

Abstract
The maturation process of green fluorescent protein (GFP) entails a protein oxidation reaction triggered by spontaneous backbone condensation. The chromophore is generated by full conjugation of the Tyr66 phenolic group with the heterocycle, a process that requires C-H bond scission at the benzylic carbon. We have prepared isotope-enriched protein bearing tyrosine residues deuterated at the beta carbon, and have determined kinetic isotope effects (KIEs) on the GFP self-processing reaction. Progress curves for the production of H2O2 and the mature chromophore were analyzed by global curve fitting to a three-step mechanism describing pre-oxidation, oxidation and post-oxidation events. Although a KIE for protein oxidation could not be discerned (kH/kD = 1.1 ± 0.2), a full primary KIE of 5.9 (± 2.8) was extracted for the post-oxidation step. Therefore, the exocyclic carbon is not involved in the reduction of molecular oxygen. Rather, C-H bond cleavage proceeds from the oxidized cyclic imine form, and is the rate-limiting event of the final step. Substantial pH-dependence of maturation was observed upon substitution of the catalytic glutamate (E222Q), indicating an apparent pKa of 9.4 (± 0.1) for the base catalyst. For this variant, a KIE of 5.8 (± 0.4) was determined for the intrinsic time constant that is thought to describe the final step, as supported by ultra-high resolution mass spectrometric results. The data are consistent with general base catalysis of the post-oxidation events yielding green color. Structural arguments suggest a mechanism in which the highly conserved Arg96 serves as catalytic base in proton abstraction from the Tyr66-derived beta carbon.
Keywords: Protein maturation, chromophore biosynthesis, fluorescent proteins, KIE, deuterium isotope effect, general base catalysis, arginine as catalyst
In recent years, the post-translational modifications yielding for the colorful chromophores of GFP-like proteins have been studied extensively (1, 2). Members of the family of fluorescent proteins are evolutionarily related to its founding member avGFP from the jellyfish Aequorea victoria (3), and are generally found in marine organisms such as reef-building corals. Fluorescent proteins (FPs) have attracted considerable interest, due to their ability to synthesize brightly fluorescing entities from intrinsic amino acid residues, such that the intense coloration of the mature protein appears to be an inherent property of the particular genetic sequence. The interior of the eleven-stranded β-barrel of FPs contains a helical peptide that spans residues 60 – 71 in avGFP, with position 66 occupied by a conserved tyrosine and 67 by a conserved glycine residue. Autocatalytic backbone cross-linking of residues 65 and 67 is followed by the net transfer of two redox equivalents to atmospheric oxygen, ultimately yielding a chromophore that remains covalently attached to the polypeptide backbone and is buried in the protein's interior (Scheme 1) (4). Substantial progress has been made in gaining a better understanding of the main chain condensation reaction that triggers the ensuing sensitivity to air oxidation (5). O2-mediated oxidation is ultimately responsible for the π-orbital extension of the Tyr66 phenolic group over the benzylic carbon center (Cβ66), such that desaturation of the Cα - Cβ bond provides conjugation with the heteroaromatic ring constructed from main chain atoms. Aside from H2O2, the product of the reaction is a two-ring chromophore embedded in the protein matrix, usually in a planar conformation (Scheme 1) (1).
Scheme 1.

Proposed three-step mechanism for GFP maturation, consisting of pre-oxidation, oxidation, and post-oxidation processes. Proposed hydration-dehydration equilibria of the α-enolate and cyclic imine forms are also shown.
Previous work has focused on the preorganization of reacting atoms by the protein fold, a feature that allows for nucleophilic addition of the amide nitrogen of Gly67 (N67) to the carbonyl carbon of residue 65 (C65), while eliminating the energetic cost associated with the disruption of hydrogen bonds typically found in buried helical structures (5). This reaction appears to be facilitated by the highly conserved Arg96 with its guanidinium group hydrogen bonded to the main chain oxygen of residue 66. Mutagenesis studies have demonstrated that Arg96 imparts electrostatic catalysis by activating the 66-67 peptide bond (6-8). In addition, Arg96 has been proposed to play a role in stabilizing the superoxide anion radical that may form upon interaction with molecular oxygen (9, 10). In previous work, we have demonstrated that the main-chain condensation reaction is facile, occurring essentially within the dead time of manual mixing and HPLC injection methods (11). These data have provided support for the existence of an equilibrium between the open and closed forms of the protein, as exposure to the acidic HPLC conditions allows for the separation of pre- and post-cyclization species of GFP (11). Likely, the closed five-membered ring is favored in the folded state of the protein. Although the aromatic α-enolate has been proposed to be the dominant pre-oxidation species (10), a mix of hydrated and dehydrated ring structures has been observed crystallographically in some GFPs (12-15), and the internal hydration-dehydration equilibria are as yet poorly understood (Scheme 1).
Several years ago, we proposed that GFP self-processing entails a protein oxidation reaction that remains centered on the five-membered ring, and does not affect the hybridization state of the benzylic Cβ66 carbon. This idea was based on the X-ray structure of the Y66L variant, where the condensed main-chain was found to be oxidized to the hydroxylated cyclic imine form (Scheme 1) (9). Complete elimination of water from the heterocycle was observed only upon desaturation of the Cα66-Cβ66 bond, which requires C-H bond cleavage at Cβ66. In the Y66L variant, re-hybridization of this carbon center is compounded by the low degree of activation of the aliphatic side chain. Desaturation was only observed upon prolonged incubation in high concentrations of a general base such as fluoride, formate or acetate (14). These compounds were able to diffuse into the protein's interior and accept a proton under physiological conditions (16, 17). Redox chemistry did not play a role in re-hybridization from sp3 to sp2, rendering a radical mechanism for Cβ66 oxidation unlikely (14). Here, we present further evidence that the chemistry observed in Y66L is analogous to the normal maturation mechanism of intact GFPs.
Like Arg96, Glu222 is completely conserved in all known fluorescent proteins, and may be involved in facilitating multiple proton transfer steps during GFP maturation (3, 18). Substitution with the sterically equivalent glutamine residue results in a variant that exhibits reduced maturation rates at physiological pH values. At pH 8, E222Q matures with a time constant of seven hours, whereas the intact parent molecule EGFP exhibits a time constant of one hour in in vitro assays (6). In previous work, we were unable to characterize any intermediate states of E222Q by trypsinolysis/MALDI (6), although an intermediate species with a reduced mass (- 2 Da) was clearly observed in mGFPsol, an enhanced variant of EGFP (11). In contrast to these intact GFPs, maturation of the E222Q variant provided substantial evidence for base catalysis (6). For this reason, we have previously interpreted the slow maturation rate at pH 8 in terms of inefficient backbone cyclization or inefficient Cα66 deprotonation (6), both pre-oxidation events. However, the data presented here have allowed us to revise our interpretation, adjusting our model for base catalysis to be focused on post-oxidation events (Scheme 1).
Recent investigations on chromophore biosynthesis in intact GFPs have provided compelling evidence that hydrogen peroxide is produced prior to the rise of green fluorescence (Scheme 2) (11). We have demonstrated that the third and final step in GFP maturation proceeds with a time constant of about 11 min and is therefore partially rate-determining. This step follows the slowest part of the overall reaction, the net transfer of two hydrogen atoms to molecular oxygen, which proceeds with a time constant of about 34 min (11). Based on trypsinolysis/MALDI data, an intermediate with a mass loss of 2 Da accumulates during the reaction. These results are consistent with dehydrogenation prior to full protein maturation, in support of a mechanism that involves a long-lived oxidized intermediate. However, the previously published data do not address the chemical nature of the final step yielding green color.
Scheme 2.

Outline of the three major GFP maturation steps modeled in global curve fitting of kinetic data.
An as yet unresolved question in GFP maturation concerns the mechanism of C-H bond scission at the benzylic CH2 group, a process essential for full π-orbital conjugation of the phenolic with the imdazolinone ring. As the O2-mediated chemistry is completed prior to this event (Scheme 2) (11), homolytic C-H bond cleavage appears unlikely. Here, we demonstrate that a proton is abstracted from Cβ66, which is acidified by the electron-withdrawing properties of the oxidized imidazolinone ring (14). Preliminary KIE data for mGFPsol maturation were reported in a recent review article (1). In the present work, we present deuterium isotope effect data for the mGFPsol and E222Q self-processing reactions, and propose that Arg96 is the general base that catalyzes proton abstraction.
