

Abstract
A high-fat diet causes activation of the regulatory protein cJun NH2-terminal kinase 1 (JNK1) and triggers the development of insulin resistance. JNK1 is therefore a potential target for therapeutic treatment of metabolic syndrome. We explored the mechanism of JNK1 signaling by engineering mice in which the Jnk1 gene was ablated selectively in adipose tissue. JNK1-deficiency in adipose tissue suppressed high fat diet-induced insulin resistance in the liver. JNK1-dependent secretion of the inflammatory cytokine IL6 by adipose tissue caused increased expression of liver SOCS3, a protein that induces hepatic insulin resistance. Thus, JNK1 activation in adipose tissue can cause insulin resistance in the liver.
Metabolic stress caused by a high-fat diet (HFD) results in activation of the regulatory protein cJun NH2-terminal kinase 1 (JNK1) (1). JNK1 is activated, in part, by increased serum free fatty acids that induce a stress signaling pathway in target tissues (2). JNK1 phosphorylates the adapter protein IRS1 at an inhibitory site that can block signal transduction by the insulin receptor (3). JNK1 may therefore directly induce insulin resistance (4). However, JNK1 may also influence insulin sensitivity indirectly. Thus, JNK1 may act in hematopoietic cells to regulate the expression of cytokines that can influence insulin sensitivity (5). Indeed myeloid cells, including macrophages, may be critical (5).
To test the role of JNK1 in myeloid cells during the development of diet-induced insulin resistance, we examined the phenotype of mice with JNK1-deficiency in myeloid cells (figs. S1,S2) and hematopoietic cells (fig. S3). No significant difference in the response of these JNK1-deficient HFD-fed mice, compared with control HFD-fed mice, was detected in glucose and insulin tolerance tests (figs S2,S3). These data indicate that, although JNK1 in hematopoietic cells may contribute to HFD-induced insulin resistance, other cell types must also participate in the development of insulin resistance. Adiposity is known to influence insulin responsiveness (6) through a mechanism that involves adipose-derived fatty acids and hormones/cytokines (collectively termed “adipokines”) that can modulate insulin sensitivity (7). We tested the role of JNK1 in adipocytes on the regulation of insulin sensitivity.
Mice lacking JNK1 in adipose tissue (FKO) were generated using animals with conditional (floxed) Jnk1 and adipose tissue-specific expression of Cre recombinase (Fabp4-Cre+ Jnk1f/-). Littermates without conditional Jnk1 (Fabp4-Cre+ Jnk1+/-) were used as control mice (FWT). The Jnk1+, Jnk1f, and deleted Jnk1 (ΔJnk1) alleles were detected by PCR amplification of genomic DNA (fig. S1A). Efficient deletion of Jnk1f was detected in the adipose tissue of FKO mice (fig. S1B). In contrast, Jnk1f was not deleted in other tissues of FKO mice, including macrophages (fig. S1C). Quantitative PCR analysis demonstrated that Jnk1 mRNA was markedly reduced in epididymal fat and brown fat of FKO animals (Fig. 1A). Immunoblot analysis confirmed the reduction of JNK1 protein in fat depots from FKO mice, while JNK1 was preserved in liver, muscle, and macrophages (Fig. 1B). JNK1 is activated in mice following exposure to metabolic stress (4). Indeed, we found that JNK1 was activated in the adipose tissue, striated muscle, and liver of HFD-fed FWT mice (Fig. 1C). In contrast, HFD-fed FKO mice exhibited JNK activation in muscle and liver, but not adipose tissue (Fig. 1C). Together, these data indicate that FKO mice are useful for studies of the role of JNK1 in adipose tissue.
Fig. 1.
