

Summary
In metazoans, different cyclin-dependent kinases (CDKs) bind preferred cyclin partners to coordinate cell division. Here we investigate these preferences in human cells, and show that cyclin A assembles with Cdk1 only after complex formation with Cdk2 reaches a plateau, in late S and G2 phases. To understand the basis for Cdk2’s competitive advantage despite Cdk1’s greater abundance, we dissect their activation pathways by chemical genetics. Cdk1 and Cdk2 are activated by kinetically distinct mechanisms, even though they share the same CDK-activating kinase (CAK), Cdk7. We recapitulate cyclin A’s selectivity for Cdk2 in extracts, and override it with a yeast CAK that phosphorylates monomeric Cdk1, redirecting Cdk1 into a pathway normally restricted to Cdk2. Conversely, upon Cdk7 inhibition in vivo, cyclin B, which normally binds Cdk1 nearly exclusively, is diverted to Cdk2. Therefore, differential ordering of common activation steps promotes CDK-cyclin specificity, with Cdk7 acting catalytically to enforce fidelity.
Introduction
The eukaryotic cell cycle is driven by sequential activation and inactivation of cyclin-dependent kinases (CDKs), helping to ensure that chromosome duplication alternates with segregation to maintain genome stability (Morgan, 2007). In yeast, the same CDK catalytic subunit triggers both DNA replication and mitosis—versatility achieved in part by binding different cyclins that confer distinct substrate specificities (Loog and Morgan, 2005; Bloom and Cross, 2007). In metazoans, multiple catalytic subunits pair with preferred cyclin partners to execute specific functions: Cdk4 and Cdk6 with D-type cyclins to control G1 progression, Cdk2 with cyclins E and A to promote S phase, and Cdk1 with cyclins A and B to trigger mitosis. These combinations are presumably optimized to regulate cell cycle events appropriately in response to internal, environmental and developmental cues.
Results of Cdk gene disruptions in mice challenged this model, and raised questions about the true extent of CDK specialization. The absence of individual or multiple “interphase” CDKs does not affect early embryonic viability or prevent mouse embryonic fibroblasts (MEFs) from proliferating in culture (Rane et al., 1999; Berthet et al., 2003; Ortega et al., 2003; Malumbres et al., 2004; Berthet et al., 2006; Santamaria et al., 2007). Compensation occurs through enhanced formation of atypical CDK/cyclin complexes, for example, increased pairing of Cdk1 with cyclins E and A in the absence of Cdk2 (Aleem et al., 2005). Cdk2-/- mice are viable but infertile due to a failure of meiosis, and MEFs are delayed in entering S phase after release from serum starvation (Berthet et al., 2003; Ortega et al., 2003). Redundancy between interphase CDKs was revealed by a Cdk2 Cdk4 double knockout, which unlike either single mutation results in embryonic lethality (Berthet et al., 2006). Nonetheless, mouse embryos lacking Cdk2, -4 and -6 survive until midgestation and, in MEFs derived from them, Cdk1 forms active complexes with cyclins D, E, A and B to drive cell division, albeit more slowly than normal (Santamaria et al., 2007).
The defects in survival, development and reproduction of Cdk knockout mice imply that non-canonical or non-physiologic CDK/cyclin pairs cannot provide full function in vivo. It follows that fidelity of binding is important for normal cell cycle control, but the rules governing partner selection remain poorly defined. Some specificity must arise from the relative affinities of different CDK-cyclin interactions (Desai et al., 1995). In addition, the dependence of certain CDKs on chaperones or assembly factors to assist in complex formation could impose a check on illegitimate pairing (LaBaer et al., 1997). In vivo, however, complexes such as Cdk1/cyclin E that do not form readily with purified proteins can be assembled and activated when Cdk2 is absent (Aleem et al., 2005; Santamaria et al., 2007). Conversely, Cdk2/cyclin B complexes are stable in vitro but relatively rare in normal cells.
Here we show that cyclin A binds preferentially to Cdk2 throughout G1 and S phase in human cells; efficient Cdk1-binding does not begin until Cdk2/cyclin A assembly is nearly complete. Cdk1 is ~10-fold more abundant than Cdk2 in human cell extracts, however (Arooz et al., 2000), and there is no assembly factor known to enforce this bias, so we considered a third way preferences could be established—through separate activation pathways. CDK activation minimally depends on two common steps: binding to cyclin and phosphorylation of the activation segment (T-loop) by a CDK-activating kinase (CAK) (Morgan, 2007). A pathway in which cyclin-binding precedes T-loop phosphorylation was deduced from the structures of human Cdk2 in monomeric form (De Bondt et al., 1993), in an unphosphorylated complex with cyclin A (Jeffrey et al., 1995), and in a fully active complex phosphorylated on T-loop residue Thr160 (Russo et al., 1996). The order of steps may vary, however, and until recently had not been determined for any mammalian CDK in vivo.
The requirement for T-loop phosphorylation is conserved in yeast and metazoan CDKs, but the CAK-CDK networks have diverged [reviewed in (Fisher, 2005)]. In mammals the CAK is itself a CDK—the Cdk7/cyclin H/Mat1 complex—required to activate both Cdk1 and Cdk2 in vivo (Larochelle et al., 2007). The budding yeast Saccharomyces cerevisiae has a single-subunit CAK, Cak1, required at both G1/S and G2/M boundaries (Sutton and Freiman, 1997). The fission yeast Schizosaccharomyces pombe has two CAKs: Mcs6 and Csk1, orthologs of Cdk7 and Cak1, respectively, both of which must be inactivated to arrest the cell cycle (Lee et al., 1999; Saiz and Fisher, 2002).
To probe the functions and substrate specificity of the human CAK, we mutated Cdk7 to enlarge the ATP-binding pocket, rendering the kinase analog-sensitive (AS)—susceptible to inhibition by bulky purine analogs (Larochelle et al., 2006). We created a Cdk7as/as HCT116 human colon carcinoma cell line and showed that inhibition of Cdk7 in vivo prevented activation of Cdk2, and blocked assembly and activation of Cdk1/cyclin B (Larochelle et al., 2007). Because Cdk7 can only phosphorylate Cdk1 in the presence of cyclin (Fisher and Morgan, 1994), a CAK requirement for complex formation in vivo implied that cyclin binding and T-loop phosphorylation are mutually dependent and must occur in concert.
Here we order the steps in the physiologic activation of Cdk2. In contrast to Cdk1, Cdk2 is phosphorylated by Cdk7 as a monomer and then binds cyclin—an activation pathway similar to the predominant one in budding yeast (Ross et al., 2000). We recapitulate the selectivity of cyclin A for Cdk2 in extracts, and overcome this preference by the addition of a CAK from yeast, which phosphorylates monomeric Cdk1 and redirects it into the activation pathway normally reserved for Cdk2. In vivo, inhibition of Cdk7 results in increased binding of cyclin B to Cdk2 at the expense of Cdk1. Therefore, the activity and substrate preferences of Cdk7 help determine pairing rules for interphase and mitotic CDK/cyclin complexes in human cells.
