

Abstract
The leukocyte-enriched p110γ and p110δ isoforms of PI3K have been shown to control in vitro degranulation of mast cells induced by cross-linking of the high affinity receptor of IgE (FcεRI). However, the relative contribution of these PI3K isoforms in IgE-dependent allergic responses in vivo is controversial. A side-by-side comparative analysis of the role of p110γ and p110δ in mast cell function, using genetic approaches and newly developed isoform-selective pharmacologic inhibitors, confirms that both PI3K isoforms play an important role in FcεRI-activated mast cell degranulation in vitro. In vivo, however, only p110δ was found to be required for optimal IgE/Ag-dependent hypersensitivity responses in mice. These observations identify p110δ as a key therapeutic target among PI3K isoforms for allergy- and mast cell-related diseases.
Mast cell activation is pivotal in the allergic cascade. Ag-dependent aggregation of the high affinity receptor for IgG (FcεRI) on mast cells leads to the activation of an intracellular signaling cascade that culminates in secretory granule exocytosis and allergic responses in vivo (1-3). PI3Ks, a group of signal transduction enzymes that produce intracellular lipid second messengers, have been implicated in signaling through the FcεRI and various other receptors in mast cells (4). The exact role of PI3K activation downstream of the FcεRI remains unclear. Most likely, PI3K action is involved in the assembly of a “signalosome” complex, which promotes, among other events, calcium mobilization and activation of protein kinase C, which together lead to mast cell exocytosis (4-8).
Mammals have eight isoforms of PI3K (5, 9). The subset of PI3K enzymes that are acutely activated by membrane-bound receptors are known as the class I PI3Ks. Of these, the class IA PI3Ks signal downstream of tyrosine (Tyr) kinases and consist of a p110 catalytic subunit (p110α, p110β, or p110δ) complexed to one of five regulatory subunits (collectively known as “p85s”). The p85s have SH2 domains, which allow the p85/p110 complex to become recruited to phospho-Tyr residues upon activation of Tyr kinase signaling.
In contrast, p110γ, the only class IB PI3K, signals downstream of G protein-coupled receptors (GPCRs).4 p110γ forms a heterodimer either with p101 or p84/p87, highly homologous regulatory subunits which are unrelated to p85 (9-11). Whereas p110α and p110β are widely distributed, p110γ and p110δ are enriched in leukocytes (12-14). Combined with the fact that mice with loss-of-function of p110γ or p110δ are viable (15), immunological studies have initially focused on these isoforms of PI3K (16).
Cross-linking of the FcεRI by multivalent Ag is known to activate a Tyr kinase signaling cascade, which provides a direct molecular link to class IA PI3K signaling (4, 8). Genetic or pharmacological inactivation of p110δ has been shown to lead to a substantial, but not complete, block in the allergic responses in mice (3, 17, 18). Surprisingly, genetic inactivation of p110γ in mice has been reported to lead to a complete block in passive cutaneous and systemic anaphylaxis responses in vivo (19). This is remarkable, given that the FcεRI Tyr kinase signaling pathway does not appear to provide a direct molecular link to this GPCR-coupled PI3K. Evidence has been presented for p110γ being part of an auto/paracrine mechanism whereby exocytosed mast cell-derived GPCR agonists, initially released by an FcεRI-dependent pathway, promote hyperactivation of mast cells through GPCR signaling to overcome inhibition by the lipid phosphatases SHIP and PTEN, which antagonize PI3K signaling (11, 19).
Differences in experimental procedures, especially when using model organisms such as mice, often make it difficult to directly compare data from different laboratories. We have therefore directly compared side-by-side the roles of the p110γ and p110δ isoforms of PI3K in mast cell signaling in vitro and in the allergic immune response in vivo. For this, we have used PI3K mutant mice on the same genetic background, as well as a panel of newly developed small molecule inhibitors against PI3K isoforms (20-22). We find that in vitro, both p110γ and p110δ are important for IgE/Ag-dependent mast cell activation. In vivo, however, IgE/Ag-triggered allergic responses appear to a large extent driven by p110δ and are not dependent on p110γ. These findings have implications for the ongoing development of small molecule-PI3K inhibitors for allergy and inflammation.
Materials and Methods
Mice
Mice in which p110γ or p110δ have been inactivated have been described previously (23, 24). Mice were backcrossed onto a C57BL/6 genetic background for >10 generations. Age-matched, 6–10-wk-old mice were used for all experiments. C57BL/6 mice (Harlan, U.K.) were used for pharmacological experiments. All protocols involving live animals were approved by the United Kingdom Home Office and local ethical review committee.
Small molecule inhibitors
Compounds used were: TGX-155 (p110β-selective; (25, 26)), IC87114 (p110δ-selective; (27)), and AS-605240, AS-604850 (22) and AS-252424 (21) (p110γ-selective). Compound(s) or vehicle (0.5% carboxy-methyl-cellulose in 0.25% Tween 20 (Sigma-Aldrich)) were administered per os 1 h before Ag challenge. PI3K inhibitors were tested at 30 mg/kg and administered 1 h before Ag challenge.
Mast cell culture
Mast cell precursors were isolated from bone marrow of 6-wk-old C57BL/6 male mice, as described (17), and maintained in RPMI 1640 medium (Invitrogen Life Technologies) containing 10% ultra-low IgG FBS (Invitrogen Life Technologies), penicillin and streptavidin, glutamine and 20 ng/ml recombinant mouse stem cell factor (SCF), and 20 ng/ml IL-3 (PeproTech) for at least 4 wk and with culture times not exceeding 8 wk. Expression of FcεRI and Kit were confirmed by flow cytometry as described (17).