Experimental Procedures
Preparation of immature mGFPsol protein
Six-His-tagged mGFPsol, a GFP variant that has also been termed GFP-trix, bears the mutations F64L/S65T/F99S/M153T/V163A/A206K in wild-type avGFP background (11). Expression in rich media was carried out in strain JM109(DE3), utilizing the pRSETB expression plasmid (Invitrogen) bearing the N-terminally 6His-tagged mGFPsol gene (12). mGFPsol inclusion bodies were prepared by bacterial expression at 42 °C, solubilized in buffer containing 8 M urea, 50 mM HEPES pH 7.9, 50 mM NaCl and 1 mM DTT, and purified by Ni-affinity chromatography in the denatured state essentially as described (9, 11). Kinetic experiments were carried out immediately after protein purification on the same day.
Preparation of immature E222Q protein
Protein folding from inclusion bodies proved to be extremely inefficient for the E222Q variant (avGFP- F64L/S65T/E222Q), termed E222Q in this paper. Therefore, E222Q was purified from soluble bacterial fractions by following a two-hour procedure as reported previously (6, 19). Rapid purification was followed by flash-freezing in 150 μl aliquots (1.2 -1.7 mg/ml protein) and storage at -80 °C. One liter of liquid media and a two-hour induction period (25 °C) yielded a total of 4 - 5 mg protein purified to homogeneity. The same yield was obtained when E222Q-expressing E. coli were grown in minimal media to incorporate a deuterium label into the expressed protein (see below).
Isotopic enrichment procedure
Cβ-di-deuterated tyrosine was purchased from Cambridge Isotope Laboratories, Inc. The pRSETB plasmid (Invitrogen) bearing DNA coding for 6His-tagged mGFPsol or E222Q was transformed into Escherichia coli strain BL21(DE3) for protein expression in minimal media. Minimal media stock solutions were prepared in the following way. The trace elements stock solution was prepared by combining 85 ml water with 0.60 g CaCl2·2H2O, 0.60 g FeSO4·7H2O, 0.115 g MnCl2·4H2O, 0.08 g CoCl2·6H2O, 0.07 g ZnSO4·7H2O, 0.03 g CuCl2·2H2O, 0.002 g H3BO3, 0.025 g (NH4)6Mo7O24·4 H2O, and 0.50 g EDTA-Na2, adjusting the volume to 100 ml, and filter-sterilizing the solution. The 5X minimal salts stock solution was prepared by the combination of 150 ml water, 3 g KH2PO4, 11.32 g Na2HPO4·7H2O, 0.5 g NaCl, and 1.0 g NH4Cl. The pH was adjusted to 7.2, the volume to 200 ml, and the solution was heat-sterilized. The 5X amino acid stock solution (without tyrosine) was prepared by dissolving 2.5 g alanine, 2.0 g arginine, 2.0 g aspartic Acid, 250 mg cysteine, 2.0 g glutamine, 3.25 g glutamic Acid, 500 mg histidine, 1.15 g isoleucine, 2.1 g lysine, 1.25 g methionine, 650 mg phenylalanine, 500 mg proline, 10.5 g serine, 1.15 g threonine, 1.15 g valine, 2.75 g glycine, and 1.15 g leucine in a total volume of 1 liter deionized water, followed by heat-sterilization (20). A 10 mg/ml tryptophan stock solution was prepared separately by dissolving 1.0 g tryptophan in 100 ml 50 mM HEPES pH 7.9, 300 mM NaCl, followed by filter-sterilization. In addition, 20% glucose, 20 mg/ml thiamine, and 1.0 M MgCl2 stock solutions were prepared in water. A 10 mM uracil stock was prepared in 50 mM HEPES pH 7.9, 300 mM NaCl.
The minimal medium was assembled to a final volume of 1 liter by combining 200 ml minimal salts stock, 20 ml glucose stock, 2.0 ml thiamine stock, 1.0 ml MgCl2 stock, 5.0 ml trace elements stock, 20.0 ml uracil stock, 25 ml tryptophan stock and 200 ml amino acid stock. 200 mg ampicillin and 170 mg Cβ-di-deuterated tyrosine (as dry powder) were added just before inoculation. One liter minimal medium was inoculated with 8 ml overnight culture (LB broth supplemented with 90 μg/mL carbenicillin). Flask cultures were grown in a shaker (300 rpm) at 37 °C until the O.D.600 reached 0.8, then the temperature was adjusted to 42 °C to prepare mGFPsol inclusion bodies and to 25 °C to express soluble E222Q, and shaking was reduced to 250 rpm. mGFPsol was induced for 4.0 h and E222Q for 2 h by addition of 250 mg IPTG, then harvested by centrifugation (6).
Trypsinolysis and peptide purification
To determine the incorporation efficiency of the deuterium label, the molecular mass of proteolytic peptides derived from isotopically labeled and unlabeled material was determined. Tryptic peptides of E222Q were generated exactly as described previously (6). For inclusion body-derived mGFPsol, 55 μL of water was added to 165 μL of 8 M urea-denatured protein to yield a final concentration of 6 M urea. 4 μL of 10 mg/ml trypsin (Sigma T-1426, TCPK-treated bovine) was added to 800 ml digest buffer (50 mM HEPES pH 8.0, 300 mM NaCl, 20 mM CaCl2) immediately before use, and combined with the protein solution. The digest was incubated for 3 min at 30 °C, dithiothreitol was added to yield a concentration of 10 mM, followed by a two-minute incubation at room temperature. Tryptic peptides were separated by reverse-phase HPLC (Waters 600 HPLC system equipped with a Waters 996 photodiode array detector) on a C18 analytical column (Vydac) utilizing a water/ACN (acetonitrile) gradient containing 0.1% TFA (trifluoroacetic acid). Peptides were eluted after column equilibration at 45% solvent B for 10 min, followed by a linear gradient from 45% to 60% solvent B at 1% per minute. The absorbance was monitored at 220, 280 and 380 nm. Five fractions were collected in eppendorf tubes, split into two equal parts of 0.75 ml each, and lyophilized immediately. One part of each HPLC fraction was subjected to MALDI mass spectrometry, the other part was saved for ESI ion trap mass spectrometric analysis.
MALDI Mass Spectrometry
Lyophilized peptides were resolubilized in 18 μl water, and 1 μl peptide solution was mixed with 3 μl of a saturated solution of α-cyano-4-hydroxycinnamic acid matrix. MALDI data were collected as described previously, utilizing a Voyager DE STR mass spectrometer equipped with a nitrogen laser (6). Masses of all peptides eluted by HPLC were determined by MALDI and matched with calculated masses obtained from theoretical tryptic digests (see Supplementary Table 1). Peptides found in the HPLC fraction eluting at 51-52% solvent B (elution time 16 to 17 min) contained the chromophore-forming residues, as well as a C-terminal peptide fragment. This fraction was submitted for ion trap mass spectrometric analysis.
