

Metals are essential nutrients that can also be toxic. Safe trafficking of metal ions is necessary inside cells, and specific metal transport pathways exist to deliver them to their destinations.1,2 In human cells, the copper chaperone Hah1 and the Wilson disease protein (WDP) constitute a copper transport pathway—Hah1 is a single-domain cytoplasmic protein; WDP is a multidomain protein anchored on organelle membranes and has a cytosolic N-terminal region consisting of six homologous metal-binding domains (MBDs). All WDP MBDs and Hah1 contain a conserved CXXC motif that binds Cu1+, and Cu1+ is transferred from Hah1 to a WDP MBD via direct and specific Hah1–MBD interactions.1,3,4
Although the MBDs of WDP have different functional roles,4,5 all of them, as well as Hah1, have similar Cu1+ binding affinities.3 This similarity indicates that the Hah1 to WDP Cu1+ transfer is under kinetic control mediated by Hah1–WDP interactions, and that the functional differences among WDP MBDs are not defined by their Cu1+ binding abilities but may be related to how each MBD interacts with Hah1. Very limited quantitative information is available, however, on the Hah1–WDP interaction dynamics. This is partly because the Hah1–WDP interactions are transient, and transient interactions are difficult to quantify in ensemble-averaged experiments.
Here we report using nanovesicle trapping and single-molecule fluorescence resonance energy transfer (smFRET) measurements to probe the transient interactions between Hah1 and the fourth MBD (MBD4) of WDP in real time. We chose MBD4 as a representative WDP MBD because it is known to interact with Hah1 directly for Cu1+ transfer.4,6 Quantification of Hah1–MBD4 interaction dynamics will help understand how Hah1 and the full length WDP interact for Cu1+ transfer.
A primary obstacle in single-molecule experiments to probe transient protein interactions is the low concentrations (10−12-10−9 M) commonly used to spatially separate molecules for detection, which limits the experiments to strong protein interactions. Weak protein interactions, including Hah1–WDP interactions, need to be studied at higher concentrations. Nonspecific protein–glass surface interactions during molecule immobilization present another challenge and must be minimized.
To overcome these challenges, we adapted a nanovesicle trapping strategy (Figure 1), which was used to study protein and RNA folding and DNA–protein interactions at the single-molecule level.7 We trapped the two interacting molecules in a 100 nm diameter lipid vesicle. Because of the confined volume (∼5 × 10−19 L), the effective concentration is ∼3 μM for each protein inside. Low concentrations of vesicles are then immobilized on a lipid bilayer or polymer-coated glass surface so protein–glass interactions are eliminated.
Figure 1.
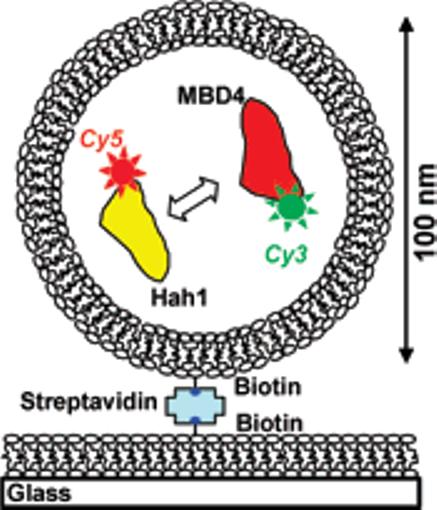
Nanovesicle trapping and smFRET for studying transient protein–protein interactions.
To report Hah1–MBD4 interactions by smFRET, we introduced a C-terminal cysteine in both Hah1 and MBD4 and labeled this cysteine of Hah1 with Cy5 and that of MBD4 with Cy3. Cy3–Cy5 form a FRET pair with a Förster radius of ∼6 nm. The cysteines in the CXXC motifs were protected specifically from labeling. We purified the labeled proteins and confirmed their Cu1+ binding and transfer functions (Supporting information (SI)).