Assessment of Akt/protein kinase B (PKB) phosphorylation in mast cells in vitro
For stimulations with adenosine or SCF, cells were starved for 3 h in serum- and cytokine-free medium. Cells (2.5 × 106 cells in a 1 ml volume) were then treated with compound or 0.5% DMSO for 15 min, followed by stimulation with SCF (20 ng/ml, 5 min) or adenosine (300 nM, 1 min). Cell stimulation was terminated by the addition of 2× Laemmli electrophoresis buffer followed by assessment of Akt/PKB phosphorylation by western blot using anti-phospho-Ser473 Akt/PKB Ab (Cell Signaling Technology) as described (17). For Ag stimulation, mast cells were sensitized overnight by incubation with 0.1 μg/ml IgE-DNP (Clone SPE-7; Sigma-Aldrich) at 37°C and challenged with DNP (30 ng/ml) the next day for the indicated periods of time.
In vitro cell adhesion of mast cells
A total of 80 μl of a mast cells suspension (6.25 × 105 cells/ml Tyrode's buffer (10 mM HEPES (pH 7.4), 130 mM NaCl, 6.2 mM D-glucose, 5.0 mM KCl, 1.4 mM CaCl2, 1.0 mM MgCl2, and 0.1% BSA)) was incubated on prewarmed fibronectin-precoated 96-well plates (human fibronectin Multiwell; BD Biosciences) containing 10 μl of inhibitor solution or 0.1% DMSO per well. To stimulate cell adhesion, 10 μl of a 200 ng/ml solution of SCF in Tyrode's buffer was added and cells were incubated at 37°C for 30 min. After washing 3 times with Tyrode's buffer to remove nonadherent cells, the adherent cells were lysed in 100 μl of Tyrode's buffer containing 0.5% Triton X-100, followed by quantification of β-hexosaminidase content as described below. Cell adhesion was expressed as the % of adhesion induced by stimulation with PMA (10 ng/ml, 60 min) in adjacent wells.
In vitro mast cell degranulation
Mast cells were sensitized overnight by incubation with 0.1 μg/ml IgE-DNP (Clone SPE-7; Sigma-Aldrich) at 37°C. The next day, cells were resuspended in Tyrode's buffer at 2 × 106 cells/ml. 105 cells were plated in 96-well plates, preincubated for 20 min with inhibitor or 0.1% DMSO, followed by stimulation for 20 min with 30 ng/ml DNP-human serum albumin (HSA) (Sigma-Aldrich), in a final volume of 100 μl following. Cell supernatant and cellular pellets were harvested by 5 min centrifugation at 1500 rpm. To measure β-hexosaminidase activity, 50 μl of supernatant or cell pellet (lysed in 100 μl of Tyrode's buffer containing 0.5% Triton X-100) were transferred to 96-well flat-bottom plates containing 50 μl of 3.7 mM p-nitrophenol-N-acetyl-β-D-glucosaminide (Sigma-Aldrich) in 100 mM Na-acetate (pH 4.5) and further incubated for 1 h at 37°C. Reaction was stopped by addition of 100 μl of 2 M NaOH, followed by measurement of absorbance at 405 nm.
Passive cutaneous anaphylaxis (PCA)
Mice were lightly anesthetized with isoflourane/oxygen in an anesthesia chamber, followed by intradermal (i.d.) injection into the pinnea of the ear. For each experimental mouse, 20 μl PBS or 50 ng anti-DNP IgE (SPE-7; Sigma-Aldrich) in 20 μl PBS were injected in the right and left ear, respectively, followed 24 h later by an i.v. injection of 100 μg DNP-HSA (Sigma-Aldrich) in 100 μl 0.5% (w/v) Evans blue dye in PBS (Evans blue was coinjected to allow visualization of vascular leakage as a measure of increased vascular permeability). Thirty minutes after the i.v. injection, the mice were sacrificed in a CO2 asphyxiation chamber. Tissue sections around the i.d. injection site were excised with a sample corer, followed by weighing and extraction of the extravasated Evans blue by incubation in 200 μl formamide at 55°C for 24 h and measurement of absorbance at 620 nm (OD620 nm). Data are expressed as OD620 nm = absorbance (at 620 nm) of IgE-injected skin biopsy minus absorbance (at 620 nm) of PBS-injected skin biopsy.
Vascular permeability assay
The procedure to determine vascular permeability was similar to that of the PCA assay. Following i.v. injection of 100 μl 0.5% Evans blue in saline, the ears were injected i.d. 1 hr later either with 20 μl volume of PBS, adenosine (5 × 10−3 M; Sigma-Aldrich), histamine (5 ng/ml; Sigma-Aldrich), or mast cell extract (obtained by sonicating 2 × 107 4-wk-old bone marrow-derived mast cells (BMMCs) (cultured as in Ref. 17) in 2 ml of ice-cold PBS). Thirty minutes later, animals were sacrificed in a CO2 asphyxiation chamber and tissue biopsies taken and processed as described above. Data are expressed as OD620 nm = absorbance (at 620 nm) of histamine/mast cell extract skin biopsy minus absorbance (at 620 nm) of PBS-injected skin biopsy.
Statistical analysis
Results from in vivo experiments (mean) were assessed using a nonparametric Mann-Whitney U test with results of analysis and animal numbers presented in the relevant figure legends. The differences between wild-type (WT) and mutant animals or untreated and treated groups were statistically not significant if p > 0.05 (labeled as n.s.), significant if p < 0.05 (*), very significant if p < 0.01 (**), and extremely significant if p < 0.001 (***). In vitro data were analyzed by nonparametric t test. GraphPad Prism software was used for all statistical analysis.