Results
Fidelity of Cyclin-CDK Pairing in Human Cells
Cdk1’s ability to compensate for genetic loss of interphase CDKs suggests plasticity—or promiscuity—in cyclin-CDK pairing. To determine how faithful this process is normally, we developed a quantitative assay to compare relative amounts of Cdk1 and Cdk2 bound to different cyclins in vivo. We fractionated K562 human leukemia cells into populations at different stages of the cell cycle by centrifugal elutriation (Figure 1A, B). Next, cyclins A, B and E were immunoprecipitated from each fraction and the immune complexes were denatured. Cdk1 and Cdk2 were then recovered with CDK-specific antibodies that precipitate their targets with similar efficiency, and detected in immunoblots with a PSTAIRE antibody that recognizes both CDKs equally well (Supplemental Figure 1A, B). In vivo, cyclin E bound almost exclusively to Cdk2, as expected because it does not readily form complexes with Cdk1, whereas cyclin B bound Cdk1 but not Cdk2, even though it binds Cdk2 efficiently in vitro (Figure 1C; Desai et al., 1995). Cyclin A assembled preferentially with Cdk2 early in the cell cycle; only after Cdk2 complexes reached a plateau in late S or G2 phase did it begin to bind Cdk1 (Figure 1C). In asynchronous cell extracts, the Cdk2:Cdk1 ratio in cyclin A complexes is ~2:1 (Figure 1D), despite a ~10-fold greater abundance of Cdk1 (Figure 1E and data not shown). These relative levels of total Cdk1 and Cdk2 are in agreement with a previous report (Arooz et al., 2000). Another study detected cyclin A binding to both Cdk2 and Cdk1 in human cells, but did not resolve a difference in timing between the two events (Pagano et al., 1992). A similar lag in Cdk1/cyclin A assembly occurred in HCT116 cells released from serum starvation (Supplemental Figure 1D). Therefore, uniquely among cyclins we tested, cyclin A undergoes a transition during interphase, from forming new complexes predominantly with Cdk2 to assembling with Cdk1.
Figure 1. Quantitative analysis of CDK/cyclin complex formation.

(A) K562 cells were fractionated by centrifugal elutriation and cellular DNA content was determined by flow cytometry.
(B) Immunoblots of asynchronous and elutriated K562 cell extracts were probed for cyclins E, A and B, and Cdk1 and -2 (A, asynchronous).
(C) Cyclin E, A, or B was immunoprecipitated from whole-cell extracts of elutriated K562 cells, the immunoprecipitates boiled in 1% SDS, the supernatants diluted in HBS + 0.5% Triton-X 100, and Cdk1 and -2 immunoprecipitated. Relative levels of Cdk1 and -2 associated with each cyclin were measured by immunoblotting with PSTAIRE antibody.
(D) Cyclin A-bound Cdk1 and -2 were recovered from asynchronous K562 cell extracts and detected as described in C; decreasing amounts of recovered Cdk2 were compared with total Cdk1 recovered to estimate fold-enrichment of Cdk2.
(E) Total Cdk1 and -2 in asynchronous cell extract were immunoprecipitated after SDS denaturation, and their relative amounts determined by titrating the anti-Cdk1 eluate against a fixed amount of anti-Cdk2 eluate in a PSTAIRE immunoblot.
A Cdk2-Specific Activation Pathway In Vivo
In vitro, Cdk2, but not Cdk1, can be phosphorylated by Cdk7 in the absence of a cyclin (Fisher and Morgan, 1994) and can bind cyclin A in the absence of T-loop phosphorylation (Desai et al., 1995). These differences suggest that the two CDKs could follow distinct paths to activation, which might favor Cdk2 binding to cyclin A. The ability to inhibit CAK specifically in Cdk7as/as HCT116 cells allowed us to test this idea, by ordering the steps of Cdk2 activation in vivo.
To ask if Cdk2 is phosphorylated as a monomer in vivo, we measured Cdk2 phospho-isoforms in monomeric and cyclin-bound populations derived from extracts of asynchronously growing cells. In sizing columns, Cdk2 migrated over a ~158-669 kDa range together with cyclin A and in a peak at ~30 kDa devoid of cyclin A, and was phosphorylated on Thr160 in both populations, albeit to different extents (Figure 2A). We treated Cdk7as/as cells with the analog 3-MBPP1 (Burkard et al., 2007), which inhibited Cdk7as but not Cdk7WT in vitro and arrested growth of Cdk7as/as but not wild-type cells (Supplemental Figure 2). This resulted in the loss of phosphorylated Cdk2 monomer within 2 hr. In contrast, T-loop phosphorylation in complexes persisted for at least 12 hr in the absence of CAK activity (Figure 2A, B and Supplemental Figure 3). Dephosphorylation was a specific consequence of Cdk7 inhibition; the phospho-Thr160 signal in all size populations was insensitive to 3-MBPP1 in wild-type cells (Supplemental Figure 4A).
Figure 2. Differential Stability of Phospho-Thr160 on Monomeric and Cyclin-Bound Cdk2.

(A) Asynchronously grown Cdk7as/as HCT116 cells were treated with DMSO or 5 μM 3-MBPP1 for 2 hr. Extracts were fractionated in a Superdex 200 sizing column, and cyclin A, Cdk2 and T-loop phosphorylated Cdk2 (T160-P) were detected by immunoblot.
(B) Pooled sizing column fractions from cells treated for indicated times with 3-MBPP1 were immunoblotted and probed for cyclin A, total Cdk2, and Cdk2 T160-P. Pool 1 contains the highest molecular weight Cdk2, pool 2 the ~200 kDa peak and pool 3 the ~30 kDa peak (see Supplemental Figure 3 for complete data set). The faster-migrating isoform detected by antibodies to total Cdk2 (middle panel, lower arrow) is phosphorylated on Thr160.
(C) Cdk2 was immunoprecipitated from pooled fractions in (B) and tested for bound cyclin A by immunoblot (top panel) and associated histone H1 kinase activity (bottom panel).
(D) Recombinant His-cyclin A was incubated with pool 3 from DMSO- or 3-MBPP1-treated cells, prior to Cdk2 immunoprecipitation and histone H1 kinase assay. Activity was quantified by phosphorimager.
To compare different Cdk2 populations directly, we pooled fractions containing: 1) a minor form at ~669 kDa, possibly aggregated or with components besides Cdk2 and cyclin (which was not characterized further); 2) the ~300 kDa peak of Cdk2 and cyclin A; and 3) ~30 kDa, presumably monomeric Cdk2 (Figure 2B and Supplemental Figure 3). In pool 1, phosphorylated Cdk2 decreased with time in proportion to total Cdk2, precluding a measurement of phosphate turnover. In pool 2, unphosphorylated Cdk2 increased after 3 hr but remained constant thereafter. In pool 3, Cdk2 phosphorylation was abolished by Cdk7 inhibition for 3 hr. Therefore, in the absence of active CAK, Cdk2-Thr160 phosphorylation seemed to decay rapidly on monomers but more slowly in complexes.
Cdk2 immunoprecipitated from pools 1 and 2 was cyclin A-bound and active as a kinase, whereas Cdk2 in pool 3 was cyclin A-free and inactive (Figure 2C). Monomeric Cdk2 from mock-treated cells was activated ~12-fold by the addition of purified cyclin A prior to immunoprecipitation, but Cdk2 from inhibitor-treated cells was only stimulated ~2-fold (Figure 2D). The phosphorylated Cdk2 monomer is therefore a potential activation intermediate, dependent on Cdk7 to maintain its steady state level.