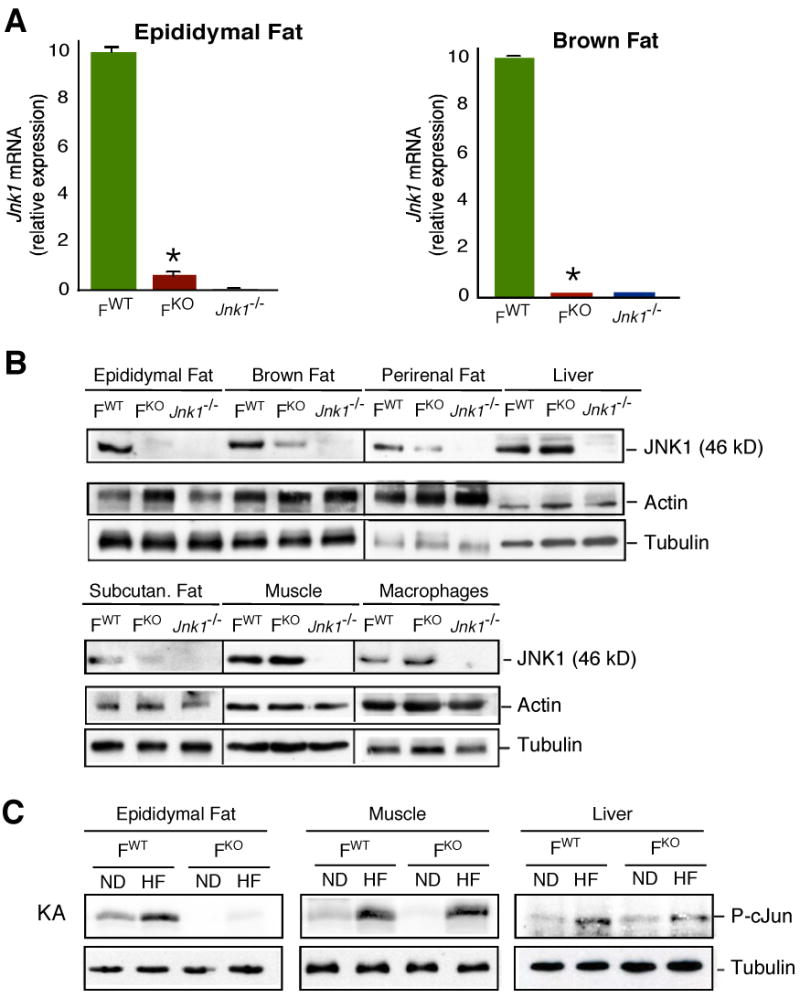
Creation of mice with adipose tissue-specific deficiency of JNK1. (A) The expression of Jnk1 mRNA in adipose tissue was examined by quantitative RT-PCR analysis (Taqman©) and is presented as relative mRNA expression (mean ± SD; n = 5). The data are normalized for the amount of Gapdh mRNA in each sample. The amount of Jnk1 mRNA was significantly reduced in the adipose tissue of FKO mice compared to FWT mice (*, P < 0.01). (B) JNK1 expression in adipose tissue, liver, muscle (quadriceps), and macrophages isolated from FWT, FKO, and Jnk1-/- mice was examined by immunoblot analysis using an antibody to JNK1. Control immunoblots were performed using antibodies to Actin and Tubulin. (C) FKO and FWT mice were maintained on a standard chow diet (ND) or on a high fat diet (HF) for 16 wk. Protein extracts were prepared from epididymal fat, muscle (quadriceps), and liver. Equal amounts of cell extract prepared from FWT and FKO mice, confirmed by immunoblot analysis using an antibody to Tubulin, were used to measure JNK activity in a kinase assay (KA) using ATP[γ-32P] and cJun as substrates.
Comparison of HFD-fed FWT and FKO mice demonstrated that these animals gained similar body mass (fig. S4) and blood lipids (fig. S5), became glucose intolerant (Fig. 2A) with reduced glucose-induced insulin secretion (Fig. 2C), and developed mild fasting hyperglycemia (Fig. 2L). In contrast, when compared to HFD-fed FWT mice, the HFD-fed FKO mice showed improved insulin sensitivity during an insulin tolerance test (Fig. 2B) and reduced hyperinsulinemia (Fig. 2K). We performed a 2-hr hyperinsulinemic-euglycemic clamp study to assess organ-specific glucose metabolism in awake FWT and FKO mice. After 3 weeks of HFD, both groups of mice developed whole body insulin resistance, as indicated by significant reductions in glucose infusion rate and whole body glucose turnover during the clamp (Fig. 2D,E). HFD-fed FWT mice developed insulin resistance in liver, as indicated by increased hepatic glucose production (HGP) during the clamp, but HFD-fed FKO mice remained insulin sensitive in liver (Fig. 2H,I). Basal HGP was not affected by feeding a HFD or by JNK1 deletion in adipose tissue (Fig. 2G). Studies of hepatic gluconeogenesis demonstrated that increased blood glucose caused by pyruvate administration was suppressed in HFD-fed FWT mice, but not HFD-fed FKO mice (fig. S6). No differences in whole body glycolysis or glycogen synthesis were detected between HFD-fed FWT and FKO mice (Fig. 2F,J). Together, these data demonstrate that adipose-specific disruption of the Jnk1 gene prevents diet-induced hepatic insulin resistance.
