

Abstract
Background
Tyrosine kinase inhibitors (TKIs) have advanced cancer treatment. Sunitinib, a recently-approved, multi-targeted TKI, prolongs survival for patients with metastatic renal cell carcinoma (RCC) and gastrointestinal stromal tumor (GIST), but concerns about cardiac safety have arisen with this agent.
Methods
To determine the cardiovascular risk associated with sunitinib, we reviewed all cardiovascular events in patients with imatinib-resistant, metastatic GIST at the Dana-Farber Cancer Institute enrolled in a Phase I/II protocol evaluating the efficacy of the drug (n=75). Sunitinib’s effects on left ventricular ejection fraction (LVEF) and blood pressure (BP) were also examined. Studies in isolated cardiomyocytes and mice investigated potential mechanisms of sunitinib-associated cardiac effects.
Findings
Eleven percent (8/75) of subjects suffered a cardiovascular event with congestive heart failure (CHF) occurring in 8% (6/75) of the population. Twenty-eight percent (10/36) of patients treated at the FDA-approved dose had LVEF declines of ≥ 10 EF%, and nineteen percent (7/36) experienced LVEF declines of ≥ 15 EF%. Sunitinib induced significant increases in mean systolic and diastolic BP in patients, and 47% (35/75) of individuals developed hypertension (HTN) (>150/100 mmHg). CHF and LV dysfunction generally responded to withholding drug and instituting medical management. In mice and cultured cardiomyocytes, sunitinib caused mitochondrial injury and cardiomyocyte apoptosis.
Interpretation
Sunitinib treatment can lead to HTN, LVEF decline, and/or CHF. Experimental studies suggest that this is due, at least in part, to direct cardiomyocyte toxicity which may be exacerbated by HTN. Patients treated with sunitinib should receive close monitoring and prompt treatment for HTN and/or LVEF decline.
Introduction
Small molecule tyrosine kinase inhibitors (TKIs), designed to inhibit kinases that are mutated or over-expressed in cancer cells, have advanced the management of certain cancers such as chronic myeloid leukemia, renal cell carcinoma (RCC), and gastrointestinal stromal tumor (GIST).1 This targeted approach has provided enhanced anti-tumor activity with less toxicity than traditional chemotherapies for many patients. However, TKIs also inhibit normal variants of these kinases in non-cancerous cells. This may lead to unanticipated toxicities including cardiotoxicity.2-5 As demonstrated by clinical experience with trastuzumab,6-9 TKI-related cardiac dysfunction may be difficult to recognize in early clinical trials. Furthermore, symptoms of congestive heart failure (CHF) are often nonspecific and can be misattributed to malignancy alone.
Sunitinib (Sutent, Pfizer) is a recently FDA-approved, multi-targeted TKI that prolongs survival in patients with GIST and RCC.10-13 It inhibits cancer and angiogenesis targets including vascular endothelial cell growth factor receptors (VEGFR) 1-3, platelet-derived growth factor receptors (PDGRF) α and β, FMS-like tyrosine kinase-3 (Flt-3), c-kit (stem cell factor receptor), colony stimulating factor 1 receptor (CSF1R), and the ret oncogene product, RET.14-17 It is currently being evaluated for activity in more than 25 different tumor types in over 120 registered clinical trials, enrolling ~20,000 patients (www.clinicaltrials.gov/beta).
Most of our knowledge on the cardiac effects of sunitinib comes from oncology efficacy trials that examine overall safety.10-13,18 Although two cases of CHF were reported in a Phase I study,18 Demetri et al reported no systematic mean decrease in left ventricular ejection fraction (LVEF) in a phase III clinical trial in GIST patients.13 Motzer et al reported modest declines in LVEF without clinical sequelae in RCC patients.12,19 While the latter study hints at the potential for cardiotoxicity, the absence of studies designed primarily to assess sunitinib-associated cardiovascular dysfunction leaves many questions about potential cardiotoxicity unanswered.
We conducted a review focused on cardiac adverse events in 75 patients with imatinib-resistant, metastatic GIST treated with sunitinib for a median of 7·8 months at the Dana-Farber Cancer Institute (DFCI) as part of an early Phase I/II clinical trial. The objectives of this retrospective, single-center review were 1) to define the incidence of CHF, myocardial infarction (MI) and cardiovascular death for all GIST patients treated with sunitinib (n=75), and 2) to examine the effects on left ventricular (LV) systolic function and blood pressure (BP) for the subset of patients treated at the FDA-approved dose (n=36). Our findings, supported by endomyocardial biopsies from two index patients who developed CHF while being treated with sunitinib and by studies in cultured cardiomyocytes and mouse models, demonstrate the potential for cardiac adverse events with this agent.
Methods
Patients
Between April 2002 and June 2004, ninety-seven patients with imatinib-resistant GIST were treated with repeating cycles of sunitinib as part of an open-label, single-arm, dose-escalation Phase I/II trial at the DFCI, Massachusetts General Hospital, and Memorial Sloan-Kettering Cancer Center to evaluate drug efficacy and tolerability. A total of seventy-five patients were enrolled at the DFCI and formed the cohort of our cardiovascular study. Patients had detailed baseline examination and weekly follow-up visits with the DFCI oncology team during the trial. Longitudinal cardiac surveillance, including serial evaluation of LVEF each cycle by radionuclide ventriculography (RVG), and weekly BP and troponin I (TnI) measurements were instituted on October 22, 2002. BNP was not routinely monitored. The study was approved by the DFCI Institutional Review Board and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Cardiovascular Review
Three cardiovascular specialists (M.A.R., T.F., M.H.C.) developed endpoint definitions as guidelines to adjudicate cardiovascular events, using the standard cardiac criteria outlined below. Given preliminary data by Fiedler et al,18 cardiovascular events of interest were defined to be CHF and MI. The electronic medical records, including outpatient and inpatient notes, emergency room visits, cardiac testing, laboratory results, and information on causes and dates of death were reviewed (T.F.C., M.A.R., M.H.C.). All deaths and potential nonfatal cardiovascular events were identified. Prior to this study, the DFCI oncologists involved in the trial adjudicated all deaths as either cardiovascular or non-cardiovascular. Given the patient population’s complex co-morbidities, we categorized a patient death as cardiovascular only when there was concordance between the oncology and cardiology reviewers.