This form could arise from Cdk7 acting directly on monomeric Cdk2 or, as an inactivation intermediate, from Cdk2/cyclin complexes by disassembly or (more likely) cyclin degradation. To investigate the latter possibility, we tested the effect of proteasome inhibition on Cdk2 phospho-isoforms. Phosphorylation of Cdk2 monomer persisted in cells treated with the proteasome inhibitor MG132 but was abolished after treatment with both MG132 and 3-MBPP1 (Figure 3A and Supplemental Figure 5). We conclude that this species is a bona fide activation intermediate arising from the phosphorylation of monomeric Cdk2 by Cdk7.
Figure 3. Thr160 Phosphate Turnover Is Restricted to Cdk2 Monomer In Vivo.
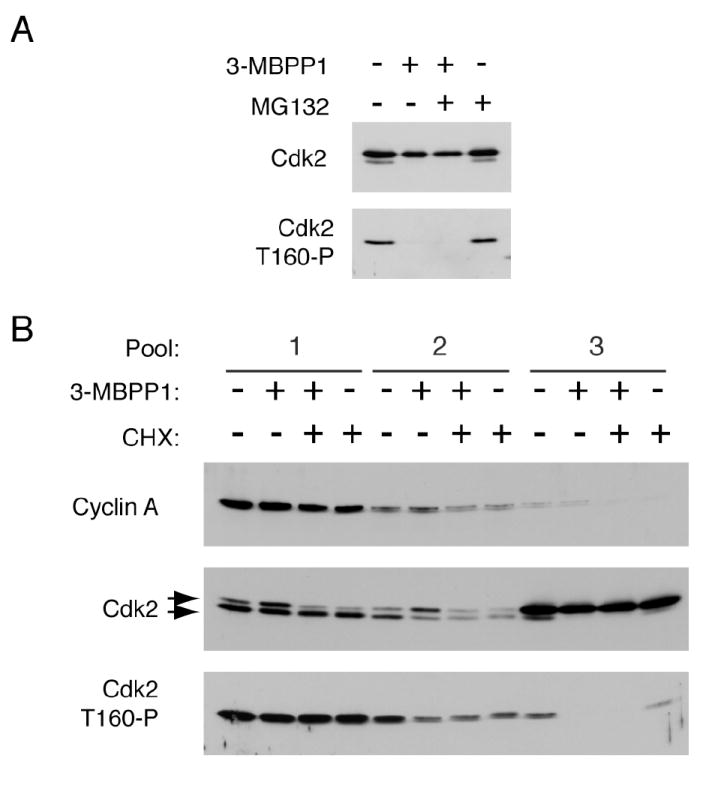
(A) Cdk7as/as HCT116 cells were treated with 5 μM 3-MBPP1, 10 μM MG132 or both, as indicated, for 2 hr. Monomeric Cdk2 was isolated by gel exclusion chromatography and probed for Cdk2 and Cdk2 T160-P (see Figure 2A and Supplemental Figure 5 for complete data set).
(B) Cdk7as/as HCT116 cells were treated with 5 μM 3-MBPP1, 100 μg/ml cycloheximide or both for 2 hr. Extracts were separated in sizing columns and fractions were pooled (see Supplemental Figure 6 for complete data set). Cyclin A, Cdk2 and Cdk2 T160-P were detected by immunoblot.
T-Loop Phosphate Turnover Is Restricted to Monomeric Cdk2 In Vivo
The proportion of slower-migrating, unphosphorylated Cdk2 increased in high molecular weight fractions after Cdk7 inhibition (Figure 2B), raising the possibility that phospho-Thr160 is also subject to turnover in Cdk2/cyclin complexes. Alternatively, newly synthesized cyclin might bind to unphosphorylated Cdk2 as the phosphorylated form becomes depleted. To distinguish between these scenarios, we compared the effects of CAK inhibition in the presence and absence of protein synthesis, in Cdk7as/as cells treated for 2 hr with 3-MBPP1, cycloheximide or both. Sizing column fractions were pooled to permit direct comparison of three Cdk2 populations: 1) co-fractionating with the peak of cyclin A (equivalent to pool 2 in Figure 2B); 2) with mobility intermediate between those of cyclin A and monomeric Cdk2; and 3) monomer (Figure 3B and Supplemental Figure 6). Treatment with 3-MBPP1 alone caused loss of phosphorylated Cdk2 monomer (pool 3) and accumulation of unphosphorylated Cdk2 in cyclin-containing pools 1 and 2. Inhibition of Cdk7 in the absence of protein synthesis still diminished phosphorylation of monomeric Cdk2, but failed to increase the amount of unphosphorylated Cdk2 in complexes (Figure 6B). Therefore, in the absence of CAK activity, unphosphorylated Cdk2/cyclin arose mainly by new complex formation, not by dephosphorylation of pre-existing complexes, indicating that turnover of T-loop phosphate occurred predominantly on monomeric Cdk2.
Figure 6. CAKs Influence Specificity of CDK/Cyclin Pairing In Vitro and In Vivo.
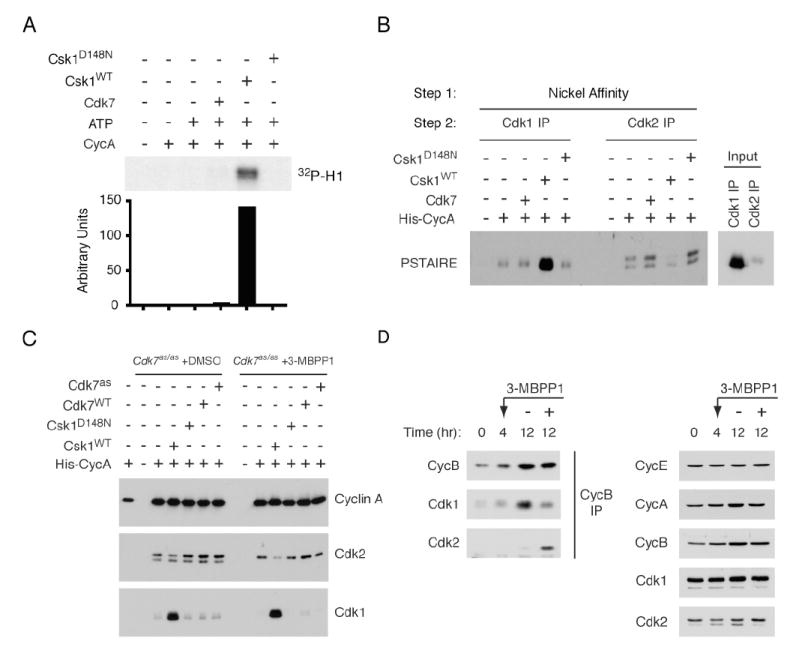
(A) Cdk1 was immunoprecipitated from pool 3 fractions of 3-MBPP1-treated cells and incubated with purified His-cyclin A without other treatments or, in indicated lanes, after pre-treatment with CAK (Cdk7 or Csk1) and ATP. Beads were washed and tested for histone H1 kinase activity, quantified by phosphorimager.
(B) Purified Cdk7 complex, or wild-type (WT) or kinase-dead (D148N) Csk1, were added as indicated to Cdk7as/as HCT116 extracts supplemented with His-cyclin A. After incubation, cyclin A-containing complexes were recovered on nickel beads, denatured and immunoprecipitated with Cdk1-or Cdk2-specific antibodies. In Input lanes, Cdk1 or Cdk2 was immunoprecipitated directly from denatured extract. Recovered CDK was detected with anti-PSTAIRE antibody.