Nano-HPLC Ion Trap Mass Spectrometry
The extent of biosynthetic incorporation of Cβ-di-deuterated tyrosine into expressed protein was determined by the Proteomics Core Facility at the University of Arizona, Tucson, AZ. Lyophilized peptides from the HPLC fraction collected at 51-52 % Solvent B (see above) were solubilized in 50 μl of water. A microbore HPLC system (Surveyor; ThermoFinnigan, Jan Jose, CA, USA) was modified to operate at capillary flow rates using a simple T-piece flow-splitter. Columns (6 cm × 100 μm ID) were prepared by packing 100 Å, 5 μm Zorbax C18 resin at 500 psi pressure into columns with integrated electrospray tips made from fused silica, pulled to a 5 mm tip using a laser puller (Sutter Instrument, Novato, CA, USA). An electrospray voltage of 1.8 kV was applied using a gold electrode via a liquid junction up-stream of the column. For each run, a 10 μl sample aliquot was injected into the analytical column using a Surveyor autosampler. The HPLC column eluent was eluted directly into the ESI (electrospray ionization) source of a ThermoFinnigan LCQ-Deca XP Plus ion trap mass spectrometer. Peptides were eluted utilizing a linear gradient from 0 – 50% Solvent B over a 30 min interval (Solvent A: water/0.1% formic acid, Solvent B: ACN/0.1% formic acid) at a flow rate of 400 nl/min. In order to resolve the isotope pattern of the doubly charged (z = 2+) peptides of interest, three separate MS runs were performed acquiring continuous zoom scans with a 10 amu (atomic mass unit) window for each of the three peptides previously characterized by MALDI (see above): m/z = 1190 (1185.5-1195.5), m/z = 1296 (1291.5-1301.5), and m/z = 1565 (1560.5-1570.5). For each doubly charged (z = 2+) peptide examined by the zoom method, the major isotopic fingerprint matched the calculated D2 peptide mass exactly (see Supplementary Table 2). However, very small peaks were observed with m/z consistent with the H2 peptide. Although these peaks were essentially at noise level, they were utilized to calculate a lower limit for the isotopic incorporation efficiency. For the H2 and D2 versions of each peptide, peak areas were determined for the second isotope peak M+1. For the peptide with experimentally determined monoisotopic m/z = 2196.6, minimum incorporation efficiency was calculated to be 97.5%, for m/z = 1565.8, 99.2%, and for m/z = 1190.6, 93.3%, providing an average of 96.6 ± 3.1 % for the lower limit of isotope incorporation efficiency in our labeled protein preparations.
Kinetic assay for hydrogen peroxide evolution during mGFPsol maturation
As described previously (11), immature urea-solubilized mGFPsol protein (approximately 0.2 mg/ml) was rapidly diluted into a stirred quartz fluorimeter cell with the thermostat set to 30 °C. To measure the generation of hydrogen peroxide, a newly purchased Amplex Red Hydrogen Peroxide Assay Kit (Molecular Probes, Inc.) was utilized. A 10 μl aliquot of denatured protein was added to a 290 μl aliquot of rapidly stirring solution consisting of 240 μl folding buffer (50 mM HEPES pH 7.9, 300 mM NaCl, 1 mM EDTA) and 50 μl Amplex Red (10-acetyl-10H-phenoxazine-3,7-diol) working solution prepared according to the manufacturer's instructions (21). The red fluorescence of the resorufin indicator dye produced in response to hydrogen peroxide was monitored via emission at 580 nm (λex = 550) as described previously (11). The conversion rate of Amplex Red to resorufin was determined by subjecting a 0.3 μM H2O2 standard solution to identical assay conditions. The amount of hydrogen peroxide in the standard was approximately equal to that generated during a typical protein maturation experiment. The pseudo-first order rate constant for this reaction, termed k4, was extracted by computer-fitting the rise of fluorescence to the equation F = Fmax − e(-kt) × Fmax, utilizing the program Kaleidagraph. As before, the extracted rate constant k4 was utilized to account for the reagent response time during protein maturation, and was determined anew for each individual experiment (11). In the current work, the reagent response time was found to be more rapid compared to previously published data (11), likely because newly purchased reagent was used. The reagent is light, air and temperature sensitive, and exhibits a limited shelf life, therefore all assays and control experiments for one kinetic data set were performed on the same day utilizing the same batch of Amplex Red working solution. Due to low levels of reagent autoxidation, all protein and hydrogen peroxide kinetic traces were baseline-corrected by subtracting the trace generated by an appropriate blank.
Kinetic assay for acquisition of green fluorescence upon mGFPsol maturation
To monitor the rise of green fluorescence upon chromophore formation, a fresh 10 μl aliquot of urea-denatured protein was diluted into folding buffer (50 mM HEPES pH 7.9, 300 mM NaCl, 1 mM EDTA) to yield a final volume of 300 μl, and fluorescence emission was monitored at 510 nm (λex = 480) in a rapidly stirring quartz cuvette at 30 °C (11). For the maturation study involving unlabeled protein, fluorimeter settings were as described previously (11). For the deuterium-labeled protein, fluorescence emission data were collected in one-minute intervals for the first 120 min, followed by two-minute intervals up to 240 min and five-minute intervals up to 360 min, to avoid photobleaching of the newly formed chromophore. All fluorescence experiments were carried out on a Jobin Ivon Horiba FluoroMax-3 instrument.
Kinetic assay for E222Q maturation
Kinetic experiments on immature E222Q were carried out at eight different pH values between pH 7.72 and 10.02. UV-Vis absorbance scans were collected on a Shimadzu UV-2401 spectrophotometer as a function of time (6), and kinetic runs were performed on both unlabeled and isotope-labeled protein. For each experiment, aliquots of flash-frozen E222Q protein were removed from the freezer, diluted 10-fold into buffer of appropriate pH (20 mM PIPES, HEPES, CHES or CAPS, 300 mM NaCl, 1 mM EDTA), and an absorbance scan was collected immediately. Subsequently, the samples were capped to avoid evaporation and incubated in a temperature-controlled chamber at 30 °C. The increase in absorbance at 483 nm (chromophore anion) was determined by collection of absorbance scans at various time points until saturation was observed. At pH 10, time points were collected over a total period of 0.73 hours for unlabeled protein and 4.0 hours for isotope-labeled protein. At pH 7.7, the data collection time ranged from 32 hours for unlabeled protein to 76 hours for labeled protein. For each kinetic experiment, the change in chromophore absorbance was plotted as a function of time, and computer-fitted to a rate equation describing a unimolecular reaction [A = Amax - e(-kt) × Amax], using the program Kaleidagraph (Figure 3 inset) (6). The extracted pseudo-first order rate constants kobs were plotted as a function of pH. The pH-rate profiles for unlabeled and isotope-labeled protein pools were each computer-fitted to an expression for a base-catalyzed reaction of the form: kobs = (kint × Ka)/(10[-pH] + Ka). According to this model, the observed rate depends on the protonation equilibrium of an ionizable group with acid dissociation constant Ka, whereas kint represents the intrinsic (pH-independent) rate constant.
Figure 3.

pH-rate profile of E222Q maturation, demonstrating the dependence of the observed rate constant on pH. Blue (-●-), unlabeled protein; Red (-■-), Cβ-di-deuterotyrosine-labeled protein. The symbols represent experimental data, and the solid lines represent curve fits to kinetic equations (see Methods). Inset: Increase in chromophore absorbance (483 nm) over time, measured at pH 8.
Ultra-high resolution mass determination of immature E222Q by LTQ-FTMS
200 μl of 1.8 mg/ml flash-frozen E222Q were removed from the -80 °C freezer, a UV/Vis absorbance scan was collected, and 160 μl were purified by reverse-phase HPLC. The eluting protein peak was collected in two fractions, frozen immediately in liquid nitrogen and lyophilized overnight. The lyophilized samples were redissolved into 49.5 : 49.5 : 1.0 MeOH/water/formic acid. The samples were infused by nano-electrospray ionization with a Biversa Nanomate (Advion Biosystems, Ithaca, NY), and analyzed on a Finnigan LTQ-FTMS (Thermo-Electron Corporation, San Jose, CA) running the Tune Plus software version 1.0 (Thermo). The data were collected with the autogain set to 5E6 and the maximum accumulation time set to 8000 ms and 200,000 resolution. The final spectrum was obtained by averaging 394 scans with the QualBrowser software version 1.4 SR1 (Thermo). The average spectrum was then subjected to deconvolution using the Extract option within the QualBrowser software. Theoretical spectra superimposed onto the experimental spectra are based on the calculated amino acid composition of the protein, and were generated with the freeware Isopro 3.0 and matched to the experimental spectra.
Results
A primary deuterium isotope effect for the post-oxidation step in GFP maturation
To better understand the maturation process of green fluorescent protein, we have examined the mechanism of chromophore biogenesis upon incorporation of an isotopic label. To investigate whether carbon-hydrogen bond cleavage at Cβ66 is an integral part of O2-mediated oxidation, or whether this bond is disrupted at a later stage, we incorporated Cβ-di-deuterated tyrosines into the intact, fast-maturing variant mGFPsol (Figure 1 inset) (11). We argued that the protons attached to the benzylic carbon of Tyr66 are likely the least acidic protons to be removed during GFP maturation, suggesting that this process may entail significant rate retardation. Therefore, we produced the denatured precursor form of mGFPsol from inclusion bodies, and carried out maturation kinetic assays with and without the tyrosine-based deuterium label.