CHF
Patients were classified as having CHF if they had 1) documented symptoms and/or signs of CHF (i.e. dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, jugular venous distention, and/or pedal edema), 2) reduction of LVEF below lower limits of normal (EF < 50%), and 3) chest X-ray demonstrating pulmonary edema or symptomatic relief in response to CHF therapy (i.e. diuretics, inotropic support and/or vasodilator).
MI
Patients were classified as having a MI if the providers caring for the patient diagnosed a MI based on a typical elevation in cardiac biomarkers (i.e. TnI > 0·10 ng/ml) associated with clinical symptoms (i.e. chest pain) and relevant electrocardiogram (ECG) changes. Asymptomatic TnI elevations without ECG changes or without deterioration in LVEF were not considered a MI and were tallied separately.
LVEF Assessment and Incidence of Hypertension (HTN)
We analyzed serial LVEF and BP data from the thirty-six patients enrolled on the FDA-approved dosing schedule. Patients received repeating six week cycles of 50 mg daily for 4 weeks followed by 2 weeks of no treatment (50 mg; 4 wks on/2 wks off). Data were collected through cycle 4 (24 weeks), after which more than 50% of patients had been started on -blocker and/or angiotensin converting enzyme inhibitor (ACEI) for HTN control, which confounded our ability to quantify sunitinib-associated effects on LVEF and BP. Baseline and serial LVEF screening by RVG were obtained at a single center (DFCI) on the last day of drug administration in each cycle during the first four cycles, and every other cycle thereafter. Inter-study variability of LVEF by RVG at DFCI is 2-3 EF% (A. Van Den Abbeele, personal communication). All patients had baseline BP determination and then at least weekly BP measurements during clinic visits. Utilizing the National Cancer Institute Common Terminology Criteria for Adverse Events version 3·0,19 HTN was defined as systolic BP >150 mmHg or diastolic BP >100 mmHg.
Statistical Analysis
Cardiovascular death, MI and CHF were used as the composite cardiovascular endpoint. Pre-treatment variables assessed by univariate analysis included age, gender, history of anthracycline exposure, history of coronary artery disease (CAD), history of HTN, β-blocker use, or ACEI use.
Repeated-measures mixed-model regression analysis was applied to determine LVEF at baseline and after each of the first four cycles of sunitinib with 95% confidence intervals for mean LVEF after each cycle. The covariance structure that best fit the longitudinal data according to Akaike’s information criterion was compound symmetry, which we used to model the data.20 Baseline patient characteristics between the total cohort and the subgroup treated at the FDA-approved dose were compared with use of Fisher’s exact test (binary variables), Student t-test (continuous variables), and Mann-Whitney U-test (Non-Gaussian data).
Mechanistic Studies
Mechanisms of sunitinib-associated cardiotoxicity were examined in mouse and cell culture models. Methods for these studies are presented in the on-line supplement.
Role of Funding Source
The Phase I/II sunitinib trial was sponsored in part by Pfizer, who had no involvement in this cardiovascular study. The cardiovascular study was designed by M.H. Chen in collaboration with her academic colleagues and funded by Children’s Hospital Boston Department of Cardiology funds. As corresponding author, M.H. Chen had full access to all data in the study and final responsibility to submit for publication.
Results
Patient Demographics
Baseline characteristics for the entire cohort are summarized in Table 1. All patients discontinued imatinib or other systemic anti-cancer agents at least 2 weeks prior to starting sunitinib. All had a baseline LVEF ≥ 50 EF% and none had a prior history of CHF. Four patients had histories of CAD (mean baseline LVEF = 56 ± 4 EF%), but were without symptoms for at least one year prior to enrollment. All patients received doses at or below the eventual FDA-approved dose of 50 mg; 4 wks on/2 wks off with the exception of 4 patients. The latter received one-to-two weeks of sunitinib 75 mg, before the dose was reduced to 50 mg; 2 wks on/2 wks off. Median time on study was 33·6 weeks (range 3-112 weeks). Forty-eight percent (36/75) of patients went off-study secondary to disease progression. Thirty-nine percent (29/75) of subjects transferred to the sunitinib continuation protocol. Thirteen percent (10/75) of patients died within 30 days of their last drug dose and were considered on-study deaths.
Table 1.
Characteristics of Patients
| Characteristic (n=75) | |
|---|---|
| number of patients (percent) | |
| Male | 51 (68) |
| Mean age - years | 54·3±11·5 |
| ECOG* performance status | |
| 0 | 43 (57) |
| 1 | 30 (40) |
| 2 | 2 (3) |
| Sunitinib Treatment | |
| Median weeks on-study | 33·6 |
| Range (weeks) | 3·3-112·4 |
| Dosage | |
| (mg; weeks on drug/weeks off drug) | |
| 25; 2/2 | 6 (8) |
| 50; 2/2 | 23 (31) |
| 50; 2/1 | 6 (8) |
| 75; 2/2 | 4 (5) |
| 50; 4/2 | 36 (48) |
| Prior Cardiac History | |
| CAD | 4 (5) |
| CHF | 0 (0) |
| Baseline Cardiac Risk Factors | |
| HTN | 22 (29) |
| Smoking | 20 (27) |
| Diabetes | 8 (11) |
| Hyperlipidemia | 8 (11) |
| Cardiac Medications at Baseline | |
| ACE inhibitor | 6 (8) |
| Beta blocker | 10 (13) |
| Statin | 5 (7) |
| Prior Thyroid Disease | |
| Hypothyroidism† | 9 (12) |
| Prior Chemotherapy | |
| Anthracycline | 15 (20) |
| Imatinib | 75 (100) |
ECOG = Eastern Cooperative Oncology Group; 0 = Fully active, able to carry on all pre-disease performance without restriction, 1= Restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature, 2 = Ambulatory and capable of all self-care but unable to carry out any work activities; up and about more than 50% of waking hours.
History of hypothyroidism or elevated TSH at baseline.
LVEF Assessment
LVEF assessment was available each cycle in all patients after institution of routine LVEF monitoring in October 2002 (n=65). Ten patients went off-protocol prior to routine LVEF monitoring. All patients treated at the FDA-approved regimen (50 mg; 4 wks on/2wks off) had baseline and serial LVEF evaluation each cycle during drug administration.