(C) Extracts from Cdk7as/as HCT116 cells, treated as indicated with DMSO or 3-MBPP1 for 3 hr, were incubated with His-cyclin A and exogenous CAKs as indicated. Cyclin A-containing complexes were recovered on nickel beads and relative amounts of recovered Cdk1 and -2 determined by immunoblot.
(D) Cyclin B was immunoprecipitated from extracts of Cdk7as/as HCT116 cells, synchronized by serum starvation and release and treated at 4 hr with DMSO or 5 μM 3-MBPP1, as indicated, for 8 hr. Associated Cdk1 and-2 were detected by immunoblot (left: anti-Cdk2 blot was exposed ~60-fold longer than was anti-Cdk1 blot). Total levels of Cdk1 and -2, and cyclins A, B, and E were determined by immunoblot (right).
Cdk7 Phosphorylates Monomeric Cdk2 and Cyclin-Bound Cdk1 in Extracts
The results thus far suggested that Cdk2 follows an activation pathway distinct from the one we described for Cdk1 (Larochelle et al., 2007). To test this idea in vitro, we first asked which CDK isoforms—complexes or monomers—were phosphorylated by Cdk7 in whole-cell extracts. Because AS kinases prefer bulky ATP analog substrates whereas wild-type kinases prefer natural ATP (Shah et al., 1997), we can detect specific protein phosphorylation by a single AS kinase in an extract containing radiolabeled N6-(benzyl)-ATP and 1 mM unlabeled ATP. Under these conditions, Cdk7as phosphorylated several proteins, including Rpb1 (the largest subunit of RNA Polymerase II), Cdk1 and Cdk2 (Larochelle et al., 2006; Larochelle et al., 2007).
To monitor the substrate specificity of endogenous Cdk7 during the cell cycle, we synchronized Cdk7as/as cells in S phase by thymidine block, released them into fresh medium containing nocodazole, and collected samples at 2-hr intervals to measure DNA content (Figure 4A) and prepare extracts for labeling (Figure 4B). Total CDK labeling by Cdk7as increased as cells progressed through S phase and G2, but declined as they accumulated in mitosis at 10 hr. In contrast, Rpb1 was labeled more uniformly, with a peak 2 hr after release. To discern specific CDK phosphorylations, we immunoprecipitated Cdk1, Cdk2, cyclin A and cyclin B after labeling. In anti-Cdk1 and -cyclin B immunoprecipitates, phosphorylation peaked at 6-8 hr (Figure 4C), correlating with the physiologic execution point of Cdk7 function in G2 (Larochelle et al., 2007). Labeling in anti-Cdk2 immunoprecipitates was relatively constant throughout S/G2, but there was almost no incorporation in complexes containing cyclin A (Figure 4D).
Figure 4. Cell-Cycle Dependent Phosphorylation of Cdk7 Substrates in Extracts.

(A) Cdk7as/as HCT116 cells were arrested in S phase by addition of thymidine for 15 hr, released into fresh medium containing nocodazole and harvested every 2 hr to determine DNA content by flow cytometry.
(B) Extracts from cells treated as in (A) were labeled by endogenous Cdk7as with [γ-32P]N6-(benzyl)-ATP. Only Rpb1 (~200 kDa) and Cdk1 + 2 (~34 kDa), were detected in this exposure.
(C) Anti-cyclin B or –Cdk1 immunoprecipitates from labeling reactions in (B) were probed for cyclin B and Cdk1 by immunoblot and for labeling by autoradiography.
(D) Anti-cyclin A or -Cdk2 immunoprecipitates from reactions in (B) were probed for cyclin A and Cdk2 by immunoblot and for labeling by autoradiography.
(E) Cyclin B and Cdk1 were immunoprecipitated after labeling with purified Cdk7as/cyclin H/Mat1 and [γ-32P]N6-(benzyl)-ATP in extracts of HeLa cells synchronized at indicated cell-cycle positions. Immunoprecipitates were probed for cyclin B and Cdk1 by immunoblot and for phosphorylation by autoradiography.
(F) Cyclin A and Cdk2 were immunoprecipitated from labeling reactions as in (E). Cdk2 protein was detected by immunoblot, and the extent of labeling by autoradiography, from single exposures of the same membrane and dried gel, respectively.
Cdk1 and Cdk2 were also phosphorylated by purified recombinant Cdk7as/cyclin H/Mat1 added to extracts of synchronized HeLa cells (Figure 4E, F). The similar labeling in G2 and mitotic extracts differed from the results with endogenous Cdk7as, which labeled CDKs more strongly in G2 (Figure 4B). As was the case in Cdk7as/as extracts, however, we detected labeling in Cdk1, cyclin B and Cdk2, but not cyclin A immunoprecipitates (Figure 4E, F). During S and G2, Cdk2 co-precipitating with cyclin A was mostly phosphorylated on the T-loop in both HCT116 and HeLa extracts (Figure 4D, F), possibly explaining why it was unavailable for labeling if no phosphate turnover occurred in the complex in vitro, as appeared to be the case in vivo. Taken together, the results in vivo and in synchronized extracts indicate that Cdk7 activity is not required to maintain phospho-Thr160 in pre-formed Cdk2/cyclin A complexes, and that Cdk2 monomer phosphorylation is likely to be a significant reaction in vivo.
Cdk7 Phosphorylates Monomeric Cdk2 Efficiently
The relative efficiencies with which human CAK phosphorylates Cdk2 monomers and complexes have been disputed (Fisher and Morgan, 1994; Kaldis et al., 1998; Wohlbold et al., 2006). To attempt to reconcile previous results, we determined apparent kinetic parameters for different Cdk2 isoforms with fully active, T-loop phosphorylated Cdk7/cyclin H/Mat1 (Figure 5A, B, Supplemental Table 1). Monomeric Cdk2 and Cdk2/cyclin A were phosphorylated with similar efficiencies (kcat/KM) but different kinetics. Cyclin A appeared to increase affinity for Cdk2, reflected in KM values of ~5.8 and ~0.59 μM for monomer and complex, respectively. At near-saturating substrate concentrations, however, the enzyme turned over ~17 times faster on the monomer (kcat 1.20 versus 0.07 s-1 for complex). (Note: KM and kcat for monomeric Cdk2 are probably underestimated because of inability to saturate CAK.) At Cdk2 concentrations well below KM, Cdk7 phosphorylated monomeric and cyclin A-bound Cdk2 to similar extents as predicted by the nearly equal kcat/KM values, in reactions performed in the presence of 200 μM ATP, i.e. above KMATP (Larochelle et al., 2006) but below physiologic ATP concentrations of ~2 mM. Only when ATP was also limiting, at 10 μM [the concentration used in (Kaldis et al., 1998)], did we observe a strong preference for complexes (Supplemental Figure 7). The kinetics suggest that Cdk7 may be adapted to phosphorylate Cdk2 as a monomer—a more abundant species in vivo than the unphosphorylated complex (Figure 2A)—in opposition to phosphatases that work better on monomeric substrates (Poon and Hunter, 1995; Cheng et al., 1999).
Figure 5. Cdk2 but not Cdk1 can be Activated by a CAK-First Pathway.

(A) Titrations of Cdk2D145N and Cdk2D145N/cyclin A to determine kinetic parameters of phosphorylation by Cdk7/cyclin H/Mat1 (Supplemental Table 1). The D145N mutation renders Cdk2 inactive, eliminating background due to autophosphorylation.
