeda H, Matsuo T, Imura H. Neurosci Lett. 1992;138:245–248. doi: 10.1016/0304-3940(92)90925-w. [DOI] [PubMed] [Google Scholar]
- 14.Nakaishi S, Nakai Y, Fukata J, Naito Y, Usui T, Imura H. Int J Obes. 1990;14:951–955. [PubMed] [Google Scholar]
- 15.Rohner-Jeanrenaud F, Walker C-D, Greco-Perotto R, Jeanrenaud B. Endocrinology. 1989;124:733–739. doi: 10.1210/endo-124-2-733. [DOI] [PubMed] [Google Scholar]
- 16.Holt S J, York D A. Physiol Behav. 1989;45:1123–1129. doi: 10.1016/0031-9384(89)90098-x. [DOI] [PubMed] [Google Scholar]
- 17.Potter E, Behan D P, Linton E A, Lowry P J, Sawchenko P E, Vale W W. Proc Natl Acad Sci USA. 1992;89:4192–4196. doi: 10.1073/pnas.89.9.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaughan J, Donaldson C, Bittencourt J, Perrin M H, Lewis K, Sutton S, Chan R, Turnbull A V, Lovejoy D, Rivier C, Rivier J, Sawchenko P E, Vale W. Nature (London) 1995;378:287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- 19.Sutton S W, Behan D P, Sabine L L, Kaiser R, Corrigan A, Lowry P, Potter E, Perrin M H, Rivier J, Vale W. Endocrinology. 1995;136:1097–1102. doi: 10.1210/endo.136.3.7867564. [DOI] [PubMed] [Google Scholar]
- 20.Behan D P, Heinrichs S C, Troncoso J C, Zi-Xin L, Ling N, De Souza E B. Nature (London) 1995;378:284–287. doi: 10.1038/378284a0. [DOI] [PubMed] [Google Scholar]
- 21.Koob G F, Heinrichs S C, Menzaghi F, Pich E M, Britton K T. Semin Neurosci. 1994;6:221–229. [Google Scholar]
- 22.Rivier C, Smith M, Vale W. In: Corticotropin-Releasing Factor: Basic and Clinical Studies of a Neuropeptide. De Souza E B, Nemeroff C B, editors. Boca Raton, FL: CRC; 1990. pp. 175–189. [Google Scholar]
- 23.Lenz H J, Raedler A, Greten H, Brown M R. Am J Physiol. 1987;252:R34–R39. doi: 10.1152/ajpregu.1987.252.1.R34. [DOI] [PubMed] [Google Scholar]
- 24.Malin D H, Lake J R, Newlin-Maultsby P, Roberts L K, Lanier J G, Carter V A, Cunningham J S, Wilson O B. Pharmacol Biochem Behav. 1992;43:779–784. doi: 10.1016/0091-3057(92)90408-8. [DOI] [PubMed] [Google Scholar]
- 25.Malin D H, Lake J R, Carter V A, Cunningham J S, Hebert K M, Conrad D L, Wilson O B. Psychopharmacology. 1994;115:180–184. doi: 10.1007/BF02244770. [DOI] [PubMed] [Google Scholar]
- 26.Levin E D, Briggs S J, Christopher N C, Rose J E. Pharmacol Biochem Behav. 1993;44:51–61. doi: 10.1016/0091-3057(93)90280-7. [DOI] [PubMed] [Google Scholar]
- 27.Winders S E, Grunberg N E. Life Sci. 1990;46:1523–1530. doi: 10.1016/0024-3205(90)90425-q. [DOI] [PubMed] [Google Scholar]
- 28.Winders S E, Dykstra T, Coday M C, Amos J C, Wilson M R, Wilkins D R. Psychopharmacology. 1992;108:501–506. doi: 10.1007/BF02247428. [DOI] [PubMed] [Google Scholar]
- 29.Flynn F W, Webster M, Ksir C. Behav Neurosci. 1989;103:356–364. doi: 10.1037//0735-7044.103.2.356. [DOI] [PubMed] [Google Scholar]
- 30.Yang C-Y, Wu W-H, Zbuzek V K. Psychopharmacology. 1992;106:417–420. doi: 10.1007/BF02245428. [DOI] [PubMed] [Google Scholar]
- 31.Hariharan M, VanNoord T, Greden J F. Clin Chem. 1988;34:724–729. [PubMed] [Google Scholar]
- 32.Behan D P, Potter E, Sutton S, Fischer W, Lowry P J, Vale W W. Ann NY Acad Sci. 1993;697:1–8. doi: 10.1111/j.1749-6632.1993.tb49918.x. [DOI] [PubMed] [Google Scholar]
- 33.Brady L S, Smith M A, Gold P W, Herkenham M. Neuroendocrinology. 1990;52:441–447. doi: 10.1159/000125626. [DOI] [PubMed] [Google Scholar]
- 34.Arase K, Shargill N S, Bray G A. Am J Physiol. 1989;256:R751–R756. doi: 10.1152/ajpregu.1989.256.3.R751. [DOI] [PubMed] [Google Scholar]
- 35.Pelleymounter M A, Cullen M J, Baker M B, Hecht R, Winters D, Boone T, Collins F. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 36.Hall S M, McGee R, Tunstall C, Duffy J, Benowitz N. J Consult Clin Psychol. 1989;57:81–86. doi: 10.1037//0022-006x.57.1.81. [DOI] [PubMed] [Google Scholar]
- 37.Arase K, York D A, Shimizu H, Shargill N, Bray G A. Am J Physiol. 1988;255:E255–E259. doi: 10.1152/ajpendo.1988.255.3.E255. [DOI] [PubMed] [Google Scholar]
- 38.Arase K, Shargill N S, Bray G A. Physiol Behav. 1989;45:565–570. doi: 10.1016/0031-9384(89)90074-7. [DOI] [PubMed] [Google Scholar]
- 39.Chalmers D T, Lovenberg T W, De Souza E B. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bray G A, York D A. Physiol Rev. 1979;59:719–809. doi: 10.1152/physrev.1979.59.3.719. [DOI] [PubMed] [Google Scholar]
- 41.Donaldson C J, Sutton S W, Perrin M H, Corrigan A Z, Lewis K A, Rivier J E, Vaughan J M, Vale W W. Endocrinology. 1996;137:2167–2170. doi: 10.1210/endo.137.5.8612563. [DOI] [PubMed] [Google Scholar]
- 42.Behan D P, Khongsaly O, Ling N, De Souza E B. Brain Res. 1996;725:263–267. doi: 10.1016/0006-8993(96)00347-2. [DOI] [PubMed] [Google Scholar]
- 43.Brown M R, Gray T S, Fisher L A. Regul Pept. 1986;16:321–329. doi: 10.1016/0167-0115(86)90032-7. [DOI] [PubMed] [Google Scholar]
- 44.Vale W, Spiess J, Rivier C, Rivier J. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 45.Milner T A, Reis D J, Pickel V M, Aicher S A, Giuliano R. J Comp Neurol. 1993;8:151–167. doi: 10.1002/cne.903330203. [DOI] [PubMed] [Google Scholar]