Results
Mouse lines used in this study were as follows. Mice which lack expression of p110γ as a consequence of gene deletion/knockout (KO) are referred to as γKO (23). Mice expressing a germline mutation encoding a kinase-dead version of p110δ (p110δD910A) are referred to as δD910A (24). Both mouse lines were backcrossed onto the C57BL/6 genetic background for >10 generations. For genetic studies, the WT control mice were derived from inter-crosses of mice heterozygous for the p110 mutations. C57BL/6 WT mice from commercial breeders were used for pharmacological experiments. Isoform-selective PI3K inhibitors and their IC50 for the different PI3Ks are listed in Table I. In vivo doses for each inhibitor were established previously taking into account pharmacokinetic profiles (data not shown).
Table I.
In vitro IC50 of compounds for inhibition of class I PI3K isoformsa
| In Vitro IC50 (μM) |
||||
|---|---|---|---|---|
| p110α | p110β | p110δ | p110γ | |
| IC87114 | >100 | 1.82 | 0.07 | 1.24 |
| AS-605240 | 0.06 | 0.27 | 0.3 | 0.008 |
| AS-252424 | 0.94 | >20 | >20 | 0.03 |
| AS-604850 | 3.4 | >20 | >20 | 0.19 |
| TGX-155 | >20 | 0.03 | 0.34 | >20 |
| LY294002 | 0.7 | 0.306 | 1.33 | 7.26 |
p110δ activity is important for the development or maintenance of tissue site-specific mast cell population(s)
We previously reported that genetic inactivation of p110δ leads to a reduction in mast cell numbers in specific tissues, such as the dermis of the ear and the submucosal and muscularis layers of the stomach (17). Mast cell numbers in other tissues, such as the dermis of the back and the mucosa layer of the stomach, were unaffected ((17); Fig. 1A). We have now also assessed the impact of p110γ deletion on mast cell numbers and found comparable mast cell numbers in γKO and WT mice at all anatomical sites assessed, in line with previously published data on a more limited set of tissues (19). Only the dermis of the back skin showed a minor reduction (∼22%) of toluidine blue-positive mast cells in p110γKO mice (Fig. 1A). These data show that p110δ, unlike p110γ, has an impact on mast cell differentiation, which should be taken into account when interpreting studies using δD910A mice.
FIGURE 1.
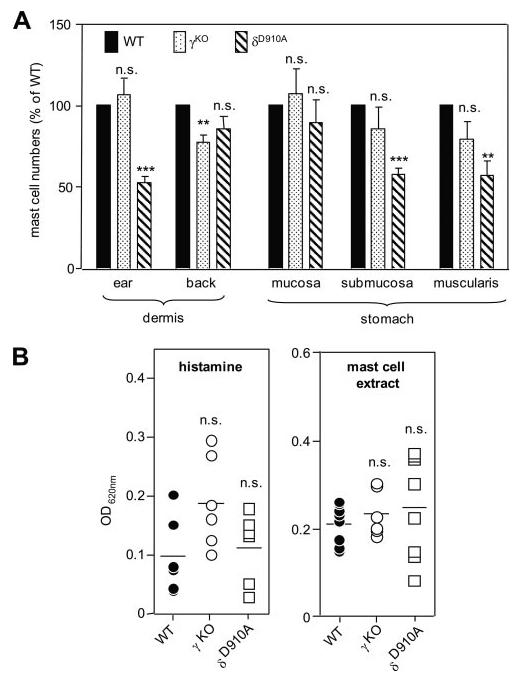
Impact of genetic inactivation of p110γ or p110δ on mast cell numbers and vascular permeability responses in vivo. A, Mast numbers in different anatomical locations. Depending on the anatomical location, the units for counting were as follows: ear dermis: per 5 mm length, beginning at the ear tip; stomach: per one complete sagittal section; and back dermis: unit is per 10 high power fields (×40 objective = ×400 magnification). Exclusively, dermal mast cells (those found among dermal collagen) were counted for each mouse; s.c. mast cells (those found among fat cells of the subcutis) were not counted, as thickness of the s.c. fat can vary greatly among animals depending on body condition. Data are presented as mast cell numbers expressed as % of WT (n = 5 for all genotypes). The mast cell distribution in δD910A mice has been published previously (17) and is presented here for comparative purposes. B, Effect of vasoactive stimuli on vascular leakage in PI3K mutant mice. Numbers of mice used were as follows: histamine: WT, γKO, and δD910A, n = 6 each; and mast cell extract: WT, n = 8; γKO, n = 6; and δD910A, n = 8.
Inactivation of p110γ or p110δ does not affect vascular responsiveness to proinflammatory stimuli
Recently, evidence has been presented for the presence of p110γ and p110δ in endothelial cells and vascular smooth muscle cells (28-31). Given that allergic responses in p110γ and p110δ mutant mice have been assessed by leakage of Evans blue out of the vessels (17, 19), it is not clear to what extent altered vascular responsiveness of PI3K mutant mice may have contributed to the observed reduced allergic responses in these mice.
To gain insight into this question, we tested the direct effect of vasoactive compounds on vascular permeability in mutant mice, again using leakage of Evans blue dye into the surrounding tissue as a read-out. Injection of histamine led to a robust increase in vascular permeability that was similar in all genotypes (Fig. 1B; note that the tendency for increased responsiveness of γKO mice is statistically not significant). Vascular permeability responses to mast cell extracts (prepared by sonication of BMMCs, as a model for a more complex agonist) were also similar in WT, γKO, and δD910A mice (Fig. 1B). Taken together, these data show an intact responsiveness of the vasculature to inflammatory stimuli upon systemic inactivation of p110γ or p110δ.
Distinct roles for p110γ and p110δ in adenosine signaling in mast cells
In line with a previous report (19), we find that adenosine-stimulated phosphorylation of Akt, a surrogate marker of PI3K activity, is abrogated in γKO BMMCs (Fig. 2A). In agreement with this observation, adenosine-induced Akt/PKB phosphorylation was very sensitive to pharmacological inhibition of p110γ, with an IC50 for AS-252424 of 85 nM, as compared with 3.6 μM for the p110δ inhibitor IC87114 (Fig. 2A).