Figure 1.

Progress curves for mGFPsol maturation of unlabeled protein (top panel) and isotopically enriched protein (bottom panel). Red (-●-), rise in red fluorescence due to resorufin production from hydrogen peroxide; Green (-■-), rise in green fluorescence due to formation of the mature GFP chromophore. The symbols represent experimental data (for clarity, only 10% of the collected data points are shown). The solid lines describe theoretical progress curves calculated from the rate constants extracted by global curve fitting of experimental data. Black, precursor protein; Blue, pre-oxidation intermediate (α-enolate); Purple, post-oxidation intermediate (cyclic imine); Red, resorufin (hydrogen peroxide indicator); Green, mature protein. Inset: Schematic of pre-cyclization structures with Cβ-di-hydro (unlabeled) and Cβ-di-deutero (labeled) Tyr66.
To incorporate deuterated tyrosine residues, mGFPsol was expressed in minimal media supplemented with a complete set of canonical amino acids, with the exception of tyrosine, which was replaced by Cβ-di-deuterated tyrosine (20). Inclusion bodies of mGFPsol containing either di-hydro- or di-deutero-tyrosines were prepared by protein expression at 42 °C. The inclusion bodies were isolated and washed extensively, followed by solubilization in 8 M urea and affinity-purification. Maturation kinetic experiments were carried out as reported previously by triggering protein folding via rapid dilution of the chaotrope (11). For unlabeled mGFPsol, four independent kinetic runs were performed in the present study, such that inclusion of the recently published data allowed for statistical evaluation of a total of five complete data sets. Similarly, five independent kinetic experiments were performed on deuterium-labeled mGFPsol. Each complete data set consisted of a series of baseline-corrected kinetic traces that were collected on the same day utilizing the same aliquot of freshly prepared urea-solubilized protein. First, the rate of hydrogen peroxide evolution upon protein maturation was monitored by employing an enzyme-linked fluorogenic assay that produces the red-fluorescent compound resorufin in the presence of H2O2 (21). Second, the rate of resorufin production from H2O2 was monitored by use of a hydrogen peroxide standard solution. Third, the progress curve for complete protein maturation was determined by monitoring the green fluorescence emission of the mature chromophore. The protein self-processing reaction was initiated by 30-fold dilution from 8 M urea, and the reaction was followed for either two hours (unlabeled protein) or six hours (labeled protein) (Figure 1).
For each of the kinetic data sets, the progress curves for resorufin production and chromophore formation were fitted to a sequential three-step mechanism (Scheme 2) using the program DynaFit (22). In the global curve-fitting procedures, the rate constant for resorufin production from H2O2 (k4) was held constant, and ranged from 1.1 to 2.3 min-1 (τ4 = 0.43 to 0.93 min), whereas the rate constants for the three major protein maturation steps were modeled by the variable parameters k1, k2, and k3. For each kinetic data set, values for these rate constants were extracted by modeling the time evolution of six species (precursor, intermediate X, intermediate Y, mature GFP, H2O2, and resorufin) as described previously (Scheme 2) (11). Average values and sample standard deviations were calculated from five individual determinations for each rate constant, obtained from curve fitting of five independent data sets each for di-hydro and di-deutero mGFPsol (Table 1).
Table 1.
Kinetic constants for mGFPsol maturation from inclusion bodies.
| Pre-oxidation τ1 (min) |
Oxidation τ2 (min) |
Post-oxidation τ3 (min) |
Overall τTotal (min) |
|
|---|---|---|---|---|
| mGFPsol, unlabeled (n = 5) | ||||
| Average (± Std. Dev.) |
1.93 (± 1.53) |
37.10 (± 3.14) |
15.28 (± 5.53) |
54.31 (± 6.54) |
| mGFPsol, deuterium-labeled (n = 5) | ||||
| Average (± Std. Dev.) |
4.99 (± 2.45) |
42.06 (± 6.45) |
90.04 (± 27.95) |
137.09 (± 28.79) |
| Deuterium isotope effects calculated for mGFPsol | ||||
| kH/kD (± Std. Dev.) |
2.58 (± 2.40) |
1.13 (± 0.20) |
5.89 (± 2.81) |
2.52 (± 0.61) |
We found that the three-step kinetic model provided an excellent fit to the data obtained for both unlabeled and deuterium-labeled protein (Figure 1). For the pre-oxidation process consisting of protein folding and main-chain cyclization, the extracted time constants τ1H = 1.93 ± 1.53 min and τ1D = 4.99 ± 2.45 min exhibit large standard deviations, such that the deuterium isotope effect, kH/kD = 2.58 ± 2.40, remains indeterminate (Table 1). Likely, the large error in τ1 is the result of inhomogeneous protein folding rates (23). The second major process, which produces hydrogen peroxide, proceeds on a much slower time scale, yielding the time constants τ2H = 37.10 ± 3.14 min and τ2D = 42.06 ± 6.45 min. From these data, the deuterium isotope effect kH/kD for oxidation is calculated to be 1.13 ± 0.20 (Table 1). Therefore, within experimental error, the oxidation rate does not appear to be modified upon deuteration, as the deviation from unity is smaller than the standard deviation determined by error propagation. In contrast, the average time constant for the post-oxidation step is extended substantially upon deuteration, with a value of τ3H = 15.28 ± 5.53 min for the unlabeled protein and a value of τ3D = 90.04 ± 27.95 min for the di-deutero protein. Therefore, τ3 is largely responsible for the overall change in maturation rate observed upon tyrosine deuteration (Figure 1). The KIE for this step is calculated to be 5.89 ± 2.81, consistent with a full primary deuterium isotope effect. Therefore, C-H bond scission at Cβ66 is part of the third step of protein maturation (Scheme 2), and constitutes the major rate-limiting event of the final process leading to green fluorescence. We conclude that desaturation of the bridging carbon Cβ66 is not an integral part of protein oxidation, but instead follows hydrogen peroxide production (Scheme 1).
Maturation of soluble E222Q proceeds from the oxidized intermediate stage
To better define the involvement of catalytic residues in the three-step self-modification process, we purified immature E222Q protein in soluble form as described previously (6). This variant is derived from EGFP as is mGFPsol, and both bear the substitutions F64L/S65T adjacent to the chromophore π-system known to enhance brightness and folding (24-26). However, E222Q does not bear the cycle 3 folding mutations F99S/M153T/V163A distant from the active site, nor does it contain the A206K substitution that eliminates dimerization (27). With respect to the arguments presented in this work, these differences are judged to be irrelevant, since immature E222Q was expressed in soluble form (this variant could not be folded efficiently from inclusion bodies (7)). We rapidly purified E222Q after a short induction period and flash-froze the partially mature protein. Absorbance measurements were utilized to determine that 33% of the freshly purified material had fully matured to the green-fluorescent stage. In previously performed trypsinolysis/MALDI experiments, we had been unsuccessful in detecting any oxidized intermediate states (6), therefore we proceeded with kinetic assays that monitor hydrogen peroxide evolution. In spite of numerous attempts under a wide variety of experimental conditions and pH values, we were unable to observe hydrogen peroxide production upon E222Q maturation, suggesting that the protein may already be oxidized.
To determine of the types of intermediates trapped in flash-frozen E222Q, we collected reverse-phase HPLC fractions of full-length protein, and analyzed them by ultra-high resolution mass spectrometry entailing nano-electrospray ionization and LTQ Fourier Transform detection. The spectra provided isotopic resolution of the intact protein chain, which has a calculated mass of 31,078 Da in the absence of any covalent modifications. We found that the early fraction (“up-slope”) of the HPLC peak consisted almost entirely of mature E222Q. This HPLC fraction provided mass spectral peaks with isotopic envelopes that fit well to species with masses 31,058 amu (- 20 Da) and 31,074 amu (- 20 + 16 = - 4 Da) (Figure 2A). However, the mass spectrum did not provide clear evidence of either the precursor form (31,078 amu) or the dehydrogenated (oxidized) form (31,076 amu). The 31,074 species is consistent with the addition of one oxygen atom (+16 Da) to the mature protein, likely due to secondary oxidative events such as methionine sulfoxide formation. In support of non-specific oxidative events, the addition of oxygen atoms is further demonstrated by LTQ-FTMS spectra collected on mature E222Q, where the observed bands are consistent with masses of 31,058 amu (mature), 31,074 amu (mature + 16 Da), and 31,090 amu (mature + 32 Da) (see supplementary Figure 1).