We used total internal reflection fluorescence microscopy with 532 nm laser excitation to measure smFRET of single interacting pairs of Cy5–Hah1 and Cy3–MBD4 trapped in nanovesicles. We only analyzed data from vesicles containing one Cy5–Hah1 and one Cy3–MBD4 (see SI). This way we also eliminated the complication of dimeric or multimeric interactions between molecules of the same type, which are unavoidable in ensemble experiments and complicate protein interaction studies. This is particularly relevant in studying Hah1–WDP interactions, as Hah1 could form homodimers.8
The fluorescence and the corresponding FRET efficiency (EFRET) trajectories of two interacting pairs of Hah1 and MBD4 molecules are shown in Figure 2A and 2B. The Cy3 and Cy5 fluorescence of each pair show anticorrelated intensity fluctuations, reporting transient Hah1–MBD4 interactions. Three EFRET states are clear in the EFRET trajectories at EFRET ∼0.2, 0.5, and 0.9, denoted as E0, E1, and E2, respectively. E0 is clearly different from that (<0.1) of the acceptor photobleached or blinked state. Considering the small dimensions of Hah1 and MBD4 (2-3 nm),4,8,9 the small value of E0 indicates Hah1 and MBD4 are distant from each other; we thus associated E0 with their dissociated state (see SI). The values of E1 and E2 indicate the two proteins are within a few nanometers in these two states; we thus associated E1 and E2 with two transient Hah1–MBD4 interaction complexes (see SI and Figure S7). The significant difference between E1 and E2 indicates the Cy3–Cy5 distances differ by nanometers between the two complexes, which can arise from their differences in overall interaction geometries or in the conformation of one or both proteins. The latter is less likely as NMR studies showed Hah1 only has angstrom-scale conformational flexibilities,10 which should not result in significant difference in EFRET that is sensitive only at the nanometer scale. The structure of WDP MBD4 has not been solved, but the NMR structure of the fourth MBD of the Menkes protein, which is homologous to WDP MBD4, has only angstrom-scale conformational flexibility, as well.9 The direct resolution of two distinct interaction complexes here is the first evidence that multiple interaction intermediates exist for metallochaperone–target protein interactions and that Hah1 can form complexes with WDP without Cu1+. We have further confirmed that nonspecific interactions between the proteins and the lipids are minimal and the protein interactions are not induced by the labeling (see SI).
Figure 2.
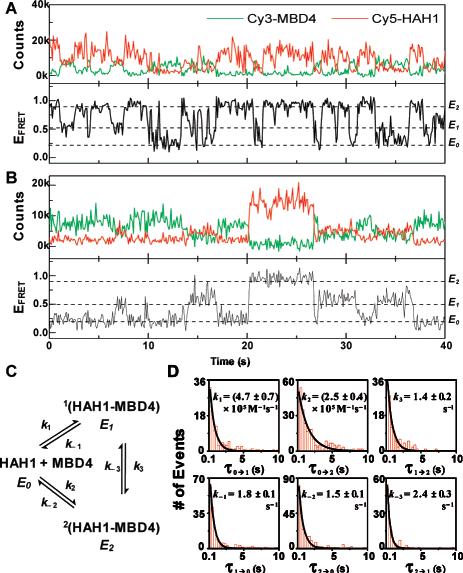
(A and B) Fluorescence and EFRET (ICy5/(ICy3 + ICy5)) trajectories of two interacting Cy5–Hah1 and Cy3–MBD4 pairs. (C) Hah1–MBD4 interaction scheme. (D) Waiting time (τ) distributions for the six kinetic steps in (C). Data compiled from 163 Hah1–MBD4 interacting pairs and total 1101 transitions. Solid lines are exponential fits.
In the EFRET trajectories, the transitions between E0 and E1 and between E0 and E2 reflect the Hah1–MBD4 association and dissociation events, while those between E1 and E2 are the interconversions between the two interaction complexes. Figure 2C shows the Hah1–MBD4 interaction scheme deduced from the smFRET data. The six rate constants in the scheme are obtainable from the statistical distributions of the six types of waiting times before each EFRET transition (Figure 2D; see SI). All waiting time distributions can be fitted with single-exponential decays. For Hah1–MBD4 complex dissociations and interconversions, which are unimolecular reactions, the decay constants are the rate constants. For protein association, the effective concentration (∼3 μM) of a molecule in a nanovesicle needs to be factored out from the decay constants to obtain the rate constants. The determined rate constants are ∼100 s−1 for complex dissociations and interconversions, and ∼105 M−1 s−1 for protein associations (Figure 2D). Particularly, we have directly resolved the interconversion dynamics between the two interaction complexes and quantified the rate constants of both the forward and the reverse reactions–ensemble studies of intermediate interconversion dynamics typically only obtain the sum of the forward and reverse rate constants. The rate constants also enabled determination of separate dissociation constants for the two interaction complexes with K1 = 3.9 ± 0.6 μM and K2 = 6 ± 1 μM (see also Figure S6).