(B) Concentration-velocity plots for reactions in (A). Error bars denote standard deviation of mean in triplicate measurements.
(C) Monomeric Cdk2 was immunoprecipitated from pool 3 fractions of DMSO-or 3-MBPP1-treated cells. In lanes 2-5, immobilized Cdk2 was incubated with or without His-cyclin A and tested directly for kinase activity. In lanes 6-9, Cdk2 was first incubated with ATP (all samples), with Cdk7as and 3-MBPP1 added as indicated. After washes, beads were incubated with cyclin A with or without 3-MBPP1, washed again and tested for kinase activity, quantified by phosphorimager.
(D) Cdk1 was immunoprecipitated from pool 3 fractions of DMSO- or 3MBPP1-treated cells. In lanes 2-5, immobilized Cdk1 was incubated with purified Myc-cyclin B, washed and tested for kinase activity. In lanes 6-9, Cdk1 was treated exactly as described for lanes 6-9 of (C), except cyclin B was added in place of cyclin A. In lanes 10-12, Cdk1 underwent a single treatment with ATP, Cdk7as, 3-MBPP1 and cyclin B as indicated. In lanes 13-15, Cdk1 was incubated first with cyclin B and, after washing, with ATP, Cdk7as and 3-MBPP1 as indicated. Complexes were then tested for kinase activity, quantified by phosphorimager.
Cdk7 Activates Cdk2 but not Cdk1 by a CAK-First Mechanism In Vitro
We next tested if CDKs isolated from human cells could be activated in vitro by sequential T-loop phosphorylation and cyclin-binding. Monomeric Cdk2 from mock- but not 3-MBPP1-treated Cdk7as/as cells could be activated by purified cyclin A (Figures 2D and 5C). The unphosphorylated monomer from inhibitor-treated cells was therefore a suitable substrate for order-of-addition experiments (Figure 5C), in which immobilized Cdk2 was incubated first with Cdk7as/cyclin H/Mat1 and ATP, then with cyclin A, and tested for kinase activity towards histone H1. This sequence resulted in a nearly 300-fold increase in Cdk2 activity (lane 7). Addition of 3-MBPP1 during the first step when no cyclin was present prevented activation (lane 8), but had no effect at the cyclin A binding step (lane 9). Therefore, endogenous Cdk2 can be phosphorylated by Cdk7 and subsequently activated by binding to cyclin.
We then asked whether Cdk1 could follow the same pathway (Figure 5D). Cdk1 monomers were not activated by incubation with purified cyclin B, regardless of whether they came from mock-or 3-MBPP1-treated cells (lanes 3 and 5; see Supplemental Figure 8 for sizing column profiles of Cdk1). Therefore, untreated cells contained little or no monomeric Cdk1 that could be activated simply by adding cyclin, in contrast to the situation for Cdk2. Simultaneous addition of Cdk7, cyclin B, and ATP to Cdk1 monomer from drug-treated cells resulted in a >2,000-fold activation, which was dependent on active Cdk7, i.e. inhibited by 3-MBPP1 (Figure 5D, lanes 11 and 12). A two-step activation, in which Cdk1 was incubated with cyclin B, washed and treated with Cdk7 (lane 14), was less efficient (<1,000-fold), possibly due to the instability of Cdk1/cyclin B complexes lacking T-loop phosphorylation (Larochelle et al., 2007). In contrast, incubating immobilized Cdk1 first with Cdk7 and then with cyclin B did not result in measurable activation (lane 7), indicating that Cdk7 is incapable of phosphorylating monomeric Cdk1 from human cells, as previously shown with recombinant proteins (Fisher and Morgan, 1994). Therefore, Cdk7 can activate Cdk2, but not Cdk1, by a “CAK-first” pathway.
The Substrate Preferences of CAK Help Determine CDK/Cyclin Pairing Rules
Cdk1 could be activated by sequential incubations with S. pombe Csk1 and cyclin A (Figure 6A), indicating that the CAK from fission yeast can phosphorylate monomeric Cdk1 from human cells. This enabled us to test whether the substrate preferences of human CAK influence pairing specificity, i.e. if the inactivity of Cdk7 towards Cdk1 monomer imposes a kinetic barrier to Cdk1/cyclin A assembly, and promotes selective binding of cyclin A to Cdk2.
To address this question, we performed competitive binding assays in which His-tagged cyclin A was incubated in HCT116 cell extracts and re-isolated by nickel affinity. Recovered proteins were denatured, subjected to a second round of precipitation with antibodies to Cdk1 or Cdk2, and detected with PSTAIRE antibody. When no exogenous CAK was added, we recovered Cdk2 preferentially, recapitulating the selective binding of cyclin A to Cdk2 in vivo despite the greater abundance of Cdk1 in the extract (Figure 6B). (Note: because a larger fraction of Cdk2 is in complexes, compared to Cdk1 [compare Figure 2A and Supplemental Figure 8], the excess of Cdk1 available to bind the added cyclin is likely to be underestimated by this method.) When reactions were supplemented with extra Cdk7, the relative amounts of Cdk2 and Cdk1 recovered did not change (nor did the absolute amount or T-loop phosphorylation of Cdk2 in complex with cyclin A increase, suggesting that CAK activity was not limiting for assembly under these conditions). However, changing the pathway by which Cdk1 was activated, by addition of Csk1, caused a switch—increased recovery of Cdk1 with a concomitant decrease in Cdk2. The gain of Cdk1-binding was dependent on the catalytic activity of Csk1, and occurred mostly at the expense of binding to unphosphorylated Cdk2 (Figure 6B, C), suggesting that phosphorylated Cdk2 still competed effectively with Cdk1. In extracts of Cdk7as/as cells pretreated with 3-MBPP1 to deplete phosphorylated CDKs, the preference for Cdk2 was maintained or enhanced, but Csk1 could still redirect cyclin A to bind Cdk1 at Cdk2’s expense (Figure 6C). Therefore, subverting the normal order of Cdk1-activating steps overcame cyclin A’s preference for Cdk2.
In contrast to cyclin A, cyclin B is selective for Cdk1 in vivo (Figure 1C), suggesting that its interaction with unphosphorylated Cdk1 is strong enough to ensure that CAK will stabilize the complex before it can dissociate and allow cyclin B to bind the less-abundant Cdk2. This led us to ask if destabilizing Cdk1/cyclin B by inhibiting CAK promoted the formation of Cdk2/cyclin B complexes in vivo. We synchronized Cdk7as/as cells by serum starvation and release into fresh medium, and monitored cyclin B-CDK assembly during subsequent cell cycle progression. Addition of 3-MBPP1 4 hr after release slows cell cycle progression (Larochelle et al., 2006). This treatment did not appreciably affect the accumulation of CDKs or cyclins, but reduced the amount of Cdk1/cyclin B measured at 12 hr. In contrast, Cdk2/cyclin B complex formation, which was nearly undetectable in mock-treated cells, was enhanced by inhibition of Cdk7 (Figure 6D). Therefore, in the absence of CAK activity, Cdk2 is the default partner of cyclins A and B. We conclude that Cdk7 normally plays a catalytic role in directing proper complex assembly. Moreover, the activation of different CDKs by distinct pathways—largely governed by the substrate recognition properties of Cdk7—is a determinant of faithful CDK-cyclin pairing in vivo.