Figure 2.

Mass spectra of full-length immature E222Q protein isolated in soluble form and purified by HPLC. The isotope-resolved experimental spectra were collected on a Finnigan LTQ-Fourier Transform Mass Spectrometer. The isotopic envelopes represented by colored dots are the theoretical spectra calculated for the following masses: Red, 31,058 amu (mature protein, - 20 Da); Green, 31,076 amu (protein oxidized via dehydrogenation, - 2 Da); Orange, 31,078 amu (protein precursor, mass unmodified); Blue, 31,092 amu (dehydrogenated protein with addition of one oxygen atom, -2 Da + 16 Da); A. Spectrum of the early-eluting HPLC fraction. B. Spectrum of the late-eluting HPLC fraction. C. Superimposition of spectra shown in A and B (red = A, blue = B).
The mass spectrum collected for the bulk of the HPLC peak (“down-slope”) entailed three main bands, each with isotopic resolution (Figure 2B). The envelope of the most prominent band was reasonably well modeled by a species of mass 31,076 amu, i.e. a 2 Da loss from the mass of the precursor form, in-line with protein oxidation via the net loss of two hydrogen atoms. A minor species with slightly larger mass appeared as a small shoulder in the isotopic envelope, consistent with the unmodified precursor form (31,078 amu). In addition, a peak consistent with 31,092 Da (- 2 +16 Da) was apparent, suggesting a secondary, non-specific oxygen atom addition to the dehydrogenated intermediate of the protein. Although this mass would also be consistent with a dehydrated peroxy intermediate (-18 + 32 Da), peroxy adducts tend to be chemically unstable and would hydrolyze rapidly under the acidic HPLC conditions, rendering this interpretation unlikely. Another species is observed slightly above noise level, providing some evidence for the mature form at 31,058 Da, although the isotopic envelope is not reliable at such low peak intensities. A species consistent with dehydration (-18 Da) that would provide a mass of 31,060 amu could not be identified clearly, as its contribution (if present) would fall within baseline noise of the spectrum (Figure 2B).
The LTQ-FTMS data demonstrate that the early-eluting protein population consists primarily of mature protein (- 20 Da), whereas the late-eluting population consists primarily of the dehydrogenated (oxidized) intermediate (- 2 Da). Trace amounts of the precursor are also observed, as indicated by the small amount of experimental peak area lying outside of the theoretical band modeled at 31,076 amu (Figure 2B, green). Based on the mass spectral analysis, the dehydrogenated protein species (with or without oxidative side reactions) dominate the immature protein population, as the peaks with monoisotopic masses of 31,076 and 31,092 amu are most pronounced (see spectral overlay shown in Figure 2C). Therefore, we estimate that the pre-oxidation species presents less than 10% of the starting material, consistent with the lack of H2O2 evolution (see above). Although the presence of small amounts of precursor could interfere with data interpretation, the time evolution of chromophore formation is well modeled by a single exponential over the entire pH range examined (Figure 3 inset). Therefore, the extracted rate constants are interpreted in terms of post-oxidation chemistry only, as the third step appears to be the dominant process under the experimental conditions employed.
Maturation of soluble E222Q involves removal of the Tyr66 Cβ-proton
Kinetic experiments on immature E222Q were carried out between pH 7.7 and pH 10.0 as described previously (6). Biosynthetic labeling of E222Q protein was performed as described for mGFPsol, and kinetic assays were carried out with both Cβ-dihydro- and Cβ-dideutero-tyrosines incorporated into the protein. The chromophore maturation rate was determined at 30 °C by collecting UV-Vis absorbance scans as a function of time, and determining the fractional increase in chromophore absorbance (483 nm) over time (Figure 3 inset). The time requirement for completion of the reaction varied substantially depending on pH and deuteration, and ranged from less than one hour to three days. Below pH 7.5, the maturation rate was too slow to allow for accurate data collection. For each kinetic experiment, the data were fit to a rate equation describing a pseudo-first order reaction (Figure 3 inset) (6), and a complete pH-rate profile was generated for the di-hydro and the di-deutero E222Q proteins (Figure 3). In both cases, the profile fit well to an expression describing a base-catalyzed reaction, in which the observed rate constant kobs depends on the acid dissociation constant Ka of a titratable group, and an intrinsic, pH-independent rate constant kint. The corresponding time constant τint was extracted to be 8.38 ± 0.55 min for unlabeled E222Q, and that for the isotopically enriched material was determined to be 48.21 ± 3.92 min (Table 2). From these values, the deuterium isotope effect kH/kD was determined to be 5.75 ± 0.35, consistent with a full primary KIE. Therefore, C-H bond cleavage at the β-carbon of Tyr66 constitutes the major rate-limiting event of the process under consideration. As proton abstraction is fully rate-determining, and the immature protein population consists primarily of oxidized species (-2 Da), we conclude that the rate constant kint determined for E222Q describes the third of the three-step maturation process. This rate constant is therefore equivalent to k3 in mGFPsol maturation (Scheme 2).
Table 2.
Kinetic constants for E222Q maturation extracted from pH-rate profiles.
| pH-rate profile | Cβ-tyrosine labeling of protein pool | Intrinsic time constant τint (min) (± Std. Dev.) |
pKa (± Std. Dev.) |
|---|---|---|---|
| present study | di-hydro-tyrosines | 8.38 (± 0.55) |
9.46 (± 0.07) |
| di-deutero-tyrosines | 48.21 (± 3.92) |
9.41 (± 0.09) |
|
| kH/kD (± Std. Dev.) |
5.75 (± 0.35) |
---- | |
| previous study (reference 6) | di-hydro-tyrosines | 14.3 (± 3.0) |
9.2 (- 0.2; +0.4) |
The substantial pH-dependence of kobs (Figure 3) leads to extraction of an apparent pKa value of 9.46 (± 0.07) for di-hydro protein, and 9.41 (± 0.09) for di-deutero protein (Table 2). These data suggest that a protein group with high pKa value functions as a general base in proton abstraction, and that the acid dissociation constant of the titrating catalyst is not affected by the deuterium label. The large deuterium isotope effect supports a model in which proton transfer occurs in the transition state of the rate-limiting event, suggesting that Arg96 may be the general base (see discussion).
Incorporation efficiency of Cβ-di-deuterated tyrosine
To quantize the level of biosynthetic incorporation of the deuterium label, we first determined the masses of proteolytic peptides by MALDI. Deuterated protein derived from inclusion bodies was digested with trypsin, and peptides were separated by reverse-phase HPLC. Masses of eluting peptides were matched with calculated masses obtained from theoretical tryptic digests. The HPLC fraction eluting at 51 - 52 % ACN contained three peptides, each bearing one tyrosine residue. Two of these peptides included the chromophore-forming residues 65-67, and one was derived from the C-terminus (peptides 53-73, 46-73, and 216-238). The monoisotopic masses of principle peaks obtained from the MALDI spectra indicated a mass increase of 2 ± 0.4 Da compared to the calculated masses in the absence of isotopic enrichment (see Supplementary Table 1). To better quantize the percent enrichment, the three peptides were subjected to nano-HPLC ion trap mass spectrometry (see Supplementary Table 2). For each peptide, the mass was shifted by 2.0 Da relative to the theoretical mass, and the observed isotope distribution agreed exactly with the expected distribution calculated for the D2 (di-deutero) version. Peak area determination of the second (major) isotope peak for the D2 and H2 (di-hydro) peptides was utilized to estimate an incorporation efficiency of 96.6 ± 3.1 percent. This value constitutes a lower limit, as the H2 peptide peak area was found to be near baseline noise. Therefore, the data are consistent with essentially complete incorporation of the deuterium label, and 100% incorporation efficiency of Cβ-di-deuterated tyrosine residues was assumed for all kinetic calculations.