The ability of Hah1 and MBD4 to form multiple interaction complexes with different interaction geometries has functional significance. First, it increases the probability of complex formation when Hah1 and WDP encounter through diffusion inside cells. The formed complex may proceed to accomplish Cu1+ transfer or, if unproductive, convert to another complex for Cu1+ transfer. Second, it raises the possibility of Hah1 interacting with two WDP MBDs simultaneously and hence cooperative effects among WDP MBDs for Cu1+ transfer. The interactions of Hah1 with different combinations of MBDs may also play a role in the functional differences of these MBDs.
In summary, this study represents the first application of nanovesicle trapping to study weak protein–protein interactions on a single-molecule basis. We observed transient copper chaperone–target protein interactions one event at a time, captured distinct protein interaction intermediates, and resolved intermediate interconversion dynamics. The quantitative dynamic information will help one to understand how metallochaperones and their target proteins collaborate for metal transfer. By studying the Cu1+ dependence of the interaction kinetics, this single-molecule approach also offers a means to probe the Cu1+ transfer process and identify the productive interaction complex. Moreover, we expect that the nanovesicle trapping strategy coupled with smFRET is applicable for studying many other weak protein interactions at the single-molecule level.
Supplementary Material
Acknowledgment
We thank NSF(CHE0645392,P.C.;CHE0645518, D.L.H.), Dreyfus Foundation (P.C.), NIH traineeship (J.J.B., A.M.K.), Wilson Disease Association (P.C., D.L.H.), and NIH GM58518 (A.C.R.).
Footnotes
Supporting Information Available: Additional results and analyses, control experiments, materials and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Huffman DL, O'Halloran TV. Annu. Rev. Biochem. 2001;70:677. doi: 10.1146/annurev.biochem.70.1.677. [DOI] [PubMed] [Google Scholar]; (b) Rosenzweig AC. Acc. Chem. Res. 2001;34:119. doi: 10.1021/ar000012p. [DOI] [PubMed] [Google Scholar]
- 2.(a) Cobine PA, Pierrel F, Winge DR. Biochim. Biophys. Acta. 2006;1763:759. doi: 10.1016/j.bbamcr.2006.03.002. [DOI] [PubMed] [Google Scholar]; (b) Donnelly PS, Xiao Z, Wedd AG. Curr. Opin. Chem. Biol. 2007;11:128. doi: 10.1016/j.cbpa.2007.01.678. [DOI] [PubMed] [Google Scholar]
- 3.Yatsunyk LA, Rosenzweig AC. J. Biol. Chem. 2007;282:8622. doi: 10.1074/jbc.M609533200. [DOI] [PubMed] [Google Scholar]
- 4.Achila D, Banci L, Bertini I, Bunce J, Ciofi-Baffoni S, Huffman DL. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5729. doi: 10.1073/pnas.0504472103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Walker JM, Huster D, Ralle M, Morgan CT, Blackburn NJ, Lutsenko S. J. Biol. Chem. 2004;279:15376. doi: 10.1074/jbc.M400053200. [DOI] [PubMed] [Google Scholar]; (b) Cater MA, Forbes J, Fontaine SL, Cox D, Mercer JFB. Biochem. J. 2004;380:805. doi: 10.1042/BJ20031804. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Banci L, Bertini I, Cantini F, Della-Malva N, Migliardi M, Rosato A. J. Biol. Chem. 2007;282:23140. doi: 10.1074/jbc.M700695200. [DOI] [PubMed] [Google Scholar]
- 6.(a) Larin D, Mekios C, Das K, Ross B, Yang A-S, Gilliam TC. J. Biol. Chem. 1999;274:28497. doi: 10.1074/jbc.274.40.28497. [DOI] [PubMed] [Google Scholar]; (b) van Dongen EMWM, Klomp LWJ, Merkx M. Biochem. Biophys. Res. Commun. 2004;323:789. doi: 10.1016/j.bbrc.2004.08.160. [DOI] [PubMed] [Google Scholar]
- 7.(a) Rhoades E, Gussakovsky E, Haran G. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3197. doi: 10.1073/pnas.2628068100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cisse I, Okumus B, Joo C, Ha T. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12646. doi: 10.1073/pnas.0610673104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wernimont AK, Huffman DL, Lamb AL, O'Halloran TV, Rosenzweig AC. Nat. Struct. Biol. 2000;7:766. doi: 10.1038/78999. [DOI] [PubMed] [Google Scholar]
- 9.Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ. Nat. Struct. Biol. 1998;5:47. doi: 10.1038/nsb0198-47. [DOI] [PubMed] [Google Scholar]
- 10.Anastassopoulou I, Banci L, Bertini I, Cantini F, Katsari E, Rosato A. Biochemistry. 2004;43:13046. doi: 10.1021/bi0487591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.