Discussion
Kinetically Distinct Cdk1 and Cdk2 Activation Pathways
By chemical genetics, we have shown that Cdk1 and Cdk2 follow different paths to activation in human cells (Figure 7A). In the prevalent mode of Cdk2 activation, phosphorylation by CAK precedes binding to cyclin. This is distinct from the concerted activation mechanism of Cdk1 (Larochelle et al., 2007), even though the individual steps are common to both CDKs. Neither kinase may adhere to the sequence of cyclin-binding followed by T-loop phosphorylation that has been most extensively characterized (Desai et al., 1992; Connell-Crowley et al., 1993; Jeffrey et al., 1995; Russo et al., 1996).
Figure 7. Ordering CDK Activation Pathways in Yeast and Metazoans.
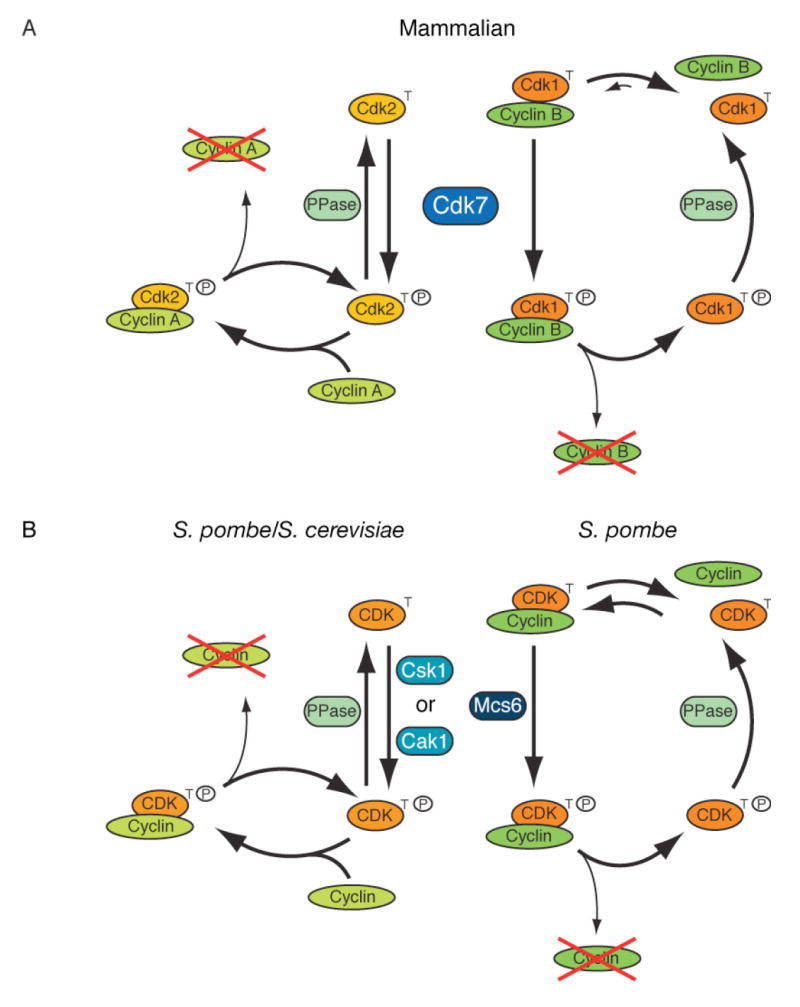
(A) In mammalian cells, CDKs follow distinct kinetic pathways to activation. Cdk2 is phosphorylated by Cdk7 before binding cyclin. Cdk7 can only phosphorylate Cdk1 in the presence of cyclin, but Cdk1/cyclin complexes are unstable unless phosphorylated by Cdk7. Upon degradation of its cyclin, Cdk2 can rejoin the monomer pool and be re-phosphorylated, whereas Cdk1 must await association with cyclin for re-phosphorylation.
(B) S. pombe could have separate mechanisms of CDK activation analogous to those in mammalian cells, by virtue of having two CAKs with different substrate specificities. The predominant activation pathway in S. cerevisiae, with only one CAK, resembles that of human Cdk2.
By kinetic criteria, monomeric and cyclin-bound Cdk2 are equally good substrates for Cdk7 in vitro. The CAK-first pathway appears to be preferred for activation in vivo, however, where unphosphorylated monomers are more abundant than unphosphorylated complexes, and where cyclin-binding traps Cdk2 in phosphorylated form. CAK activity is not needed to maintain phosphorylation of pre-formed complexes in HCT116 cells, in which no turnover of phospho-Thr160 occurred in cyclin-bound Cdk2 populations. In whole-cell extracts, we could not detect phosphorylation of Cdk2 complexes by Cdk7as. Finally, we could recapitulate distinct, Cdk1-and Cdk2-specific activation pathways in vitro, with CDK monomers isolated from cells in which Cdk7 was inhibited. We conclude that Cdk2 is activated predominantly by an ordered, CAK-first pathway in vivo.
Selectivity in Cyclin-CDK Assembly Is a Function of Cdk7 Substrate Specificity
A quantitative analysis revealed Cdk2 and Cdk1 to be nearly exclusive partners of cyclins E and B, respectively, in vivo. In contrast, partner selection by cyclin A is dynamic; Cdk2 is preferred early in the cell cycle whereas Cdk1/cyclin A begins to accumulate only as cells reach late S or G2 phase. Therefore, Cdk2 is likely to be the major CDK responsible for controlling S phase in wild-type cells, even though Cdk1 might be able to take over that function when Cdk2 levels are artificially reduced.
By ordering the steps in Cdk1 and Cdk2 activation, we uncovered a kinetic basis for the temporal sequence of cyclin A-binding; Cdk2 has priority because it can be phosphorylated as a monomer and forms stable complexes without phosphorylation. We infer the binding hierarchy to be phospho-Cdk2 > phospho-Cdk1 ≈ Cdk2 > Cdk1, because: 1) phospho-Cdk2 still competes effectively with phospho-Cdk1 generated in vitro by Csk1; 2) the phospho-Cdk1 “wins” over unphosphorylated Cdk2, but this might reflect higher concentration rather than affinity; and 3) unphosphorylated Cdk1 cannot stably bind cyclin A. Therefore, phosphorylation confers an advantage on monomeric Cdk2, revealed by the ability of phospho-Cdk1 to exclude unphosphorylated but not phosphorylated Cdk2 from cyclin A complexes.
Switching Cdk1 to a CAK-first activation pathway overrode the normal binding preference of cyclin A in vitro. This suggests a role for T-loop phosphorylation in ensuring fidelity of complex formation, and a rationale for why metazoans lack a Cak1 ortholog (Murray and Marks, 2001). Other factors, such as subcellular localization of the relevant CDKs, cyclins and CAK, could also influence pairing. We have so far been unable to express Csk1 in active form, or Cak1 to high levels, to test whether the kinetic model can completely account for cyclin A partner selection in human cells (unpublished observations).
What causes cyclin A to start binding Cdk1? By late S and G2, the majority of Cdk2 in K562 and HCT116 cells was phosphorylated (Figures 1B, 4D and Supplemental Figure 1C), suggesting that it was sequestered by cyclin, which might then allow newly synthesized cyclin A to bind Cdk1. Consistent with Cdk2 becoming saturated in vivo, the ability to divert cyclin B into Cdk2 complexes by inhibiting Cdk7 was diminished at later times in the cell cycle (data not shown), and lowering Cdk2 levels by RNA interference can increase Cdk1/cyclin A assembly (L’Italien et al., 2006). Thus passive depletion of monomeric Cdk2 might suffice to explain the transition, but we cannot rule out a more active, switch-like process. The nearly exclusive binding of cyclin B to Cdk1 in unperturbed cells suggests preferential association prior to T-loop phosphorylation. That phosphorylation is required, however, to lock the proteins in a stable complex; inhibiting Cdk7 liberated cyclin B to bind Cdk2. CAK therefore acts catalytically to ensure that cyclin B forms complexes with Cdk1 rather than Cdk2, and might do the same for Cdk1/cyclin A during G2.