Fig. 2.

JNK1-deficient adipose tissue prevents diet-induced insulin resistance. (A-C) FKO and FWT mice were maintained on a standard chow diet (ND) or on a high fat diet (HF) for 16 wk. (A) Glucose tolerance test (GTT). Mice fasted overnight were injected intraperitoneally with glucose (1 mg/g). Blood glucose concentration was measured at the indicated times (mean ± SD; n = 14). (B) Insulin tolerance test (ITT). Mice fed ad libitum were injected intraperitoneally with insulin (0.75 mU/g). Blood glucose concentration was measured at the indicated times (mean ± SD; n = 14). (C) Glucose-induced insulin release. Mice fasted overnight were injected intraperitoneally with glucose (2 mg/g). Blood insulin concentration was measured at the indicated times (mean ± SD; n = 14). No statistically significant differences between FKO and FWT mice were detected (P > 0.05). (D-J) FKO and FWT mice were maintained on a standard chow diet or on a high fat (HF) diet for 3 wk. (D) Steady-state glucose infusion rates to maintain euglycemia during the hyperinsulinemic-euglycemic clamps. (E) Insulin-stimulated whole body glucose turnover. (F) Whole body glycolysis. (G) Basal hepatic glucose production (HGP). (H) Insulin-stimulated rates of HGP during clamps. (I) Hepatic insulin action, expressed as insulin-mediated percent suppression of basal HGP. (J) Glycogen synthesis. The data presented are the mean ± SE for 6 to 8 experiments. Statistically significant differences are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (K-L) Resting blood insulin and glucose were examined in mice that were fasted overnight (mean ± SD, n =10). Statistically significant differences between FKO mice and FWT mice are indicated (*, P < 0.01).
To confirm the effect of adipose JNK1-deficiency on insulin sensitivity, we tested insulin-stimulated Ser/Thr phosphorylation and activation of AKT. HFD-fed FWT mice exhibited reduced insulin-stimulated AKT activation in adipose tissue (Fig. 3B), liver (Fig. 4B), and muscle (fig. S7). In contrast, FKO mice exhibited HFD-induced inhibition of insulin-stimulated AKT activation in muscle (fig. S7), but not in adipose tissue or liver (Figs. 3B & 4B). This effect of adipose-specific JNK1-deficiency on hepatic AKT activation (Fig. 4B) is consistent with the observation that HFD-fed FKO mice showed improved hepatic insulin sensitivity compared with HFD-fed FWT mice (Fig. 2I). Interestingly, HFD-fed FKO mice also exhibited reduced hepatic steatosis compared to HFD-fed FWT mice (Fig. 4A). These data confirm that JNK1 in adipose tissue is, at least in part, required for HFD-induced insulin resistance in both adipose tissue and liver.
Fig. 3.
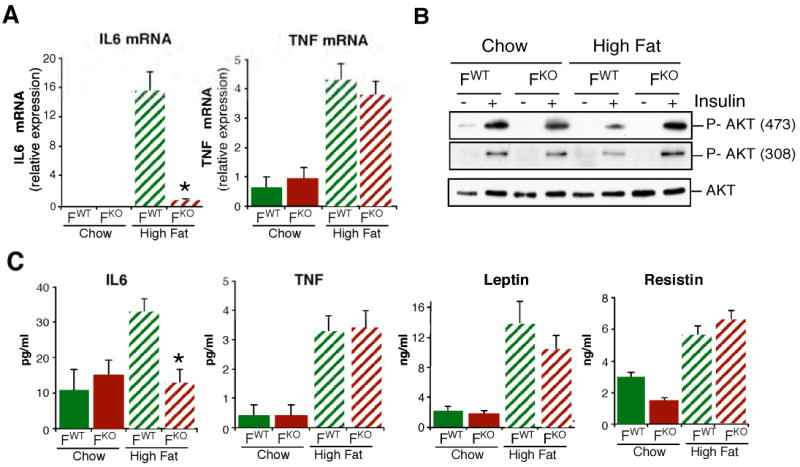
JNK1-deficient adipose tissue causes reduced diet-induced expression of IL6. (A) The expression of IL6 and TNFα mRNA in adipose tissue was examined by quantitative RT-PCR analysis (Taqman©) and is presented as relative mRNA expression (mean ± SD; n = 6). Statistically significant differences between FKO mice and FWT mice are indicated (*, P < 0.01). (B) FWT and FKO mice were fasted overnight and treated with insulin (1.5 U/kg body mass) by intraperitoneal injection. The epididymal fat pads were isolated after 30 min and examined by immunoblot analysis using antibodies to AKT and phospho-AKT. (C) Mice were fasted overnight and the amount of IL6, TNFα, Leptin and Resistin in plasma were measured (mean ± SD; n=10). Statistically significant differences between FKO mice and FWT mice are indicated (*, P < 0.05).