FIGURE 2.
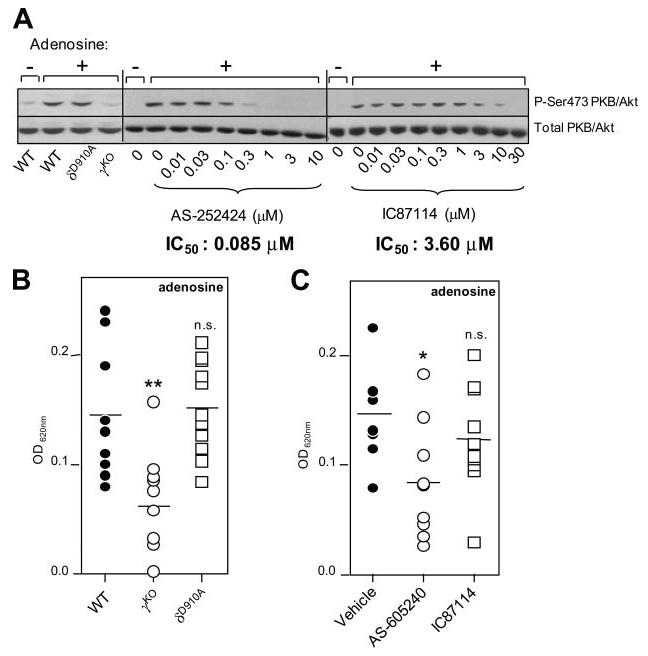
Effect of p110γ or p110δ inhibition on adenosine-dependent Akt/PKB phosphorylation in mast cells and on adenosine-dependent vascular permeability. A (Left panel), γKO and δD910A BMMCs were stimulated with adenosine or vehicle (control) and Akt/PKB phosphorylation assessed by Western blotting, as described in Materials and Methods. A representative blot of two independent experiments is shown. Middle and right panels, BMMCs were pretreated for 30 min with varying concentrations of inhibitors, followed by adenosine stimulation for 1 min and immunoblotted for Akt/PKB (phospho-Ser473 or total). IC50 values were determined by ratiometric analysis of immunoblots, for which Akt/PKB phosphorylation was calculated as the ratio between phosphorylated Akt/PKB and total Akt/PKB for each lane and expressed as the percentage of Akt/PKB phosphorylation in the absence of inhibitor (data not shown). A representative immunoblot of three independent experiments is shown. B, Impact of genetic inactivation of p110γ or p110δ on adenosine-induced PCA response in vivo. Number of mice used: WT and γKO, n = 10 each; and δD910A, n = 11. C, Impact of pharmacological inactivation of p110γ or p110δ on adenosine-induced PCA response in vivo. Number of WT mice dosed with AS605240, n = 9 or IC87114, n = 9.
We next assessed the in vivo impact of PI3K deficiency on adenosine-stimulated mast cell-dependent vascular permeability. Adenosine-stimulated increases in vascular permeability have been reported to be mast cell-dependent (32), and γKO mice have been reported to be completely resistant to adenosine-stimulated increases in vascular permeability (19). Using a similar protocol as was used in Ref. 19, we found a severe, but not complete, reduction in adenosine-stimulated vascular permeability upon genetic or pharmacological inactivation of p110γ (Fig. 2B). δD910A mice (Fig. 2A) and WT mice treated with the p110δ-selective inhibitor IC87114 (Fig. 2C) remained sensitive to this type of stimulation. The observation that IC87114, at the doses tested in these experiments, did not affect the adenosine response suggests that IC87114 has no off-target effects on p110γ under these conditions in vivo. Together with the in vitro data described above, these data confirm that p110γ plays an important role in adenosine-stimulated vascular permeability.
Distinct roles for p110γ and p110δ in Kit receptor signaling in mast cells
We have previously shown that p110δ is the main source of PI3K activity downstream of the activated Kit Tyr kinase receptor for SCF and largely controls SCF-stimulated proliferation, migration, and adhesion (17). SCF can also potentiate FcεRI-activated mast cell degranulation, a response which can be attenuated by the p110δ-selective inhibitor IC87114 (17). Indeed, SCF-stimulated Akt/PKB phosphorylation is very sensitive to IC87114 (IC50 of 0.27 μM) compared with the p110γ-selective compound AS-252424 (IC50 of 4.29 μM) (Fig. 3A). These data confirm and extend our previous data on the critical role of p110δ in SCF/Kit signaling in BMMCs (17). This is further corroborated by the blockade of SCF-induced mast cell adhesion upon genetic (Fig. 3B) or pharmacological (Fig. 3C) inactivation of p110δ. This biological response is refractory to genetic or pharmacological blockade of p110γ. These data further demonstrate the functional distinction which can exist between different PI3K isoforms in a specific biological response.
FIGURE 3.