Discussion
Proton transfer steps in GFP maturation, and models for exocylic C-H bond scission
The overall GFP maturation reaction requires several proton transfer steps, such as deprotonation of the Gly67 amide nitrogen during ring closure, and deprotonation of the Tyr66 α-carbon to promote enolization of the heterocycle (Scheme 1) (6). Neither the cyclization reaction nor the associated proton transfer steps limit the overall rate of chromophore biosynthesis, as they appear to proceed within the first few minutes of the reaction (Table 1) (11). However, to produce the mature chromophore, a carbon-hydrogen bond must be broken at the β-carbon of Tyr66 (Cβ66), as this carbon center changes its hybridization state from sp3 to sp2 upon maturation. A reasonable mechanism may involve homolytic bond cleavage, which would place an unpaired electron onto the exocyclic carbon center upon H-atom transfer to an activated oxygen species (28). Nonetheless, data presented here support heterolytic bond scission with proton abstraction from the exocyclic carbon center by a near-by base. This event could be mediated by a peroxy anion species generated upon reduction of dioxygen (9, 14). However, the kinetic data demonstrate that deprotonation of Cβ66 occurs after the release of H2O2 from the active site (Scheme 3). Evidently, the nascent carbanion triggers a series of bond rearrangements that result in the expulsion of the hydroxyl leaving group from the heterocycle. Cβ66 may be activated by the electron withdrawing properties of the imidazolinone ring, in combination with π-orbital delocalization over the phenolic group. Glu222 may assist in this process by donating a proton to the hydroxyl group attached to C65, making use of a proton relay system that involves two ordered water molecules in highly conserved positions (9, 10) (Figure 3).
Scheme 3.

Proposed mechanism for stereospecific deprotonation of Cβ66 by general base catalysis involving Nη2 of Arg96. Carbanion formation is thought to trigger dehydration of the heterocycle, a process that may be facilitated by the Glu222 carboxylic acid as a proton donor.
Chromophore biosynthesis proceeds via oxidation of the five-membered ring to the cyclic imine
Results presented here provide strong support for a mechanism in which the hybridization state of the bridging Cβ66 is not affected by the reduction of molecular oxygen. Specifically, H2O2 evolution is not accompanied by a detectable deuterium isotope effect for C-H bond cleavage at Cβ66 (Table 1). Based on these data, protein oxidation appears to solely involve the heterocyclic functionality (9, 14). The absence of a KIE on oxidation confirms a mechanistic scheme in which the protein is first oxidized to yield the cyclic imine form (Scheme 1), as originally proposed based on the X-ray structure of the Y66L variant (9). In support of this idea, we have previously identified a - 2 Da intermediate generated during mGFPsol maturation (11). Here, we present ultra-high resolution mass spectral evidence that a - 2 Da intermediate of the E222Q variant can be trapped by flash freezing rapidly purified protein. The isotopically resolved mass spectrum of full-length E222Q indicates that the majority of the immature protein population has lost the mass equivalent of two hydrogen atoms (Figure 2).
The final maturation process consists of rate-limiting proton abstraction from the bridging carbon
In this work, we provide direct evidence that bond rearrangement at Cβ66 occurs post-oxidation, as a six-fold primary deuterium isotope effect is observed for this step upon deuteration of the exocyclic carbon (Table 1). The kinetic data provide nearly identical KIE values for τ3 and τint, determined for mGFPsol and E222Q to be 5.8 and 5.9 respectively (Tables 1 and 2). As these values refer specifically to the third and final step in GFP maturation (Scheme 1), a proton must be removed from the bridging carbon in the transition state of the rate-determining event of this process. Evidently, proton transfer initiates bond rearrangements that lead to full π-orbital conjugation of the phenolic group with the heterocycle, while the hydroxyl adduct is ejected from the five-membered ring (Scheme 3) (1, 9). The pKa of Cβ66 likely resides above physiological pH values, as carbon acids tend to bear pKa values of 20 or higher (29). For this reason, proton abstraction in the third and final step contributes substantially to the overall rate-retardation observed in chromophore biogenesis.
In the present study, the intrinsic time constant τint for full E222Q maturation from the oxidized intermediate state is extracted to be 8.4 (± 0.6) min, as determined by curve fitting of the pH-rate profile (Figure 3 and Table 2). The kinetic model entails a base catalyzed reaction, in which the observed rate constant is a function of an intrinsic time constant and an acid dissociation constant. In a previously published study, the time constant was extracted to be 14.3 (± 3.0) min, although fewer data points were collected over the relevant pH range (Table 2) (6). A precise determination of the intrinsic time constant depends critically on the collection of high pH data, however, the pH-rate profile levels off above pH 11, where protein denaturation interferes with kinetic measurements. Taken together, the E222Q data support an intrinsic time constant in the range of 8 - 14 min, a value comparable in magnitude to that determined for τ3 in mGFPsol, 15.3 (± 5.5) min (Tables 1 and 2). The similarity in magnitudes suggests that the same protein group is responsible for catalyzing C-H bond cleavage at Cβ66 in both variants.
Catalysis involves a protein group with high pKa value, suggesting Arg96 as general base
The pH-rate profile for E222Q maturation from the oxidized intermediate to the green-fluorescent form supports general base catalysis with a conjugate acid pKa value of 9.4 (Figure 3). The introduction of Cβ-di-deuterated tyrosine residues does not modify the extracted dissociation constant (Table 2). As a primary isotope effect is observed, proton abstraction must occur in the transition state of this step, requiring that the titrating group is positioned in close vicinity to the carbon acid. Inspection of high-resolution crystal structures such as that of GFP-S65T (12) allows for the identification of only one protein-based group fitting these requirements, the guanidine/guanidinium group of Arg96. Therefore, we propose here that proton abstraction from the exocyclic carbon is facilitated by Arg96. We note that the kinetically equivalent mechanism, whereby catalysis is carried out by a hydroxide ion, may also be consistent with the data (30), because the slope of the curve fitted to a log-log plot is close to unity between pH 7.5 and 8.5. However, the curve levels off above pH 9.0, and the extracted pKa of 9.4 is judged to be too low to support hydroxide ion catalysis (pKa of water ∼ 16).
Arg96 appears well positioned to activate a water molecule for proton abstraction from Cβ66 (Scheme 3). One face of the mature chromophore typically entails a series of hydrophobic contacts, such as Phe165 in GFP or Pro63 in Anthozoa fluorescent proteins (31). Positioned near this face is the guanidinium group of Arg96, with each of its terminal nitrogen atoms Nη1 and Nη2 involved in two hydrogen bonding interactions. These include the highly conserved interaction between Nη2 and the carbonyl oxygen of the chromophore's imidazolinone ring, as well as a hydrogen bond between Nη1 and the carbonyl oxygen of Thr62. Geometric considerations render a solvent-mediated interaction of Nη1 with Cβ66 unlikely (distance 4.9 Å in S65T), as solvent access is blocked by the carbonyl oxygen of Thr62. In contrast, Nη2 is found within van der Waals distance to Cβ66, with a separation of 3.9 Å in S65T (Figure 4) (12). Strikingly, in the post-oxidation intermediate trapped in Y66L, this separation is further reduced to 3.7 Å (9), and the hydrogen bonding geometry observed for Nη2 is more in line with a tetrahedral amino group, a geometry that would stabilize the neutral guanidine form (see below). Therefore, we propose that proton transfer may be mediated by a water molecule serving as a proton shuttle to Nη2. The structural arrangement predicts that the pro-S proton may be abstracted from the cyclic imine intermediate in a stereospecific manner (Scheme 3). Although an appropriately positioned solvent molecule is not observed crystallographically, several ordered waters are found in the vicinity of the β-methylene bridge, suggesting that diffusion of trapped solvent may aid in full maturation. In support of this notion, the crystal structure of the chemically reduced α-enolate form of Y66H demonstrates that access of solvent to Cβ66 is aided by the non-planar geometry of the nascent chromophore, as three ordered water molecules are observed within 3.3, 3.7, and 3.9 Å of Cβ66 (10) (Figure 4). In addition, previous work has provided evidence that small molecules such as iodide are able to access Cβ66 and Arg96 (16), in line with the observation that formate catalyzes deprotonatation at the exocylic carbon in the Y66L variant (14).
Figure 4.