Fatal and Nonfatal Cardiovascular Events
Eleven percent (8/75) of subjects suffered a cardiovascular event during the trial (Table 2A). One patient had a cardiac-related death due to peri-operative MI, pulmonary edema, and cardiovascular collapse, after palliative debulking surgery complicated by multi-organ system compromise. Death occurred five days after surgery. An additional patient with a history of CAD suffered a non-fatal, non-ST elevation MI. New York Heart Association (NYHA) class III-IV CHF developed in eight percent (6/75) of patients.21 All six CHF patients were being treated with the FDA-approved dose or lower at the time of the event. One patient had received one cycle of 75 mg; 2 wks on/2 wks off, followed by 81 weeks of 50 mg; 2 wks on/2 wks off prior to developing CHF. Clinical details of patients with CHF are contained in the On-line Supplement – Table 1. The median time to cardiovascular event was 30·5 weeks (range 10·7 - 84·9).
Table 2A.
Incidence of Cardiovascular and Fatal Events
| End Point | |
|---|---|
| number of patients (percent) | |
| Death from any cause | 10(13) |
| Death from cardiovascular causes | 1 (1) |
| Death from noncardiovascular causes | 9 (12) |
| Nonfatal cardiovascular events | |
| Myocardial infarction | 1 (1) |
| Heart failure | 6 (8) |
Other Cardiovascular Abnormalities
Weekly TnI levels were available in 68 patients (seven patients were enrolled prior to routine TnI monitoring). TnI elevations above normal developed in eighteen percent (12/68) of patients (Table 2B). Most elevations were modest (mean 0·74 ± 0·98 ng/ml, nl < 0·10 ng/ml). Investigation is ongoing to determine whether these TnI elevations can serve as a biomarker of sunitinib-associated myocardial injury, as has been shown for anthracyclines.22
Table 2B.
Incidence of LVEF < Lower Limits of Normal, Elevated Troponin, and Hypertension
| Cardiac Finding | ||
|---|---|---|
| number of patients (percent) | n | |
| LVEF < 50% | 13 (20) | 65 |
| Elevated troponin (> 0.10 ng/ml) | 12 (18) | 68 |
| Hypertension (> 150/100 mmHg) | 35 (47) | 75 |
Forty-seven percent (35/75) of the entire cohort developed HTN (systolic BP > 150 mmHg and/or diastolic BP >100 mmHg) (Table 2B).
Predictors of Cardiac Events
The only significant univariate associations for the composite cardiovascular endpoint of cardiovascular death, MI, and CHF were history of HTN (p=0·04) and history of CAD (p=0·003) (Table 3). Seventy-five percent (3/4) of the patients with a history of CAD suffered a cardiovascular event versus 7% (5/71) of those without CAD. Multivariable logistic regression analysis indicated that history of CAD was the only significant independent predictor (adjusted odds ratio = 39·6, 95% CI: 3·5 - 454, p=0·0004).
Table 3.
Predictors of Cardiovascular Events: Univariate Associations based on Logistic Regression
| Composite Cardiac Endpoint | CHF | |||||
|---|---|---|---|---|---|---|
| Variable | OR | 95% CI | P value | OR | 95% CI | P value |
|
|
|
|
||||
| Age, years | 1·04 | 0·97-1·11 | 0·30 | 1·06 | 0·97-1·14 | 0·17 |
| Gender | 0·43 | 0·10-1·87 | 0·26 | 0·44 | 0·08-2·35 | 0·34 |
| Anthracycline | 1·38 | 0·25-7·66 | 0·72 | 0·79 | 0·09-7·28 | 0·83 |
| CAD | 39·6 | 3·46-453·8 | 0·003* | 16·75 | 1·85-151·8 | 0·03* |
| HTN | 4·90 | 1·06-22·71 | 0·038* | 2·63 | 0·49-14·19 | 0·27 |
| β-blocker | 5·14 | 0·99-26·3 | 0·06 | 1·33 | 0·14-12·76 | 0·81 |
| ACEI | 0·01 | 0·00-ND | 0·23 | 0·00 | 0·00-ND | 0·31 |
Composite cardiac endpoint = cardiovascular death, MI, and CHF.
ND = upper limit of 95% CI is not determinable because none of the six patients taking ACEI at baseline had a cardiac event.
Statistically significant univariate predictor.
The only significant univariate association for predictors of CHF was history of CAD (p=0·03) (Table 3). Fifty percent (2/4) of patients with CAD developed heart failure, compared to 6% (4/71) without CAD. Multivariate logistic regression analysis indicated that CAD was the only significant independent predictor (adjusted odds ratio = 16·8, 95% CI: 1·9 - 152, p=0·012).
LV Systolic Dysfunction and HTN at the FDA-Approved Dose
Baseline characteristics of patients treated with 50 mg; 4 wks on/2 wks off were not significantly different from the total cohort (On-line Supplement - Table 2). None had a prior history of CAD or CHF. Mean baseline LVEF was 64·5 ± 6·5 EF%, and all patients had a baseline LVEF ≥ 50%. Eight percent (3/36) of patients treated at the FDA-approved dose developed NYHA class III-IV heart failure.21 Most patients experienced LVEF decline from baseline on sunitinib (Figure 1). Six percent (2/36) and nineteen percent (7/36) of patients developed LVEF decline ≥ 20 EF% and ≥ 15 EF%, respectively. Eleven percent (4/36) developed either CHF or LVEF decline of ≥ 20 EF% to an EF < 50%. Mean LVEF change by cycle for all patients was determined (Figure 2). Compared to baseline, there was a significant drop in mean LVEF after each of the four cycles. Mean decline over the four cycles was 5 EF%.
Figure 1.
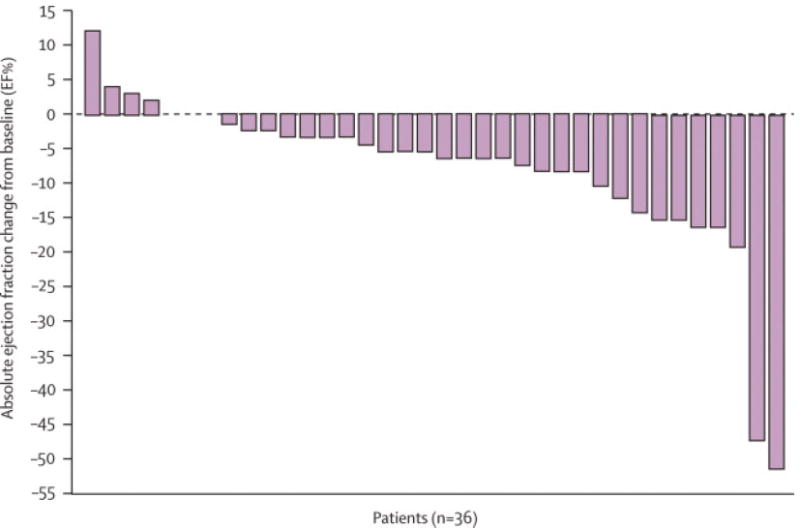
Maximal change in LVEF from baseline in patients treated at the FDA-approved dose. Individual data regarding absolute maximal change in LVEF from baseline to on-treatment with sunitinib 50 mg; 4 wks on/2 wks off is shown for all 36 patients. Three patients had no change in LVEF from baseline and are represented without bars.