Fig. 4.
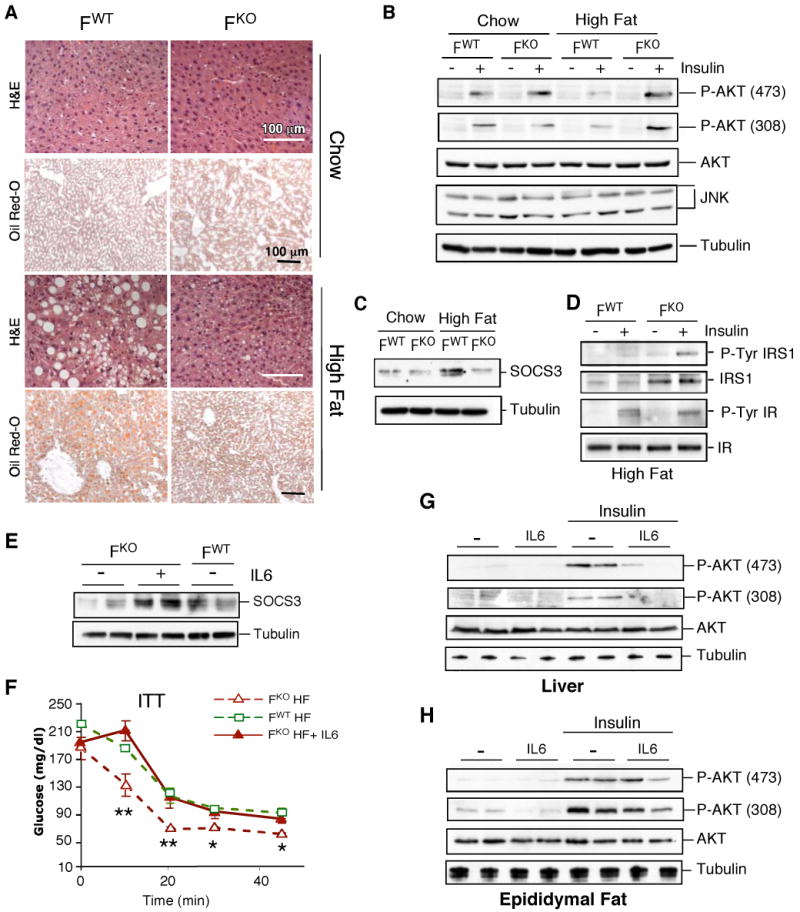
JNK1-deficiency in adipose tissue causes increased hepatic insulin sensitivity. (A) Fabp4-Cre+ Jnk1+/- (FWT) and Fabp4-Cre+ Jnk1f/- (FKO) mice were fed a standard chow diet or a high fat diet for 16 wk. Representative histological sections of liver stained with H&E or Oil Red-O are presented. (B) Mice were fasted overnight and treated with insulin (1.5 U/kg body mass) by intraperitoneal injection. Livers were isolated after 30 min and examined by immunoblot analysis to detect AKT, phospho-AKT, JNK1/2, and Tubulin. (C) The expression of SOCS3 in the liver was examined by immunoblot analysis. Control immunoblots were performed using an antibody to Tubulin. (D) The tyrosine phosphorylation and expression of the insulin receptor (IR) and IRS1 in the liver were examined by immunoblot analysis. (E) FWT and FKO mice were fed a HFD (16 wks). FKO mice were treated with IL6 (1μg/kg) by sub-cutaneous injection. At 90 mins post-injection, the blood IL6 concentration was 44 ± 9.1 pg/ml (mean ± SD; n = 8), the liver was isolated, and the expression of SOCS3 and Tubulin were examined by immunoblot analysis. (F) Insulin tolerance test (ITT). Mice were treated by sub-cutaneous injection with IL6 (1μg/kg) and then treated (after 90 mins) with insulin (0.75 mU/g) by intraperitoneal injection. Blood glucose concentration was measured at the indicated times. Statistically significant differences between FKO and FWT mice are indicated (*, P < 0.05; **, P < 0.01). (G,H) HFD-fed FKO mice were treated by sub-cutaneous injection with IL6 (1μg/kg) and then treated (after 90 mins) with insulin (0.3 mU/g) by intravenous injection. The liver (G) and epididymal fat pads (H) were isolated after 5 mins and examined by immunoblot analysis to detect AKT, phospho-AKT, and Tubulin.