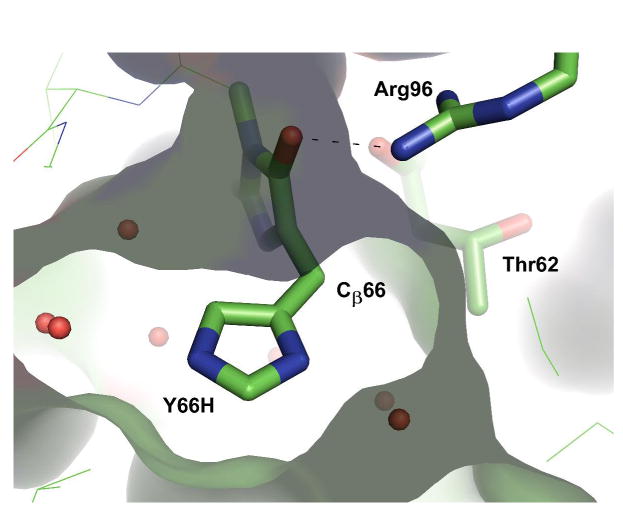
Structural arrangement of Arg96 Nη2 in relation to Cβ66, as exemplified by the X-ray structure of the chemically reduced Y66H variant (10). The image was generated from pdb ID code 2fwq (10) using the program PyMol (36). The molecular surface of the chromophore binding pocket is also shown.
pKa depression of Arg96 during GFP maturation?
Typically, arginine residues titrate with a pKa value of about 12.5 in aqueous solution, suggesting that the acid dissociation constant of Arg96 may be depressed by 3 pKa units in E222Q. Substantial pKa perturbations may occur upon protein folding due to the enclosure of the guanidinium group by the protein matrix, and sequestration away from bulk solvent. Based on theoretical calculations, the guanidine/guanidinium group is thought to adopt a series of low-energy conformations that may provide a variety of hydrogen bonding geometries, as energetically favored structures include both pyramidal and planar Nη1 and Nη2 centers (29). Rapid conformational interconversions could facilitate transient interactions that differ from those observed in mature GFPs, where Arg96 is almost certainly cationic. Distortion towards a pyramidal amino geometry, which is favored by the neutral form, could be enforced by appropriately positioned hydrogen bonding partners (29). Most importantly, the protonation equilibria in play while maturation is in progress may not reflect the final pKa value of Arg96 in the green-fluorescent state. The polarizability of the mature chromophore's π-system may lead to electrostatic stabilization of the cationic form by allowing for increased negative charge density at the imidazolinone oxygen.
Previously, we have demonstrated that in intact GFPs, the total in vitro processing time is essentially invariant over the pH range of 8 - 10, where overall maturation proceeds with a time constant of 1.0 (± 0.1) hour (6). These results suggest that the pKa of Arg96 may drop below 8 while the biosynthetic reaction is in progress. However, it is difficult assess the specific pH-dependence of step 3 in relation to all other steps, as the overall process includes protein folding in addition to the chemical steps. Remarkably, at pH 7.0, the time constant for EGFP maturation was observed to be 1.3 (± 0.6) hours, suggesting slower chromophore biosynthesis at lower pH values (6). We speculate that these data reflect a rate reduction in step 3 upon protonation of Arg96, however, the measurement error prohibits a detailed mechanistic interpretation. Compared to intact GFPs, the pKa value of Arg96 may be modulated by the E222Q protein environment such that its value is elevated to 9.4.
Inefficient maturation in Arg96 mutants of GFP
Arg96 has been demonstrated to fulfill catalytic functions in chromophore biogenesis, as methionine and alanine substitutions are known to slow down maturation substantially. In particular, the R96M variant exhibits a 5000-fold rate reduction in acquisition of green fluorescence (6). Evidently, this variant remains trapped in the pre-cyclization state, complicating an analysis of the specific involvement of arginine in the post-oxidation step. Similar to R96M, the side chain truncation variant R96A exhibits an extremely slow main-chain cyclization reaction as well (5). Although it may be feasible to test the catalytic role of Arg96 by subjecting R96A to chemical rescue experiments involving guanidine (32), this approach could be complicated by the non-native packing arrangements observed for Tyr66 in the truncation variant (5).
Importantly, the activity of Arg96 can be compensated for by a lysine residue (7). The R96K variant is able to mature to the green fluorescent state, albeit with reduced efficiency, as the ε-amino group is not directly hydrogen bonded to the chromophore's carbonyl oxygen (8). Although the R96K variant generates visible fluorescence upon overnight expression, only 30 to 50% of the protein population bears a mature chromophore. In addition, the protein fold is destabilized, as indicated by increased trypsin sensitivity (7).
Arginine as a general base in enzymatic reactions
Several enzymatic reactions have been described in which arginine residues are thought to play the role of acid-base catalyst in proton transfer reactions. Likely the best-studied example is the hydrolysis reaction of inosine monophosphate dehydrogenase (IMPDH) (30), where it has been demonstrated that guanidine derivatives can rescue the activity of the active-site R418A mutant in a pH-dependent manner (32). In this system, a lysine residue is able to replace the function of arginine with only a 10-fold rate reduction (33). The IMPDH hydrolysis reaction presents an important analogy to the mechanism proposed here for GFP, as a water molecule appears to be activated by the catalytic Arg418 in the transition state of proton transfer. The appropriately placed solvent molecule was only observable in an inhibitor complex (34), indicative of intermediate states that exhibit altered solvent positions. Surprisingly, kinetic data have suggested that the general base titrates with a pKa of about 8, in line with a substantially perturbed arginine pKa in the enzyme's active site (33). Deprotonation of a carbon acid by an arginine residue is further exemplified by the pectate/pectin lyases and by fumarate reductase (29). In pectate lyase, a catalytic arginine residue has been implicated in direct proton abstraction, and theoretical calculations have provided an estimate of 9.5 for its pKa value (35). However, to-date, no direct experimental measurements of pKa values are available for any of the proposed active site arginines in these enzyme systems.
A proposed role for Glu222 in dehydration
Our data do not support a direct role for Glu222 in proton abstraction from the exocyclic Cβ66. In the E222Q variant, the rate of the post-oxidation step is reduced by a factor of 7 near physiological pH values (6), an impairment that seems insufficient to suggest a basic function for Glu222 in the fully rate-determining process (29). Also, the position of the Glu222 carboxyl group in relation to the nascent β-methylene bridge provides an inappropriate geometry for proton removal. A more reasonable role for Glu222 would be that of proton donor to the hydroxyl leaving group, a function that would facilitate the ejection of water from the heterocycle by making use of a proton relay involving two water molecules (Scheme 3).
Conclusions
In this work, we provide several lines of evidence that GFP maturation involves base-catalyzed proton abstraction at Cβ66, and that this process succeeds heterocyclic oxidation. Ultra-high resolution mass spectrometric data on E222Q demonstrate that the observed acid dissociation constant of 9.4 cannot be ascribed to the Gly67 amide nitrogen as originally proposed (6), as the majority of the protein population is already oxidized to the cyclic imine form and therefore post-cyclization. Instead, we propose that this pKa value may be ascribed to an active site arginine residue, Arg96. To-date, Arg96 has been suggested to play a variety of roles in chromophore biosynthesis, ranging from conformational backbone pre-organization to electrostatic catalysis, with effects such as pKa depression of main chain atoms and stabilization of activated oxygen species. The interpretation presented here adds the novel function of general base catalyst for this versatile and indispensable residue found in all fluorescent proteins.
Supplementary Material
MALDI and ESI data on tryptic peptides with and without isotope enrichment; LTQ-FTMS spectrum of full-length mature E222Q protein. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Linda Breci at the Proteomics Core Facility of the University of Arizona for the collection of mass spectrometric data to estimate the efficiency of deuterium incorporation.
Abbreviations
- KIE
kinetic isotope effect
- FP
fluorescent protein
- GFP
green fluorescent protein
- MALDI
matrix-assisted laser desorption ionization mass spectrometry
- ESI
electrospray ionization mass spectrometry
- LTQ-FTMS
linear ion trap quadrupole Fourier transform mass spectrometry
- HPLC
high pressure liquid chromatography
- ACN
acetonitrile
- TFA
trifluoroacetic acid
- amu
atomic mass units
Footnotes
This work was supported by a grant from the National Science Foundation (NSF MCB-0615938) and a grant from the National Institutes of Health (NIH RO3-EB006413) to R. M. W. NSF Grant CHE-0131222 provided funds to purchase the MALDI instrument.