Figure 2.
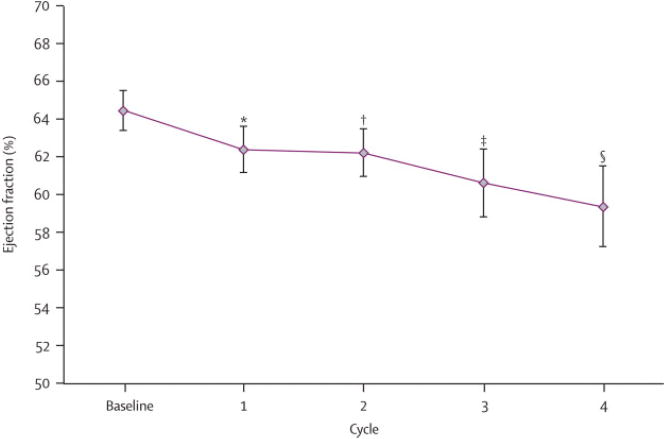
Model of predicted change in mean LVEF by treatment cycle. Modeling was performed using repeated-measures mixed-model regression analysis of LVEF data derived from the 36 patients treated over the first four cycles of treatment. There was a progressive decrease in mean LVEF with increasing cycles on sunitinib. The model predicts an initial 2% decline from baseline, followed by 1·5% decline per cycle for each subsequent cycle. Predicted LVEF % at baseline = 64·5; after cycle 1 = 62·4; after cycle 2 = 62·3; after cycle 3 = 60·6; and after cycle 4 = 59·4. * P=0·048 (cycle 1); * P=0·044 (cycle 2); * P=0·013 (cycle 3); ** P=0·007 (cycle 4).
Sunitinib induced significant BP elevation for the total cohort within the first 4 weeks on drug (cycle1) (mean increase in SBP = 21 ± 15 mmHg, DBP =14 ± 10mmHg) (Figure 3A). During sunitinib treatment, the mean maximum increase in SBP was 30 ± 15 mmHg and in DBP was 17 ± 12 mm Hg. Forty-seven percent (17/36) of patients treated with sunitinib at 50 mg; 4 wks on/2 wks off developed a SBP > 150 mmHg and/or DBP > 100 mmHg by cycle 4. Grade III HTN (defined as requiring more than one drug or more intensive therapy than previously) occurred in seventeen percent (6/36) of patients (Figure 3B).19 Even with half the cohort being treated with ACEI or -blockers for HTN by the end of cycle four, mean systolic and diastolic BP for the group remained elevated above baseline (Figure 3A). Furthermore, mean LVEF continued to decline despite the initiation of ACEI or -blockers (Figure 3B, Figure 2).
Figure 3.
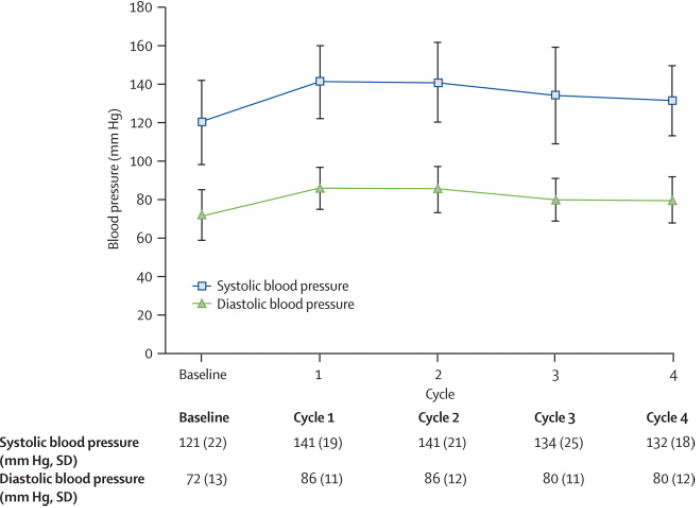
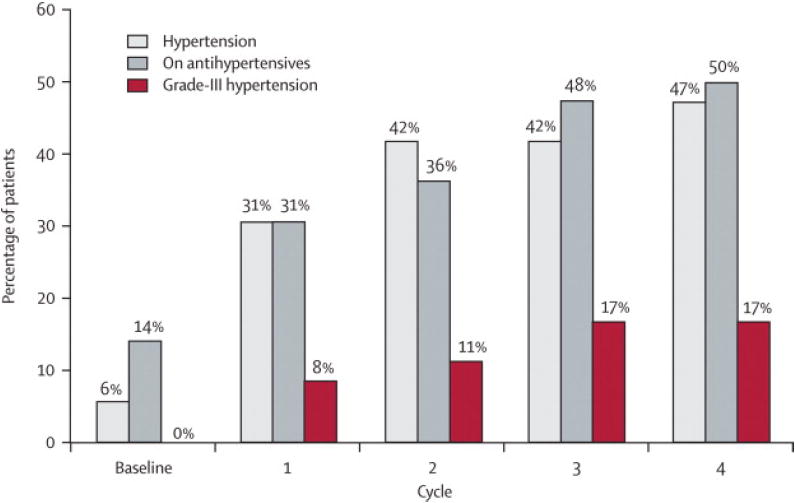
Effect of sunitinib 50 mg; 4 wks on/2 wks off on blood pressure.
A. Changes in mean systolic (SBP) and diastolic blood pressure (DBP) during the first 4 cycles of therapy. By the first cycle, SBP and DBP had significantly increased from baseline, and remained elevated through cycle 4.