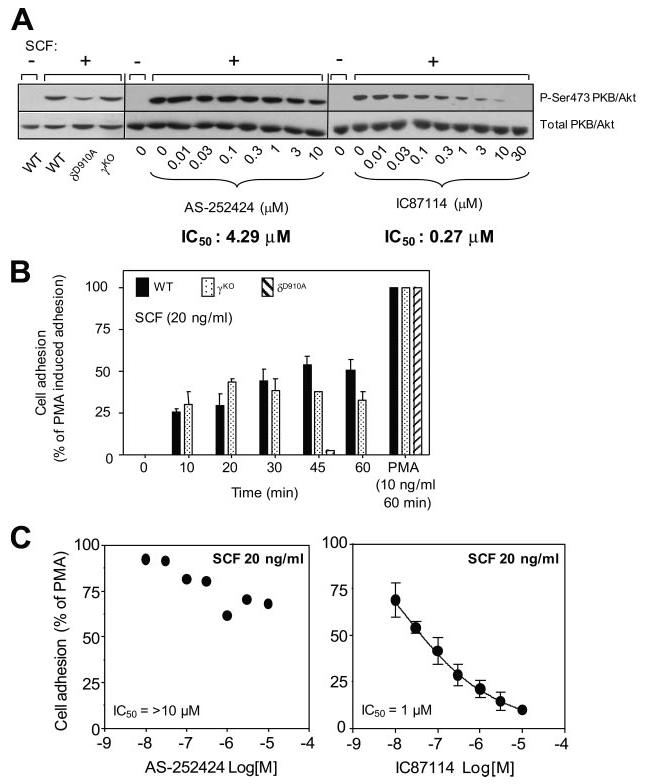
Effect of p110γ or p110δ inhibition on SCF-dependent Akt/PKB phosphorylation and adhesion of mast cells. A, (Left panel) γKO and δD910A BMMCs were stimulated with SCF or vehicle (control) and Akt/PKB phosphorylation assessed by western blotting as described in Materials and Methods. A representative blot of two independent experiments is shown. (Middle and right panels), BMMCs were pretreated for 30 min with varying concentrations of inhibitors, followed by stimulation with SCF for 5 min and immunoblotted for Akt/PKB (phospho-Ser473 or total). IC50 values were determined by ratiometric analysis of immunoblots (data not shown), as described in the legend to Fig. 2. A representative immunoblot of three independent experiments is shown. B, Impact of genetic inactivation of PI3K isoforms on SCF-dependent mast cell adhesion. The experiment shown is representative of five independent experiments. C, Impact of pharmacologic inactivation of PI3K isoforms on SCF-dependent mast cell adhesion. Graphs show data from a representative experiment done at least three (AS-2252424) or two (IC87114) times, with identical results.
Both p110γ and p110δ play important roles in FcεRI-driven mast cell degranulation in vitro
Reduced IgE/Ag-induced degranulation upon genetic or pharmacological inactivation of p110δ, or genetic inactivation of p110γ, has been reported in separate studies (17, 19). We have now tested BMMCs under the same experimental conditions and also used newly developed inhibitors against p110γ (21, 22) (Table I). We confirm that genetic inactivation of p110γ or p110δ impairs in vitro degranulation (Fig. 4A) and show that acute PI3K inactivation using isoform-selective inhibitors mirrors this response (Fig. 4B).
FIGURE 4.
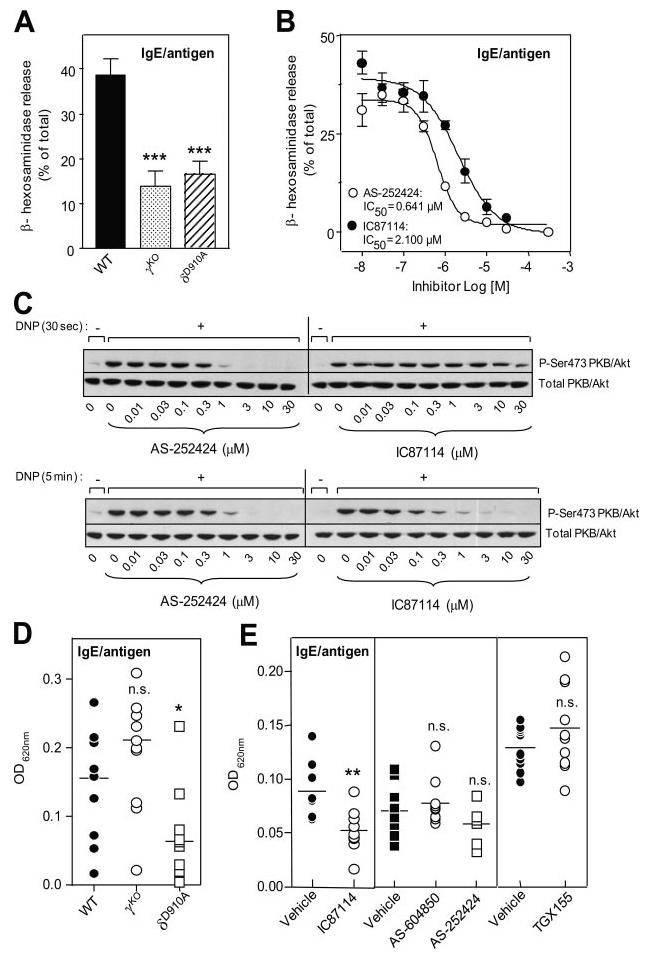
Effect of p110γ or p110δ inhibition on IgE-dependent in vitro mast cell degranulation, Akt/PKB phosphorylation, and PCA response in vivo. A, Effect of genetic inactivation of p110γ or p110δ on IgE/Ag-induced BMMC degranulation in vitro. Graph shows the mean ± SD of the following number of independent experiments: WT, n = 10; δD910A, n = 10; and γKO, n = 8; done in quadruplicates. Mean ± SD spontaneous hexosaminidase release for experiments was as follows: WT, 8.23% ± 1.83 (n = 10); δD910A, 11.88% ± 1.9 (n = 10); and γKO, 8.0% ± 1.4 (n = 8). B, Effect of isoform-selective PI3K inhibitors on IgE/Ag-induced BMMC degranulation in vitro. Graph shows data from two independent experiments ± SEM. C, Impact of PI3K inhibitors on IgE/Ag-induced Akt/PKB phosphorylation upon 30-s (top panel) and 5-min (bottom panel) stimulation of IgE-sensitized mast cells with Ag. A representative blot from three independent experiments is shown. D, PCA response of WT and gene-targeted mice (WT, γKO, and δD910A; n = 11 each). E, PCA responses of WT mice treated with PI3K inhibitors. Numbers of mice used were as follows: left panel: IC87114 (p110δ inhibitor): vehicle, n = 8 and IC87114, n = 9; middle panel: AS-604850 and AS-252424 (p110γ inhibitors): vehicle, n = 9, AS-604850, n = 10, and AS-252424, n = 8; and right panel: TGX-155 (p110β inhibitor): vehicle, n = 10 and TGX-155, n = 10.