We have characterized different activation pathways for Cdk1 and Cdk2, but the basis for that distinction—the differential ability of Cdk7 to recognize the two CDKs as monomers—has no obvious explanation. In the crystal structure of monomeric Cdk2, Thr160 is buried (De Bondt et al., 1993). The T-loop undergoes a conformational change upon cyclin A binding, which was thought to be required for access by CAK (Jeffrey et al., 1995). Cdk7 phosphorylates monomeric Cdk2 efficiently in solution, however, suggesting that the T-loop is accessible in the absence of cyclin. Csk1 can phosphorylate Cdk1 as a monomer, indicating that the Cdk1 T-loop is also accessible. Why then is monomeric Cdk1 not a Cdk7 substrate? Discrimination is unlikely to be due to differences in the T-loops, which are nearly identical. Cdk7, moreover, can recognize CDK substrates with divergent sequences surrounding the phosphorylation site (Garrett et al., 2001) and cannot phosphorylate a Cdk2 T-loop-derived peptide in vitro (Larochelle et al., 2006), arguing that recognition is not primarily determined by the activation segment itself. Proposed docking interactions between Cdk7 and Cdk2 could facilitate phosphorylation in the absence of cyclin (Lolli and Johnson, 2007). To make similar contacts, Cdk1 might require conformational changes that occur only when cyclin is bound.
Yeast and Mammalian CDK Activation Harmonized?
In S. cerevisiae, Cdk1 is phosphorylated throughout the cell cycle as a monomer, which becomes active upon binding cyclin (Ross et al., 2000). This unitary mechanism might have arisen because, with only a single CDK to drive the cell cycle, there is no need for discrimination by cyclins. Alternatively, the concerted assembly and phosphorylation of Cdk1/cyclin B in metazoans could serve to enforce a sharp G2/M transition, which is absent in budding yeast (Morgan, 2007). In favor of the second interpretation, S. pombe, which does have switch-like control over G2/M, might also activate CDKs by two different pathways that resemble Cdk1-and Cdk2-specific mechanisms in human cells. However, instead of one CAK to activate different CDKs, fission yeast have one effector CDK and two CAKs: Csk1, which prefers CDK monomers (Tsakraklides and Solomon, 2002); and Mcs6, which by analogy with Cdk7 might prefer complexes. Therefore, similar kinetic strategies could have evolved within diverged CDK networks, perhaps to insulate interphase and mitotic functions (Figure 7A, B).
Specialization of CDKs based on recognition by upstream regulatory enzymes is an emerging theme. In budding yeast, the S-phase form of Cdk1 is less prone to inhibitory Tyr19 phosphorylation than the mitotic form, due to differences conferred by binding different cyclins (Keaton et al., 2007). In addition, the CDK inhibitor (CKI) Sic1 protects S-phase but not mitotic Cdk1 from Tyr19 phosphorylation. Similarly in mammalian cells, Cdk2 is not regulated by tyrosine phosphorylation in an unperturbed cell cycle (Chow et al., 2003), possibly due to protection by the CKI p27 (Keaton et al., 2007). These studies, and ours, hint at multiple modalities to coordinate activities and insulate regulatory inputs of interphase and mitotic CDKs. Reliance on a single CDK places greater emphasis on properly timed cyclin accumulation in yeast, whereas metazoans might depend on making correct CDK/cyclin pairs. Under certain conditions, normal pairing rules can be broken, but not without detrimental effects on cell cycle control and viability. For example, our results suggest that an important function of Cdk2 in mammals might be to delay the onset of Cdk1/cyclin assembly until near the end of S phase; loss of that timing mechanism might contribute to defective cell-cycle coordination or meiotic failure in the absence of Cdk2.
Experimental Procedures
Chemical Genetic Methods
Labeling of whole cell extracts with purified or endogenous Cdk7as and [γ-32P]N6-(benzyl)-ATP was performed as described previously (Larochelle et al., 2006; Larochelle et al., 2007). 3-MBPP1 and 1-NMPP1 were dissolved in DMSO and used at the indicated concentrations.
Immunological Methods
Immunoblots, immunoprecipitations, and kinase assays were carried out as previously described (Wohlbold et al., 2006) with the antibodies: anti-Cdk1 (C19), -Cdk2 (D-12 and M2), -cyclin B1 (GN51 and H-433) and- cyclin A (H-432 and BF683) from Santa Cruz Biotechnology; anti-Cdk1-Thr161, -Cdk2-Thr160 and -Cdc2 POH-1 from Cell Signaling Technologies; anti-PSTAIRE from Sigma.
Size Exclusion Chromatography
Asynchronously growing wild-type or Cdk7as/as HCT116 cells were treated with DMSO or indicated drug(s), collected and lysed in HoB [25 mM HEPES (pH 7.4), 150 mM NaCl, 50 mM NaF, 80 mM β-glycerophosphate, 0.1 % (v/v) Triton-X 100, 1 mM EDTA, 1 mM DTT] plus complete protease inhibitor cocktail (Roche). After a 1-hr centrifugation at 100,000 × gav, protein concentrations were normalized to 5-10 mg/ml with HoB. Aliquots (0.5 ml) were loaded on a Superdex 200 10/300 GL column (Amersham Pharmacia Biotech) equilibrated with 25 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM EDTA, 10 % (v/v) glycerol. Fractions (0.5 ml) were collected and 25 μl of each used for immunoblots. Equal parts of indicated fractions were combined to form pools.
Enzyme kinetics
Kinase assays were carried out in triplicate, as described (Larochelle et al., 2001), with ~6 nM Cdk7/Cyclin H/Mat1 in 25 mM Hepes (pH 7.4), 150 mM NaCl, 10 mM MgCl2, 0.5 mM DTT, 200 μM ATP, 5 μCi [γ-32P]ATP, for 5 min at 22°C. Cdk2 or Cdk2/cyclin A was added at 0.025, 0.05, 0.1, 0.25, 0.5, 1.0 or 2.0 μM. Kinetic parameters were determined with GraphPad Prism software for Macintosh.
CDK Activation Assays
Cdk1 or Cdk2 was immunoprecipitated from Superdex 200 pool 3, washed twice with HBS [10 mM HEPES (pH 7.4), 150 mM NaCl] + 0.1 % (v/v) Triton-X 100, twice with HBS and once with HBS + 10 mM MgCl2. To allow T-loop phosphorylation, immobilized CDKs were incubated with 200 ng Cdk7as/cyclin H/Mat1 or 70 ng Csk1 in HBS + 10 mM MgCl2 + 1 mM ATP, with DMSO or 1 μM 3-MBPP1, for 45 min at room temperature. To allow cyclin-binding, CDKs were incubated 45 min with 600 ng recombinant Myc-cyclin B or His-cyclin A with 200 μg BSA in 40 μl HBS. Activation steps were separated by washes. Finally, immunoprecipitates were washed and tested for histone H1 kinase activity. Incorporation was visualized by autoradiography and quantified by phosphorimager.