B. Cumulative percentage of patients diagnosed with hypertension and on anti-hypertensive medication during the first four cycles (24 weeks) of sunitinib treatment (n=36). Hypertension was defined as > 150 mmHg systolic and/or > 100 mmHg diastolic. Grade III hypertension denotes patients who required more than one anti-hypertensive medication and/or required an increase in anti-hypertensive medication (National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0), prescribed at the discretion of the caring physician. By Cycle 4, almost half the cohort developed HTN, and 50% of patients were on anti-hypertensive medication.
Other Potential Cardiac Function Modulators
Serum electrolytes including phosphorous and calcium were monitored for all patients every two weeks and repleted as necessary. Hypocalcemia and hypophosphatemia was not seen in the patients with CHF or LVEFs below the lower limits of normal (< 50 EF%), and thus not believed to contribute to cardiac dysfunction.
Thyroid stimulating hormone (TSH) levels were available in 67 of 75 patients. Routine monitoring of thyroid function at baseline and at the beginning of each treatment cycle was initiated in all patients enrolled after October 2002 (n=53). Thyroid hormone supplementation was initiated as needed. We compared the mean times to onset of hypothyroidism versus CHF in patients treated with the FDA-approved dose of 50 mg; 4 wks on/2 wks off (n=36). Fourteen percent (5/36) of patients developed hypothyroidism after a mean of 54 weeks on drug. In contrast, the mean time to CHF was 27 weeks. Since CHF preceded hypothyroidism for all patients, hypothyroidism did not appear to significantly contribute to initial LV systolic dysfunction.
Endomyocardial Biopsies
Endomyocardial biopsies were obtained in two index patients who developed CHF and LV systolic dysfunction on sunitinib. Cardiomyocyte hypertrophy was present on light microscopy (Figure 4). Transmission electron microscopy demonstrated aberrantly-shaped, swollen mitochondria with effaced cristae, and membrane whorls. No inflammation, edema or fibrosis was seen (Figure 4).
Figure 4.
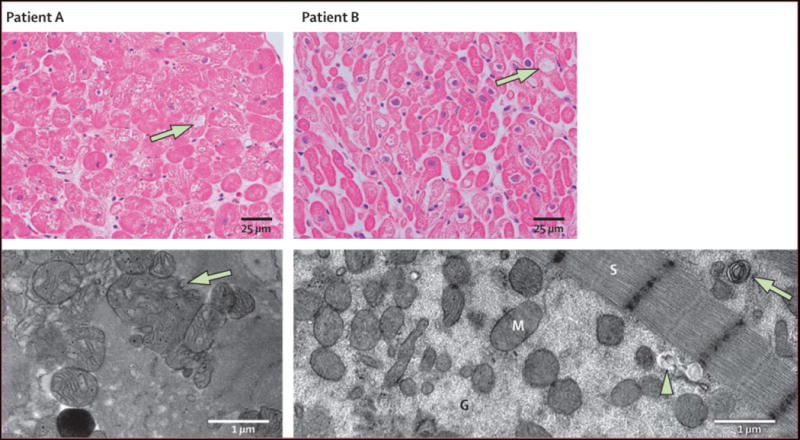
Endomyocardial biopsies obtained from two patients who developed CHF on sunitinib. Representative light photomicrographs (Patient A - top panels) show cardiomyocyte hypertrophy with mild degenerative changes and diffuse, moderate myocyte vacuolization (arrows). There was no edema, interstitial or replacement fibrosis, regional infarct or focal cell necrosis, or myocarditis or inflammation. TEM (Patient B - bottom left) demonstrates swollen, abnormal mitochondrial configurations (arrow) with effaced cristae. TEM (Patient B - bottom right) shows abundant cytoplasmic granular densities consistent with glycogen accumulation (G), membrane whorl (arrow), and lysosomal precipitates (arrowhead). Sarcomeres (S) appear well-organized and structurally normal in both patients. (Light microscopy, stained with hematoxylin and eosin, 400X; transmission EM, bar = 1 μm.)
Mechanisms of Sunitinib Cardiotoxicity
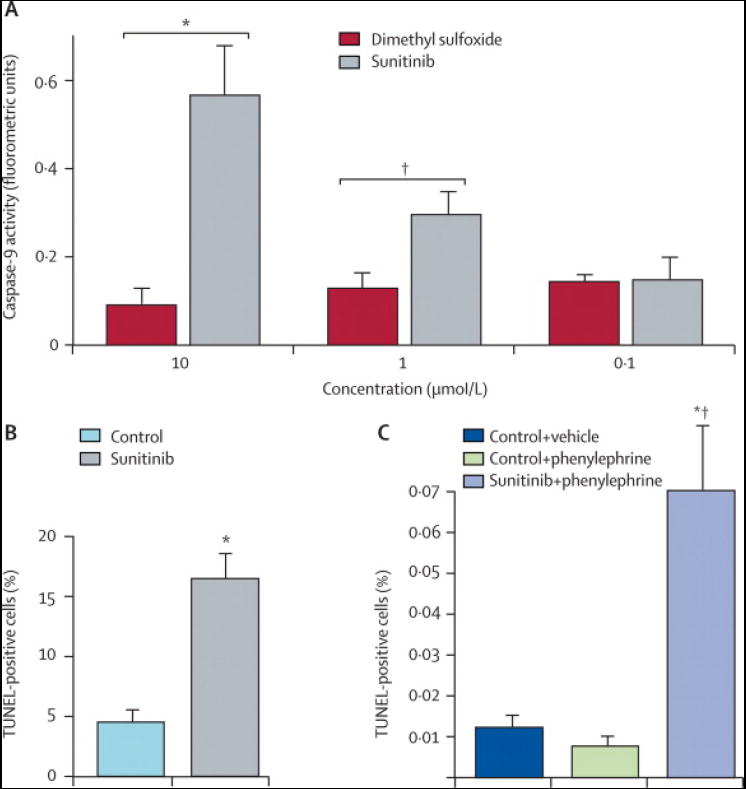
The clinical data suggest that sunitinib may be associated with cardiotoxicity. To determine if sunitinib is directly cardiotoxic, we examined sunitinib’s effects in mouse models and cultured cardiomyocytes. Mice treated for 12 days with sunitinib (40 mg/kg/d), a dose that produces blood levels comparable to those in patients,13,15,17,23 had striking abnormalities of cardiomyocytes on transmission electron microscopy with mitochondrial swelling and degenerative changes including membrane whorls and effaced cristae (Figure 5A). Sunitinib also targeted mitochondria in cultured cardiomyocytes, leading to cytochrome c release into the cytosol (Figure 5B). Cytochrome c release can activate the mitochondrial pathway for cell death or apoptosis.24 Consistent with this mechanism, sunitinib led to activation of caspase-9, an initiator caspase of the mitochondrial apoptotic pathway (Figure 5C, top). Sunitinib also induced apoptosis in cardiomyocytes as determined by TUNEL staining (Figure 5C, bottom).