References
- 1.Wachter RM. Chromogenic cross-link formation in green fluorescent protein. Acc Chem Res. 2007;40:120–127. doi: 10.1021/ar040086r. [DOI] [PubMed] [Google Scholar]
- 2.Wachter RM. The family of GFP-like proteins: Structure, function, photophysics and biosensor applications. Photochem Photobiol. 2006;82:339–344. doi: 10.1562/2005-10-02-IR-708. [DOI] [PubMed] [Google Scholar]
- 3.Tsien RY. The Green Fluorescent Protein. Ann Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 4.Remington SJ. Fluorescent proteins: maturation, photochemistry and photophysics. Curr Opin Struct Biol. 2006;16:1–8. doi: 10.1016/j.sbi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Barondeau DP, Putnam CD, Kassmann CJ, Tainer JA, Getzoff ED. Mechanism and energetics of green fluorescent protein chromophore synthesis revealed by trapped intermediate structures. Proc Natl Acad Sci USA. 2003;100:12111–12116. doi: 10.1073/pnas.2133463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sniegowski JA, Lappe JW, Patel HN, Huffman HA, Wachter RM. Base catalysis of chromophore formation in Arg96 and Glu222 variants of green fluorescent protein. J Biol Chem. 2005;280:26248–26255. doi: 10.1074/jbc.M412327200. [DOI] [PubMed] [Google Scholar]
- 7.Sniegowski JA, Phail ME, Wachter RM. Maturation efficiency, trypsin sensitivity, and optical properties of Arg96, Glu222, and Gly67 variants of green fluorescent protein. Biochem Biophys Res Comm. 2005;332:657–663. doi: 10.1016/j.bbrc.2005.04.166. [DOI] [PubMed] [Google Scholar]
- 8.Wood TI, Barondeau DP, Hitomi C, Kassmann CJ, Tainer JA, Getzoff ED. Defining the role of arginine 96 in green fluorescent protein fluorophore biosynthesis. Biochemistry. 2005;44:16211–16220. doi: 10.1021/bi051388j. [DOI] [PubMed] [Google Scholar]
- 9.Rosenow MA, Huffman HA, Phail ME, Wachter RM. The crystal structure of the Y66L variant of green fluorescent protein supports a cyclization-oxidation-dehydration mechanism for chromophore maturation. Biochemistry. 2004;43:4464–4472. doi: 10.1021/bi0361315. [DOI] [PubMed] [Google Scholar]
- 10.Barondeau DP, Tainer JA, Getzoff ED. Structural evidence for an enolate intermediate in GFP fluorophore biosynthesis. J Am Chem Soc. 2006;128:3166–3168. doi: 10.1021/ja0552693. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Patel HN, Lappe JW, Wachter RM. Reaction progress of chromophore biogenesis in green fluorescent protein. J Am Chem Soc. 2006;128:4766–4772. doi: 10.1021/ja0580439. [DOI] [PubMed] [Google Scholar]
- 12.Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria Green Fluorescent Protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 13.Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Understanding GFP chromophore biosynthesis: Controlling Backbone cyclization and modifying post-translational chemistry. Biochemistry. 2005;44:1960–1970. doi: 10.1021/bi0479205. [DOI] [PubMed] [Google Scholar]
- 14.Rosenow MA, Patel HN, Wachter RM. Oxidative chemistry in the GFP active site leads to covalent cross-linking of a modified leucine side chain with a histidine imidazole: Implications for the mechanism of chromophore formation. Biochemistry. 2005;44:8303–8311. doi: 10.1021/bi0503798. [DOI] [PubMed] [Google Scholar]
- 15.Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Understanding GFP posttranslational chemistry: Structures of designed variants that achieve backbone fragmentation, hydrolysis, and decarboxylation. J Am Chem Soc. 2006;128:4685–4693. doi: 10.1021/ja056635l. [DOI] [PubMed] [Google Scholar]
- 16.Wachter RM, Yarbrough D, Kallio K, Remington SJ. Crystallographic and energetic analysis of binding of selected anions to the yellow variants of green fluorescent protein. J Mol Biol. 2000;301:159–173. doi: 10.1006/jmbi.2000.3905. [DOI] [PubMed] [Google Scholar]
- 17.Wachter RM, Remington SJ. Sensitivity of the yellow variant of green fluorescent protein to halides and nitrate. Curr Biol. 1999;9:R628–R629. doi: 10.1016/s0960-9822(99)80408-4. [DOI] [PubMed] [Google Scholar]
- 18.Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. Fluorescent proteins from nonbioluminescent Anthozoa species. Nature Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]
- 19.Remington SJ, Wachter RM, Yarbrough DK, Branchaud BP, Anderson DC, Kallio K, Lukyanov KA. zFP538, a yellow fluorescent protein from Zoanthus, contains a novel three-ring chromophore. Biochemistry. 2005;44:202–212. doi: 10.1021/bi048383r. [DOI] [PubMed] [Google Scholar]
- 20.McIntosh LP, Dahlquist FW. Biosynthetic incorporation of 15N and 13C for assignment and interpretation of nuclear magnetic resonance spectra of proteins. Q Rev Biophys. 1990;23:1–38. doi: 10.1017/s0033583500005400. [DOI] [PubMed] [Google Scholar]
- 21.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: Applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 22.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV protease. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 23.Steiner T, Hess P, Bae JH, Wiltschi B, Moroder L, Budisa N. Synthetic biology of proteins: Tuning GFPs folding and stability with fluoroproline. PLoS ONE. 2008;3:e1680. doi: 10.1371/journal.pone.0001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 25.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the Green Fluorescent Protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 26.Crameri A, Whitehorn EA, Tate E, Stemmer WPC. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 27.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 28.Walsh CT. Posttranslational modification of proteins: Expanding nature's inventory. Roberts and Company Publishers; Greenwood Village, CO: 2006. [Google Scholar]
- 29.Schlippe YVG, Hedstrom L. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch Biochem Biophys. 2005;433:266–278. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 30.Hedstrom L, Gan L. IMP dehydrogenase: structural schizophrenia and an unusual base. Curr Opin Chem Biol. 2006;10:520–525. doi: 10.1016/j.cbpa.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Malo GD, Wang M, Wu D, Stelling AL, Tonge PJ, Wachter RM. Crystal structure and Raman studies of dsFP483, a cyan fluorescent protein from Discosoma striata. J Mol Biol. 2008;378:869–884. doi: 10.1016/j.jmb.2008.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlippe YVG, Hedstrom L. Guanidine derivatives rescue the Arg418Ala mutation of Tritrichomonas foetus IMP Dehydrogenase. Biochemistry. 2005;44:16695–16700. doi: 10.1021/bi051603w. [DOI] [PubMed] [Google Scholar]
- 33.Schlippe YVG, Hedstrom L. Is Arg418 the catalytic base required for the hydrolysis step in the IMP dehydrogenase reaction? Biochemistry. 2005;44:11700–11707. doi: 10.1021/bi048342v. [DOI] [PubMed] [Google Scholar]
- 34.Gan L, Seyedsayamdost MR, Shuto S, Matsuda A, Petsko GA, Hedstrom L. The immunosuppressive agent mizoribine monophosphate forms a transition state analogue complex with inosine monophosphate dehydrogenase. Biochemistry. 2003;42:857–863. doi: 10.1021/bi0271401. [DOI] [PubMed] [Google Scholar]
- 35.Scavetta RD, Herron SR, Hotchkiss AT, Kita N, Keen NT, Benen JAE, Kester HCM, Visser J, Jurnak F. Structure of a plant cell wall fragment complexed to pectate lyase C. Plant Cell. 1999;11:1081–1092. doi: 10.1105/tpc.11.6.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeLano WL. The PyMOL molecular graphics system. 2002 World Wide Web http://www.pymol.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MALDI and ESI data on tryptic peptides with and without isotope enrichment; LTQ-FTMS spectrum of full-length mature E222Q protein. This material is available free of charge via the Internet at http://pubs.acs.org.