We next examined the kinetics of IgE/Ag-induced PI3K activation using isoform-selective PI3K inhibitors. Previous genetic studies have suggested that phosphatidylinositol (3,4,5)-triphosphate production, the product of class I PI3K activity, is unaffected in p110γ KO mast cells activated through FcεRI in the absence of any costimulation but is strongly reduced upon costimulation of FcεRI with adenosine (19). Using Akt/PKB phosphorylation as a surrogate marker of PI3K activation, we found that the early phase of PI3K activity (30 s) downstream of activated FcεRI was, surprisingly, refractory to IC87114 inhibition and dependent on p110γ (Fig. 4C, top panel), with an IC50 of 327 nM (data not shown). The later phase (5 min), which remained equally sensitive to AS-252424, became more sensitive to IC87114 (IC50 of 86 nM; Fig. 4C and data not shown). Our findings suggest that PI3K activation downstream of the activated FcεRI in vitro is biphasic, with p110γ being activated before p110δ upon FcεRI engagement.
p110γ, but not p110δ, is dispensable for allergic responsiveness in vivo
Mast cells in vivo are exposed to stimuli from the microenvironment other than Ag which can modulate the FcεRI response, and it is therefore not always possible to extrapolate in vitro observations such as those shown in Fig. 4, A and B, to the organismal context. We therefore tested the in vivo allergic response of γKO and δD910A mice, side-by-side in the same experiment and using mice on the same genetic background (C57BL/6). Mice were sensitized locally (either in the dermis of the ear or the back) by injection of Ag-specific IgE (directed against the hapten DNP) and challenged systemically 24 h later with DNP-HSA (administered together with Evans blue). Thirty minutes later, the mast cell response was quantified by measuring extravasated Evans blue.
In line with our previously published results in δD910A mice on the BALB/c genetic background (17), inactivation of p110δ on the C57BL/6 background led to a significant (∼70%) reduction in IgE/Ag-dependent vascular permeability in the ears of sensitized mice (Fig. 4D). Similar results (∼69% reduction) were observed in the back dermis (data not shown). Surprisingly, γKO mice did not show reduced in vivo allergic responses (Fig. 4D).
To exclude that altered PCA responses in gene-targeted mice are related to developmental defects, we next pharmacologically intervened with PI3K function using isoform-selective PI3K inhibitors. Treatment of WT mice with the p110δ-selective inhibitor IC87114 at doses which do not affect p110γ (as implied by the findings shown in Fig. 2C) consistently diminished the allergic immune response by ∼40% (Fig. 4E). This milder reduction upon pharmacological, compared with genetic, inactivation of p110δ (Fig. 4D) most likely relates to the reduced number of mast cells in the ears of δD910A mice (Fig. 1A), as previously discussed (17), and the notion that IC87114, in contrast to genetic inactivation, is not expected to provide full inhibition of p110δ as is the case in homozygous δD910A mice. In contrast to IC87114, the p110γ-selective compounds AS-604850 and AS-252424 had no significant impact on the allergic response (Fig. 4E), in line with our observations in γKO mice (Fig. 4D). Administration of the p110β-selective compound TGX-155 also did not impact on the acute allergic response (Fig. 4E).
Discussion
In this manuscript, we report that we have found no evidence that p110γ, in isolation, plays a significant role in the in vivo allergic cascade. This appears to be in contradiction with previous work, which suggested that p110γ is essential for and is the only PI3K subunit which drives the in vivo IgE/Ag-triggered allergic response (19). It is possible that the proposed GPCR-driven auto/paracrine signaling amplification mechanism, largely based on in vitro observations on cultured mast cells (19), may not be operational in vivo. This conclusion is in line with the observation that KO mice for A(3), the main adenosine receptor, retain normal IgE/Ag-dependent PCA responses, despite a complete abrogation of adenosine responsiveness (32). Differences in genetic backgrounds of mice could also contribute to the discrepancies between our studies and earlier work (19). Indeed, previous studies in which p110γ function was assessed used mice bred onto the 129sv background, in contrast to our studies in which we used C57BL/6 mice (for p110γ and p110δ; this work) and BALB/c (for p110δ; data shown in Ref. 17). However, why a reduced sensitivity of γKO mice to adenosine would be retained across genetic backgrounds, in contrast to responsiveness to allergic responses, is difficult to explain. For a molecule to have an “essential” role in a process such as allergy, we believe it must have a function across genetic backgrounds, similar as what is observed for p110δ. Other experimental differences to measure the allergic response may also contribute to the observed discrepancies. Indeed, whereas both studies used vascular permeability as a measure of mast cell activation, a different sensitization protocol was applied, namely intradermal local sensitization (PCA; this study) vs i.v. systemic sensitization (passive systemic anaphylaxis; (19)). We have found the i.v. sensitization procedure in passive systemic anaphylaxis experiments to give extremely variable results in WT mice, for reasons unclear to us, but apparently unrelated to age or sex of the mice (data not shown). Other than being more robust, we also believe that the PCA protocol is a more accurate measure of mast cell contribution in allergy, given that it assesses the function of tissue-resident mast cells as the primary targets of the intradermal sensitization step, unlike in systemic sensitization protocols which also sensitize (and in the Ag challenge phase activate) other FcεRI-expressing cells, including basophils and eosinophils.