Figure 5.
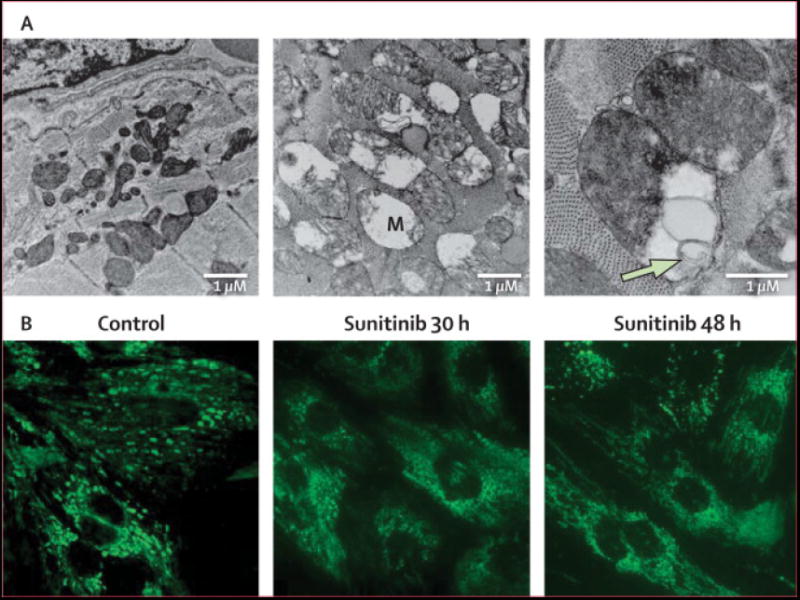
Effect of sunitinib on cardiac structure, mitochondrial function and apoptosis.
A. TEM of cardiac tissue from mice treated with control (left panel) or sunitinib (center and right panels) at 40 mg/kg/d for 10 days. Images from treated animals are notable for swollen mitochondria with disrupted cristae (M). Higher magnification image (right panel) illustrates membrane whorls (arrow) within the mitochondria of myocytes from sunitinib-treated mice.
B. Sunitinib induces cytochrome c release from mitochondria. Neonatal rat ventricular myocytes (NRVMs) in culture were incubated in media (control) or media containing sunitinib (1 μM) for the times shown. Cells were then stained for cytochrome c. The punctate staining in the control is consistent with mitochondrial-localized cytochrome c, while the diffuse staining in the sunitinib-treated cells reflects release of cytochrome c from the mitochondria into the cytosol.
C. Sunitinib activates Capase-9 and induces apoptosis.
Top: NRVMs were treated with vehicle (DMSO) or sunitinib at the concentrations noted for 40 hours. Caspase-9 activity was examined. Sunitinib resulted in significant increases in caspase-9 activity at 1 μM (** P=,0·02) and 10 μM (* P=0·006). Data are mean ± SD.
Bottom: Cardiomyocytes were treated with vehicle (control) or sunitinib (1 μM) for 44 hours, followed by TUNEL staining to detect apoptotic cells. * P=0·01 vs. control.
D. Sunitinib induces apoptosis in vivo in the setting of increased blood pressure. Mice were fed normal chow (control) or chow containing sunitinib (10mg/kg/d) for 2 weeks. During the second week, phenylephrine (PE; 30 mg/kg/d) vs. vehicle was administered via subcutaneous mini-pump. Heart sections were then stained for TUNEL. Graph depicts percent TUNEL positive cells for the various treatments. * P=0·02 vs. control + vehicle; # P = 0·002 vs. control + PE; n = 4 for control + vehicle; n = 6 for control + PE; n = 3 for sunitinib + PE.
Surprisingly, given the cardiomyocyte mitochondrial damage induced by sunitinib in vivo and in vitro, apoptosis was not significantly increased in hearts of sunitinib-treated mice (data not shown). In contrast to patients, however, sunitinib did not induce BP increases in the mice at the doses administered. Therefore, to study the effect of the interaction of sunitinib treatment and HTN on cardiac pathology, we treated mice for 2 weeks with sunitinib and added the pressor, phenylephrine (PE), an α-adrenergic agent, during the second week to mimic the BP increases seen in sunitinib-treated patients. SBPs increased from normal values of 90-100 mmHg to 127 ± 24 mmHg in mice treated with sunitinib + PE vs. 132 ± 14 mmHg in mice treated with PE alone (p=NS). Cardiomyocyte apoptosis was increased 7-fold in the hearts of the sunitinib + PE-treated mice compared to those treated with PE alone (p=0·002, Figure 5D).
Discussion
The principal finding of our study is that eleven percent of patients with imatinib-resistant, metastatic GIST treated with sunitinib for a median of 33·6 weeks in a Phase I/II clinical trial at the DFCI developed cardiac adverse events (i.e. CHF, MI or cardiac death). The most common event was NYHA class III-IV CHF, found in eight percent (6/75) of patients. Analysis of patients treated at the FDA-approved dose showed a steady decrease in LVEF over the first four cycles (24 weeks). Sunitinib also induced significant BP elevations with approximately 47% (35/75) of patients developing HTN (> 150/100 mmHg). In addition, sunitinib-associated cardiac dysfunction was associated in humans with abnormal histopathology involving myocyte hypertrophy and alterations in mitochondrial structure without inflammatory or fibrotic changes.
In contrast to our findings, later multi-center Phase III trials demonstrated less cardiotoxicity with sunitinib. Demetri et al reported no systematic mean LVEF decrease in imatinib-resistant GIST patients receiving the FDA-approved dose of sunitinib in a multi-center Phase III, placebo cross-over trial.13 Our data demonstrating sunitinib-associated LVEF decline is supported, in part, by Motzer et al, which reported a 10% incidence of LV systolic dysfunction in RCC patients treated for a median of 6 months. However, only 2% of RCC patients had grade III declines in LVEF (i.e. to < 40%) and none developed heart failure.12,19
Differences in follow-up period may partially explain the discrepancy between our findings to those of Demetri et al. This Phase III trial was prematurely unblinded after a median of 10 weeks, when the first interim analysis demonstrated improved survival with sunitinib. In our study, the median time to development of all cardiac adverse events was 30·5 weeks (range 10·7-84·9), with median time to onset of CHF at 33·4 weeks (range 10·7-84·9). Our data suggests further study should be undertaken to determine whether the longer exposure to sunitinib in our study may have allowed more opportunity for patients to develop cardiovascular sequelae.