The total fat mass, weight of the epididymal fat pads, and the size of adipocytes were not significantly different between HFD-fed FWT and FKO mice (fig. S4). The HFD increased Tnfα and Il6 mRNA expression in adipose tissue of FWT mice, but only increased Tnfα mRNA expression was detected in FKO mice (Fig. 3A). Moreover, the HFD caused a similar increase in the serum concentration of TNFα in FWT and FKO mice, but increased serum IL6 was only detected in FWT mice (Fig. 3C). Thus, JNK1-deficiency in adipocytes prevented the HFD-induced increase in the expression of the inflammatory cytokine IL6. This effect on IL6 expression was selective because no significant differences in circulating leptin or resistin concentrations were detected between FWT and FKO mice (Fig. 3C). Furthermore, no significant differences in the serum concentration of other interleukins and adipokines were detected between FWT and FKO mice (figs. S8-S10). The inflammatory cytokines TNFα and IL6 can cause insulin resistance (8, 9), and JNK can regulate the expression of both cytokines (4). However, JNK1-deficiency in adipose tissue selectively prevented HFD-induced IL6 expression (Fig. 3A,C). This finding suggests that adipocytes play a primary role in obesity-induced IL6 expression (10). In contrast, macrophages may represent the major source of TNFα expression (11). No differences in macrophage infiltration of the liver and adipose tissue were detected between HFD-fed FWT and FKO mice (figs. S6E,S9D,S11). Moreover, no defects in IL6 or TNFα expression by macrophages isolated from FKO mice were detected (fig. S12).
IL6 can induce hepatic insulin resistance (12, 13), and loss of IL6 selectively improves hepatic insulin action in obese mice (14). IL6-induced hepatic insulin resistance is mediated, in part, by increased expression of SOCS3 (15,16), a protein that binds and inhibits the insulin receptor (17, 18) and also targets insulin receptor substrate (IRS) proteins for proteosomal degradation (19). Expression of SOCS3 was increased and IRS1 was decreased in the liver of HFD-fed FWT mice, but not HFD-fed FKO mice (Fig. 4C,D). Dysregulated expression of SOCS3 and IRS1 in the liver of HFD-fed FKO mice is consistent with the observation that HFD-fed FKO mice exhibit a low circulating concentration of IL6 (Fig. 3C) and improved hepatic insulin sensitivity (Fig. 2I) compared to HFD-fed FWT mice.
To test whether the defect in adipose tissue expression of IL6 contributes to the improved hepatic insulin sensitivity of HFD-fed FKO mice, we examined whether administration of IL6 would restore HFD-induced insulin resistance phenotypes in FKO mice. Acute IL6 treatment increased hepatic SOCS3 expression in HFD-fed FKO mice to the same amount that was detected in HFD-fed FWT mice (Fig. 4E). Insulin tolerance tests demonstrated that IL6-treated HFD-fed FKO mice became equally insulin resistant as HFD-fed FWT mice (Fig. 4F). Moreover, IL6 treatment reduced insulin-stimulated AKT activation in the liver of HFD-fed FKO mice (Fig. 4G). In contrast, only a moderate effect of IL6 on AKT activation in the adipose tissue of HFD-fed FKO mice was detected (Fig. 4H). These data demonstrate that the effect of JNK1-deficiency in adipose tissue on hepatic insulin sensitivity is, at least in part, mediated by a requirement of JNK1 for HFD-induced expression of IL6.
Adipose tissue plays a critical role in glucose homeostasis by releasing adipokines that regulate insulin sensitivity in other organs (8, 20). One of these, IL6, is elevated in obese, diabetic subjects (9), and regulates glucose metabolism in multiple cell types (21-23). However, the role of IL6 in whole body insulin resistance has been debated because IL6 alters insulin signaling differently in individual tissues (24, 25). Furthermore, IL6 regulates the hypothalamic-pituitry-adrenal axis (26) and the IL6/Stat3 pathway is required for the action of insulin signaling in the brain on hepatic gluconeogenesis (27). Thus, IL6 has both central and peripheral roles on metabolism and its effects on systemic insulin resistance are complex. Nevertheless, neutralization of IL6 selectively improves obesity-induced hepatic insulin resistance and treatment with IL6 increases hepatic insulin resistance (12-15). Moreover, ablation of the IL6 target gene Socs3 in the liver of young mice causes improved hepatic insulin sensitivity (16). Together, these data and our study demonstrate that adipose tissue-derived IL6 is an important mediator of hepatic insulin resistance and that JNK1 is a component of a metabolic stress signaling pathway that regulates IL6 expression in adipose tissue. The serum concentration of IL6 represents a possible biomarker for the evaluation of the efficacy of drugs that target JNK1 and may be useful for the treatment of metabolic diseases.