Formation of CDK/Cyclin Complexes in Crude Extracts
To test CDK-binding preference of cyclin A in vitro, 500 μg of asynchronous, Cdk7as/as HCT116 whole-cell extract was incubated 1 hr at room temperature with 65 ng His-cyclin A and an ATP-regenerating system under conditions used for labeling by Cdk7as (Larochelle et al., 2006). Where indicated, 200 ng of T-loop phosphorylated Cdk7/cyclin H/Mat1 or 70 ng Csk1 was added. His-cyclin A-containing complexes were recovered on Nickel NTA Superflow beads (Qiagen) and boiled in 20 mM Tris pH 7.0 + 1 % SDS (v/v). Supernatants were diluted 10-fold with HBS + 0.5 % Triton-X 100 (v/v) and Cdk1 and Cdk2 immunoprecipitated and detected by immunoblotting with anti-PSTAIRE antibody. Alternatively, proteins bound to nickel beads were solubilized and probed directly by anti-Cdk1 and -Cdk2 immunoblot.
Supplementary Material
Acknowledgments
We thank D. Morgan and L. Wohlbold for critical review of the manuscript, P. Jallepalli for helpful discussions, A. Koff for help with elutriation and D. José for preliminary dose-response measurements with 3-MBPP1. This work was supported by U.S. National Institutes of Health grants GM056985 to R.P.F. and EB001987 to K.M.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aleem E, Kiyokawa H, Kaldis P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol. 2005;7:831–836. doi: 10.1038/ncb1284. [DOI] [PubMed] [Google Scholar]
- Arooz T, Yam CH, Siu WY, Lau A, Li KK, Poon RY. On the concentrations of cyclins and cyclin-dependent kinases in extracts of cultured human cells. Biochemistry. 2000;39:9494–9501. doi: 10.1021/bi0009643. [DOI] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P. Cdk2 knockout mice are viable. Curr Biol. 2003;13:1775–1785. doi: 10.1016/j.cub.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Berthet C, Klarmann KD, Hilton MB, Suh HC, Keller JR, Kiyokawa H, Kaldis P. Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev Cell. 2006;10:563–573. doi: 10.1016/j.devcel.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci U S A. 2007;104:4383–4388. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Ross KE, Kaldis P, Solomon MJ. Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes Dev. 1999;13:2946–2957. doi: 10.1101/gad.13.22.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow JP, Siu WY, Ho HT, Ma KH, Ho CC, Poon RY. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J Biol Chem. 2003;278:40815–40828. doi: 10.1074/jbc.M306683200. [DOI] [PubMed] [Google Scholar]
- Connell-Crowley L, Solomon MJ, Wei N, Harper JW. Phosphorylation-independent activation of human cyclin-dependent kinase 2 by cyclin A in vitro. Mol Biol Cell. 1993;4:79–92. doi: 10.1091/mbc.4.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim S-H. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- Desai D, Gu Y, Morgan DO. Activation of human cyclin-dependent kinases in vitro. Mol Biol Cell. 1992;3:571–582. doi: 10.1091/mbc.3.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai D, Wessling HC, Fisher RP, Morgan DO. The effect of phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol Cell Biol. 1995;15:345–350. doi: 10.1128/mcb.15.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell. 1994;78:713–724. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- Garrett S, Barton WA, Knights R, Jin P, Morgan DO, Fisher RP. Reciprocal activation by cyclin-dependent kinases 2 and 7 is directed by substrate specificity determinants outside the T-loop. Mol Cell Biol. 2001;21:88–99. doi: 10.1128/MCB.21.1.88-99.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massagué J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- Kaldis P, Russo AA, Chou HS, Pavletich NP, Solomon MJ. Human and yeast cdk-activating kinases (CAKs) display distinct substrate specificities. Mol Biol Cell. 1998;9:2545–2560. doi: 10.1091/mbc.9.9.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaton MA, Bardes ES, Marquitz AR, Freel CD, Zyla TR, Rudolph J, Lew DJ. Differential susceptibility of yeast S and M phase CDK complexes to inhibitory tyrosine phosphorylation. Curr Biol. 2007;17:1181–1189. doi: 10.1016/j.cub.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Italien L, Tanudji M, Russell L, Schebye XM. Unmasking the redundancy between Cdk1 and Cdk2 at G2 phase in human cancer cell lines. Cell Cycle. 2006;5:984–993. doi: 10.4161/cc.5.9.2721. [DOI] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13:55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Chen J, Knights R, Pandur J, Morcillo P, Erdjument-Bromage H, Tempst P, Suter B, Fisher RP. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 2001;20:3749–3759. doi: 10.1093/emboj/20.14.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25:839–850. doi: 10.1016/j.molcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Saiz JE, Barton WA, Fisher RP. Cdc2 activation in fission yeast depends on Mcs6 and Csk1, two partially redundant Cdk-activating kinases CAKs) Curr Biol. 1999;9:441–444. doi: 10.1016/s0960-9822(99)80194-8. [DOI] [PubMed] [Google Scholar]
- Lolli G, Johnson LN. Recognition of Cdk2 by Cdk7. Proteins. 2007;67:1048–1059. doi: 10.1002/prot.21370. [DOI] [PubMed] [Google Scholar]
- Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Morgan DO. The Cell Cycle: Principles of Control. London: New Science Press Ltd; 2007. [Google Scholar]
- Murray AW, Marks D. Can sequencing shed light on cell cycling? Nature. 2001;409:844–846. doi: 10.1038/35057033. [DOI] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon RYC, Hunter T. Dephosphorylation of Cdk2 Thr160 by the cyclin-dependent kinase-interacting phosphatase KAP in the absence of cyclin. Science. 1995;270:90–93. doi: 10.1126/science.270.5233.90. [DOI] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- Ross KE, Kaldis P, Solomon MJ. Activating phosphorylation of the Saccharomyces cerevisiae cyclin-dependent kinase, Cdc28p, precedes cyclin binding. Mol Biol Cell. 2000;11:1597–1609. doi: 10.1091/mbc.11.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Pavletich NP. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nature Struct Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- Saiz JE, Fisher RP. A CDK-activating kinase network is required in cell cycle control and transcription in fission yeast. Curr Biol. 2002;12:1100–1105. doi: 10.1016/s0960-9822(02)00903-x. [DOI] [PubMed] [Google Scholar]
- Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- Shah K, Liu Y, Deirmengian C, Shokat KM. Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc Natl Acad Sci USA. 1997;94:3565–3570. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton A, Freiman R. The Cak1p protein kinase is required at G1/S and G2/M in the budding yeast cell cycle. Genetics. 1997;147:57–71. doi: 10.1093/genetics/147.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakraklides V, Solomon MJ. Comparison of Cak1p-like cyclin-dependent kinase-activating kinases. J Biol Chem. 2002;277:33482–33489. doi: 10.1074/jbc.M205537200. [DOI] [PubMed] [Google Scholar]
- Wohlbold L, Larochelle S, Liao JC, Livshits G, Singer J, Shokat KM, Fisher RP. The cyclin-dependent kinase (CDK) family member PNQALRE/CCRK supports cell proliferation but has no intrinsic CDK-activating kinase (CAK) activity. Cell Cycle. 2006;5:546–554. doi: 10.4161/cc.5.5.2541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.