The divergent findings from Demetri et al and Motzer et al may arise from differences in patient populations and data collection methods. Motzer’s study excluded all patients with prior anti-cancer therapies. All of our patients were previously treated with imatinib and twenty percent (15/75) were also treated with anthracycline, which may have contributed to the higher incidence of sunitinib-associated CHF in our study. Further investigation is warranted to determine the impact of drug history on cardiovascular susceptibility to sunitnib. That said, our studies with cultured cardiomyocytes and mice demonstrate that sunitinib can be cardiotoxic in the absence of prior imatinib exposure, at least in these models. Finally, both Phase III trials assessed LVEF by diverse non-invasive modalities (i.e. echo, RVG) at different centers in several countries. This would result in larger measurement variability compared with our study which used the same modality, imaging protocol, and interpretation team at a single center.
Cardiotoxicity assessment methodologies also differed between our study and later Phase III trials. The retrospective cardiovascular-focused record review conducted here proved to be a sensitive approach for detecting cardiac adverse events in this oncology population. While oncology trial data collection forms comprehensively capture patient symptoms, diagnoses such as CHF, that are made based on symptom complexes within a clinical context, may be challenging to extract. The common triad of CHF symptoms – dyspnea, fatigue, and peripheral edema – is categorized under three different organ systems (respiratory, constitutional symptoms, and lymphatics/cardiovascular, respectively), making it difficult to diagnose CHF. Significant percentages of the RCC patients developed dyspnea (15%), pitting peripheral edema (11%), and fatigue (58%).25 It is possible that some of these symptoms could have been due to unrecognized heart failure.
The Phase I/II study from which we derived our cardiovascular data included individuals with co-morbidities (i.e. CAD and HTN). This may have predisposed our patient population to LV dysfunction and CHF. Patients with uncontrolled HTN were excluded from later Phase III trials.12,13 Now that sunitnib is FDA-approved, the treated patient population is likely to include older adults with a history of CAD and other cardiac risk factors. Thus, we believe, our trial, in contrast to later Phase III trials with more selective entry criteria, may better reflect the patient population that will be receiving this drug.
Our study was limited by the small patient cohort derived from an early Phase I/II clinical trial at a single center. The lack of a placebo arm limits our ability to determine the incidence of CHF in untreated GIST patients. The generalizability of the findings from our protocol needs to be validated in larger prospective cardiovascular-focused studies in broader patient populations. The 120+ ongoing sunitinib trials could provide an opportunity to define further the nature and incidence of cardiac events with this agent.
Several angiogenesis inhibitors including sunitinib have been shown to cause HTN.19,26-28 This Phase I/II trial was among the first to identify the induction of HTN associated with sunitnib administration. The degree and rapid onset of HTN in our study were surprising, given later Phase III studies reporting a 15 to 24 % incidence of HTN with sunitinib,12,13 compared to the 47% we observed. The lower incidence of HTN observed in the Phase III trials may be because patients with uncontrolled HTN were excluded at trial entry.12,13 Our patient population also had weekly BP monitoring and therefore, more data points from which to assess incidence. In light of our mouse model findings suggesting HTN may play a role in myocardial injury and apoptosis, the contribution of HTN to sunitinib-associated LV dysfunction needs to be further examined.
LV dysfunction and symptoms improved in 5 out of 6 CHF patients after dose interruption, dose modification, and/or initiation of heart failure therapy (On-line Supplement – Table 1). Sunitinib was restarted in all five patients without recurrence of CHF. However, four patients experienced episodic LVEF declines after restarting. Whether restoration of normal LV function reflects true recovery at the cardiomyocyte level or compensatory cardiac remodeling remains to be seen. Transmission electron micrographs of patient endomyocardial biopsies and the mouse hearts which showed mitochondrial injury with no apoptosis (at least in normotensive mice) and no replacement fibrosis suggest potentially reversible injury. Indeed, it is possible that some of the contractile dysfunction in patients may be due to impaired ATP generation secondary to mitochondrial dysfunction, and not to irreversible damage and myocyte loss. Optimally, identification of sunitinib-associated LVEF decline would prompt medical intervention prior to the onset of symptomatic events (i.e. CHF).
In summary, the findings of our study reveal evidence of sunitinib-associated heart failure, LV systolic dysfunction, and/or HTN in imatinib-resistant GIST patients. Cardiovascular adverse events were medically manageable in the majority of cases. Close monitoring may be a prudent approach until larger studies further define the nature and incidence of sunitinib-associated cardiovascular effects, particularly in patients with cardiac risk factors and/or history of CAD. Further studies are also needed to define the most efficacious interventions for cardioprotection in these patients to avoid adverse cardiovascular events and enable uninterrupted, long-term sunitinib administration. Such understanding may guide clinical practice with other TKIs for which the spectrum of cardiac effects are not yet fully known.
Supplementary Material
Acknowledgments
We thank Dr. Joseph Loscalzo for his guidance with this work and Dr. Elliott Antman for helpful comments on the manuscript. We appreciate the assistance of Drs. Mami Hirata and Elaine Monteiro in data collection. We would also like to thank Dr. Yung Chang and Shao Meng Chen for their support.
Funding Source T.F.C. and M.H.C. were supported by the Translational Research Fund for Cancer and Cardiology at Children’s Hospital Boston. This work was also supported by grants from the National Heart Lung and Blood Institute (HL61688 and HL67371 to T.F.) and grants from the Finnish Heart Foundation and the Finnish Cultural Foundation (to R.K.). M.A.R. is supported by a grant from the NIH-NHLBI, American Heart Association, and Thomas Smith Award and S.M.D. is supported by NIH-NIDDK and David M. Bray Scholars in Medicine Award. G.D. is supported by Pfizer.