In this study we show that specific signaling and biological responses are, to a large extent, selectively driven by a single PI3K isoform. This is the case for SCF and adenosine, which are controlled by p110δ and p110γ, respectively. In constrast, the FcεRI enlists both p110γ and p110δ. Kinetic studies measuring FcεRI-associated PI3K activation show that p110γ and p110δ PI3Ks are activated sequentially downstream of the activated FcεRI with p110γ being activated before p110δ. It is puzzling how the FcεRI, which is considered to signal intracellularly mainly through tyrosine kinases (7), activates the GPCR-coupled p110γ so early, even before p110δ. However, despite the apparent importance of p110γ in FcεRI-activated mast cell exocytosis in vitro, our work indicates that this need for p110γ activity does not translate to the in vivo situation, where p110γ appears to be dispensable. It is also possible that the density of mast cells in an in vitro Ag-activated exocytosis experiment may produce a substantially greater concentration of adenosine (or other factor(s)) in the immediate environment than may be seen in vivo where mast cells are more diffusely distributed in the tissues. Furthermore, unlike in tissue culture, adenosine would be rapidly metabolized in vivo. It is also possible that in tissues, agonists other than adenosine may override the necessity for p110γ.
In contrast to p110γ, disruption of p110δ signaling has an inhibitory effect on the allergic response across different genetic backgrounds and in WT mice treated with a p110δ-selective inhibitor. This most likely relates to the fact that blockade of p110δ has effects beyond the inhibition of activated FcεRI. Indeed, p110δ function is critical for signaling through the Kit receptor (Fig. 3), known to potentiate allergic responses in vitro and in vivo (17). Mast cells actively participate in allergy and allergic airway inflammation, and our data provide a partial mechanism for the observation that genetic or pharmacological inactivation of p110δ impairs airway hyperresponsiveness in murine models (3, 18, 33). Unfortunately, despite the availability of several strains of p110γ-deficient mice and small molecule inhibitors to p110γ, there are as yet no published reports to suggest a role for p110γ in allergic airway inflammation.
Intracellularly, class IA PI3Ks couple to the FcεRI via the adaptor protein Gab2, which recruits class IA PI3Ks to the activated FcεRI signaling complex. Deletion of Gab2 in BMMCs has a severe negative impact on both PI3K activation downstream of Kit and FcεRI, and Gab2-deficient mice have an almost complete block in the allergic response (34). This reduction is more severe than that observed in p110δ-deficient mice (17), possibly because Gab2 also binds other class IA PI3Ks, including p110α and p110β. We have previously reported that a high dose of IC87114 (40 μM serum dose) could completely wipe out the PCA response (17). We presumed at the time that this was due to possible off-target effects of this compound on p110γ (17). Our current data show that this is not the case and that other PI3K isoforms, either on their own or in combination, account for the PI3K-dependent fraction of the IgE/Ag-dependent allergic response. Taken together, it is therefore possible that the p110α and p110β isoforms of PI3K together contribute to the residual PI3K-dependent PCA response observed upon p110δ inactivation (Fig. 4, D and E). However, on its own, p110β does not significantly contribute to the PCA response (Fig. 4E). Unfortunately, selectivity of inhibitors for p110α cannot be achieved at present without resulting in many off-target effects, so that the currently available p110α inhibitors also inhibit other relevant kinases including isoforms of protein kinase C (35). Genetic investigation of the role of p110α (and p110β) PI3K isoforms has thus far also been precluded due to the embryonic lethality of homozygous p110α and p110β gene-targeted mice and the incapacity to derive cell lines from these mice (36-38). The creation of mice with conditional p110α and p110β alleles and the development of small molecule inhibitors with higher p110α isoform-selectivity will be critical to gain insight into which other PI3K isoforms may complement p110δ in controlling the IgE/Ag-dependent allergic response.
Acknowledgments
We thank Carol See for genotyping and Klaus Okkenhaug (Babraham Institute, Cambridge) and members of the Cell Signaling Laboratory for critical comments on the manuscript. We thank Emilio Hirsch (Torino University, Torino, Italy) for the p110γ KO mice.
Footnotes
This work was supported by the Biotechnology and Biological Science Research Council U.K. (Grant BB/C505659/1), the European Union FP6-502935, Barts and the London Charity, and the Ludwig Institute for Cancer Research.
Abbreviations used in this paper: GPCR, G protein-coupled receptor; BMMC, bone marrow-derived mast cell; HSA, human serum albumin; i.d., intradermal; KO, knockout; PCA, passive cutaneous anaphylaxis; SCF, stem cell factor; WT, wild type; Tyr, tyrosine; PKB, protein kinase B.
Disclosures
Bart Vanhaesebroeck is a consultant for PIramed (Slough, U.K.) and Montserrat Camps, Hong Ji, Thomas Rückle, Christian Chabert, and Christian Rommel are employees of MerckSerono (Geneva, Switzerland). Christian Rommel is an employee of Intellikine (La Jolla, CA).