Supplementary Material
References and Notes
- 1.Hirosumi J, et al. Nature. 2002;420:333. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 2.Jaeschke A, Davis RJ. Mol Cell. 2007;27:498. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguirre V, Uchida T, Yenush L, Davis RJ, White MF. J Biol Chem. 2000;275:9047. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 4.Weston CR, Davis RJ. Curr Opin Cell Biol. 2007;19:142. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Solinas G, et al. Cell Metab. 2007;6:386. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Carey DG, Jenkins AB, Campbell LV, Freund J, Chisholm DJ. Diabetes. 1996;45:633. doi: 10.2337/diab.45.5.633. [DOI] [PubMed] [Google Scholar]
- 7.Waki H, Tontonoz P. Annu Rev Pathol. 2007;2:31. doi: 10.1146/annurev.pathol.2.010506.091859. [DOI] [PubMed] [Google Scholar]
- 8.Spiegelman BM, Flier JS. Cell. 1996;87:377. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 9.Bastard JP, et al. Eur Cytokine Netw. 2006;17:4. [PubMed] [Google Scholar]
- 10.Mohamed-Ali V, et al. J Clin Endocrinol Metab. 1997;82:4196. doi: 10.1210/jcem.82.12.4450. [DOI] [PubMed] [Google Scholar]
- 11.Weisberg SP, et al. J Clin Invest. 2003;112:1796. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Diabetes. 2003;52:2784. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 13.Kim HJ, et al. Diabetes. 2004;53:1060. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- 14.Klover PJ, Clementi AH, Mooney RA. Endocrinology. 2005;146:3417. doi: 10.1210/en.2004-1468. [DOI] [PubMed] [Google Scholar]
- 15.Senn JJ, et al. J Biol Chem. 2003;278:13740. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- 16.Torisu T, et al. Genes Cells. 2007;12:143. doi: 10.1111/j.1365-2443.2007.01044.x. [DOI] [PubMed] [Google Scholar]
- 17.Emanuelli B, et al. J Biol Chem. 2001;276:47944. doi: 10.1074/jbc.M104602200. [DOI] [PubMed] [Google Scholar]
- 18.Emanuelli B, et al. J Biol Chem. 2000;275:15985. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- 19.Rui L, Yuan M, Frantz D, Shoelson S, White MF. J Biol Chem. 2002;277:42394. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 20.Mauvais-Jarvis F, Kulkarni RN, Kahn CR. Clin Endocrinol (Oxf) 2002;57:1. doi: 10.1046/j.1365-2265.2002.01563.x. [DOI] [PubMed] [Google Scholar]
- 21.Kishimoto T. Blood. 1989;74:1. [PubMed] [Google Scholar]
- 22.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Diabetes. 2002;51:3391. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 23.Pedersen BK, Steensberg A, Schjerling P. Curr Opin Hematol. 2001;8:137. doi: 10.1097/00062752-200105000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Mooney RA. J Appl Physiol. 2007;102:816. doi: 10.1152/japplphysiol.01208a.2006. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen BK, Febbraio MA. J Appl Physiol. 2007;102:814. doi: 10.1152/japplphysiol.01208.2006. [DOI] [PubMed] [Google Scholar]
- 26.Wallenius V, et al. Nat Med. 2002;8:75. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 27.Inoue H, et al. Cell Metab. 2006;3:267. doi: 10.1016/j.cmet.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 28.We thank V. Benoit, J. Cavanagh-Kyros, K. Gemme, N. Kennedy, J. Liu, and J. Reilly for expert assistance. These studies were supported by grants from the National Institutes of Health (to R.J.D.), the American Diabetes Association (1-04-RA-47 to J.K.K.), and the Pennsylvania State Department of Health (to J.K.K.). Core facilities at the University of Massachusetts used by these studies were supported by the NIDDK Diabetes and Endocrinology Research Center (DK52530). R.J.D. is an Investigator of the Howard Hughes Medical Institute.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.