Authors’ Contributions
T.F. Chu participated in clinical study conception and design, collection and review of clinical data, clinical data analysis and interpretation, clinical figure and table conception and creation, manuscript writing, and project coordination. M.A. Rupnick participated in clinical study design, data collection, data analysis and interpretation, supervision of basic science studies in her lab, and manuscript writing. R. Kerkela performed mice and cardiomyocytes studies in T.F.’s lab including data analysis and interpretation, created basic science figures and tables, and assisted with manuscript writing. S. M. Dallabrida conceived, performed and analyzed the basic science studies in M.A.R.’s lab, constructed the figures and methods for those studies, and reviewed the text. D. Zurakowski performed the biostatistical analysis and interpretation for the clinical portion of the paper, constructed the figures, tables, and methods for those analyses, and assisted in manuscript writing. L. Nguyen participated in data collection, review, and analysis. K. Woulfe assisted with mice and cardiomyocyte studies in T.F.’s lab. E. Pravda conducted imaging for the basic science studies in M.A. Rupnick’s lab. F. Cassiola conducted mouse studies in M.A.R.’s lab. J. Desai participated in clinical study conception, provision of study material and patients, collection and assembly of data, data analysis and interpretation, and manuscript writing. S. George assisted in the initial conception of the study, provision of study material and patients, and collection and assembly of clinical data. J.A. Morgan participated in provision of study material and patients and collection and assembly of clinical data. D. Harris assisted with mice and cardiomyocyte studies in T.F.’s lab. N.S. Ismail conducted the mouse and cardiac myocyte studies in M.A.R.’s lab. J.H. Chen performed the pathology analysis with additional sectioning and constructed the pathology figures. F.J. Schoen supervised J.H.C. and analyzed the pathology sections in human and mice. G. Demetri was the Prinicipal Investigator of the Phase I/II trial and participated in study conception and design, financial support, provision of study material and patients, and manuscript writing. T. Force participated in clinical study design, supervised animal and cardiomyocyte studies in his lab, wrote the basic methods with R.K. and assisted in data analysis and interpretation and manuscript writing. M.H. Chen originally conceived of the manuscript and study design, collected, assembled, and reviewed the clinical data, analyzed and interpreted the results, wrote the manuscript, and was responsible for assembly of the clinical and basic teams and oversight of the entire project.
Footnotes
Conflicts of Interest Statement J. Desai has received speaker’s honoraria from and has served as a consultant for Pfizer. S. George has served as a consultant to Pfizer. G. Demetri has served as a consultant for Pfizer, Novartis, Bristol-Myers Squibb, Ariad, Johnson & Johnson, Genentech, Infinity Pharmaceuticals, ZymoGenetics, Alnylam, Idera, Bayer, and Serono, is a member of the Scientific Advisory Board for Plexxikon and ZioPharm, and has received research support from Daiichi-Sankyo. T. Force has served on the Speakers Bureau for Merck and Co., Inc. None of the other authors have potential conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Krause D, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 2.Fabian MA, Biggs WH, 3rd, Treiber DK, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lélias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23(3):329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 3.Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12(8):908–16. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 4.Mann DL. Targeted cancer therapeutics: the heartbreak of success. Nat Med. 2006;12(8):881–2. doi: 10.1038/nm0806-881. [DOI] [PubMed] [Google Scholar]
- 5.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7(5):332–44. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 6.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 7.Pietras RJ, Pegram MD, Finn RS, Maneval DA, Slamon DJ. Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene. 1998;17(17):2235–49. doi: 10.1038/sj.onc.1202132. [DOI] [PubMed] [Google Scholar]
- 8.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 9.Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, Rosen PP, Twaddell T, Henderson IC, Norton L. Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER 2/neu-overexpressing metastatic breast cancer. Semin Oncol. 1999;26(4 Suppl 12):78–83. [PubMed] [Google Scholar]
- 10.Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G, Ginsberg MS, Bacik J, Kim ST, Baum CM, Michaelson MD. Sunitinib in patients with metastatic renal cell carcinoma. Jama. 2006;295(21):2516–24. doi: 10.1001/jama.295.21.2516. [DOI] [PubMed] [Google Scholar]
- 11.Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol. 2006;24(1):16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- 12.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 13.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 14.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3(1):24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 15.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–37. [PubMed] [Google Scholar]
- 16.Morabito A, De Maio E, Di Maio M, Normanno N, Perrone F. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist. 2006;11(7):753–64. doi: 10.1634/theoncologist.11-7-753. [DOI] [PubMed] [Google Scholar]
- 17.Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24(1):25–35. doi: 10.1200/JCO.2005.02.2194. [DOI] [PubMed] [Google Scholar]
- 18.Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O’Farrell AM, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–93. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 19.Cancer Therapy Evaluation Program. Common terminology criteria for adverse events version 3.0 (CTCAE) Bethesda: National Cancer Institute; 2003. [accessed Oct 3, 2007]. http://ctep.cancer.gov/forms/CTCAEv3.pdf . [Google Scholar]
- 20.Lindstrom M, B D. Newton-Raphson and EM algorithms for linear mixed-effects models for replicated measures data. J Am Stat Assn. 83:1014–1022. [Google Scholar]
- 21.ACC/AHA 2005 guideline update for the diagnosis and managment of chronic heart failure in the adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure) American College of Cardiology Web Site. 2005. (Accessed at http://www.acc.org/qualityandscience/clinical/measures/HF/HFPerfMeasFinal2[1]032726.pdf) [DOI] [PubMed]
- 22.Urbanova D, Urban L, Carter A, Maasova D, Mladosievicova B. Cardiac troponins--biochemical markers of cardiac toxicity after cytostatic therapy. Neoplasma. 2006;53(3):183–90. [PubMed] [Google Scholar]
- 23.Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7(3):225–33. doi: 10.1007/s10456-004-3149-y. [DOI] [PubMed] [Google Scholar]
- 24.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38(1):47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Sutent (sunitinib malate) [U.S. package insert] New York, NY: Pfizer, Inc.; 2006. [Google Scholar]
- 26.Veronese ML, Mosenkis A, Flaherty KT, Gallagher M, Stevenson JP, Townsend RR, O’Dwyer PJ. Mechanisms of hypertension associated with BAY 43-9006. J Clin Oncol. 2006;24(9):1363–9. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 27.Sica DA. Angiogenesis inhibitors and hypertension: an emerging issue. J Clin Oncol. 2006;24(9):1329–31. doi: 10.1200/JCO.2005.04.5740. [DOI] [PubMed] [Google Scholar]
- 28.Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–73. doi: 10.1158/1078-0432.CCR-06-2328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.