References
- 1.Boyce JA. The biology of the mast cell. Allergy Asthma Proc. 2004;25:27–30. [PubMed] [Google Scholar]
- 2.Wedemeyer J, Galli SJ. Mast cells and basophils in acquired immunity. Br. Med. Bull. 2000;56:936–955. doi: 10.1258/0007142001903616. [DOI] [PubMed] [Google Scholar]
- 3.Nashed BF, Zhang T, Al-Alwan M, Srinivasan G, Halayko AJ, Okkenhaug K, Vanhaesebroeck B, Hayglass KT, Marshall AJ. Role of the phosphoinositide 3-kinase p110δ in generation of type 2 cytokine responses and allergic airway inflammation. Eur. J. Immunol. 2007;37:416–424. doi: 10.1002/eji.200636401. [DOI] [PubMed] [Google Scholar]
- 4.Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 2006;6:218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- 5.Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu. Rev. Immunol. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- 6.Blank U, Rivera J. The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 2004;25:266–273. doi: 10.1016/j.it.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Rivera J, Gilfillan AM. Molecular regulation of mast cell activation. J. Allergy Clin. Immunol. 2006;117:1214–1225. doi: 10.1016/j.jaci.2006.04.015. quiz 1226. [DOI] [PubMed] [Google Scholar]
- 8.Okkenhaug K, Ali K, Vanhaesebroeck B. Antigen receptor signalling: a distinctive role for the p110δ isoform of PI3K. Trends Immunol. 2007;28:80–87. doi: 10.1016/j.it.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem. Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 11.Wymann MP, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr. Opin. Cell Biol. 2005;17:141–149. doi: 10.1016/j.ceb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, et al. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- 13.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J, Waterfield MD. P110δ, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA. 1997;94:4330–4335. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, Cooper JA, Hoekstra MF. p110δ, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J. Biol. Chem. 1997;272:19236–19241. doi: 10.1074/jbc.272.31.19236. [DOI] [PubMed] [Google Scholar]
- 15.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003;3:317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 16.Rommel C, Camps M, Ji H. PI3K δ and PI3K γ: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Rev. Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- 17.Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan AM, Tkaczyk C, Kuehn N, Gray A, Giddings J, Peskett E, et al. Essential role for the p110δ phosphoinositide 3-kinase in the allergic response. Nature. 2004;431:1007–1011. doi: 10.1038/nature02991. [DOI] [PubMed] [Google Scholar]
- 18.Lee KS, Lee HK, Hayflick JS, Lee YC, Puri KD. Inhibition of phosphoinositide 3-kinase δ attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J. 2006;20:455–465. doi: 10.1096/fj.05-5045com. [DOI] [PubMed] [Google Scholar]
- 19.Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, Hirsch E, Wymann MP. Phosphoinositide 3-kinase γ is an essential amplifier of mast cell function. Immunity. 2002;16:441–451. doi: 10.1016/s1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 20.Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE. Essential role of phosphoinositide 3-kinase δ in neutrophil directional movement. J. Immunol. 2003;170:2647–2654. doi: 10.4049/jimmunol.170.5.2647. [DOI] [PubMed] [Google Scholar]
- 21.Pomel V, Klicic J, Covini D, Church DD, Shaw JP, Roulin K, Burgat-Charvillon F, Valognes D, Camps M, Chabert C, et al. Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase γ. J. Med. Chem. 2006;49:3857–3871. doi: 10.1021/jm0601598. [DOI] [PubMed] [Google Scholar]
- 22.Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, et al. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 23.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 24.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, et al. Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science. 2002;297:1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 25.Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, et al. PI 3-kinase p110β: a new target for antithrombotic therapy. Nat. Med. 2005;11:507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 26.Jackson SP. Preparation of morpholinyl- and pyridinyl-substituted heterobicyclic ketones as selective inhibitors of phosphoinositide 3-kinase β for use against thrombosis. 2004. International, WO 2003-IB4177, 2004.
- 27.Sadhu C, Dick K, Tino WT, Staunton DE. Selective role of PI3K δ in neutrophil inflammatory responses. Biochem. Biophys. Res. Commun. 2003;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- 28.Puri KD, Doggett TA, Douangpanya J, Hou Y, Tino WT, Wilson T, Graf T, Clayton E, Turner M, Hayflick JS, Diacovo TG. Mechanisms and implications of phosphoinositide 3-kinase δ in promoting neutrophil trafficking into inflamed tissue. Blood. 2004;103:3448–3456. doi: 10.1182/blood-2003-05-1667. [DOI] [PubMed] [Google Scholar]
- 29.Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG. The role of endothelial PI3Kγ activity in neutrophil trafficking. Blood. 2005;106:150–157. doi: 10.1182/blood-2005-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Northcott CA, Hayflick JS, Watts SW. PI3-kinase upregulation and involvement in spontaneous tone in arteries from DOCA-salt rats: is p110δ the culprit? Hypertension. 2004;43:885–890. doi: 10.1161/01.HYP.0000118518.20331.e8. [DOI] [PubMed] [Google Scholar]
- 31.Vecchione C, Patrucco E, Marino G, Barberis L, Poulet R, Aretini A, Maffei A, Gentile MT, Storto M, Azzolino O, et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kγ. J. Exp. Med. 2005;201:1217–1228. doi: 10.1084/jem.20040995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tilley SL, Wagoner VA, Salvatore CA, Jacobson MA, Koller BH. Adenosine and inosine increase cutaneous vasopermeability by activating A(3) receptors on mast cells. J. Clin. Invest. 2000;105:361–367. doi: 10.1172/JCI8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee KS, Park SJ, Kim SR, Min KH, Jin SM, Puri KD, Lee YC. Phosphoinositide 3-kinase-δ inhibitor reduces vascular permeability in a murine model of asthma. J. Allergy Clin. Immunol. 2006;118:403–409. doi: 10.1016/j.jaci.2006.04.041. [DOI] [PubMed] [Google Scholar]
- 34.Gu H, Saito K, Klaman LD, Shen J, Fleming T, Wang Y, Pratt JC, Lin G, Lim B, Kinet JP, Neel BG. Essential role for Gab2 in the allergic response. Nature. 2001;412:186–190. doi: 10.1038/35084076. [DOI] [PubMed] [Google Scholar]
- 35.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110α subunit of phosphoinositide 3-kinase. J. Biol. Chem. 1999;274:10963–10968. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 37.Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110β catalytic subunit of PI 3-kinase. Mamm. Genome. 2002;13:169–172. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 38.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJ, Withers DJ, Vanhaesebroeck B. Critical role for the p110α phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441:366–370. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 39.Bilancio A, Okkenhaug K, Camps M, Emery JL, Ruckle T, Rommel C, Vanhaesebroeck B. Key role of the p110δ isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110δ function in B cells. Blood. 2006;107:642–650. doi: 10.1182/blood-2005-07-3041. [DOI] [PubMed] [Google Scholar]