

Abstract
ATP-gated ion channels (P2X receptors) are abundantly expressed in both neuronal and nonneuronal tissues, where they can serve as postsynaptic receptors. The response to ATP shows marked desensitization in some tissues but not others. Currents induced by ATP in Xenopus oocytes expressing cloned P2X1 (or P2X3) receptors had strong desensitization, whereas currents in cells expressing P2X2 receptors desensitized relatively little (90% vs. 14% decline of current in a 10-s application). In chimeric receptors, substitution into the P2X1 receptor of either one of two 34-residue segments from the P2X2 receptor removed the desensitization; these segments included the first or the second hydrophobic domain. In contrast, desensitization was introduced into the P2X2 receptor only by providing both these segments of the P2X1 (or P2X3) receptor. This suggests that desensitization requires interaction between the two hydrophobic domains of the receptor, and supports the view that these are membrane-spanning segments.
Keywords: ATP-gated channels, chimeric receptors
P2X receptors are cation-selective channels gated by extracellular ATP. In the past few years it has become clear that P2X receptors on various mammalian neurons and other cells are heterogeneous. Three main phenotypes have been distinguished with selective agonists, antagonists, and desensitization (reviewed in refs. 1 and 2). For example, P2X receptors in rat pheochromocytoma (PC12) cells are insensitive to the ATP analog α,β-methylene-ATP ([αβ-CH2]ATP), whereas in vas deferens smooth muscle cells [αβ-CH2]ATP is almost as effective an agonist as ATP itself (see ref. 2). A second difference is exemplified by the P2X receptor in the submandibular gland, where responses to ATP are not blocked by concentrations of the antagonists suramin and pyridoxalphosphate-6-azo-2,4-phenyldisulfonic acid (PPADS), which give complete block in smooth muscles or PC12 cells (3). The third difference is in desensitization; this is striking in some sensory neurons and in smooth muscles, where the current elicited by ATP declines in tens or hundreds of milliseconds, but in most other cells the current induced by ATP is sustained throughout applications lasting for several seconds (1).
The differences in desensitization are reminiscent of those seen among subtypes of other ligand-gated ion channels, and might thus be expected to have important physiological sequela. For example, at glutamate-mediated synapses, desensitization can have a significant effect on synaptic transmission (4). The time constant of decay of synaptic currents mediated by ATP is in the range of 10–20 ms (5–7), suggesting that desensitization may play a role. Desensitization may also limit neurotoxicity. Those neurons that express slowly desensitizing glutamate receptors are more susceptible to excitotoxicity (8). Some P2X receptors have a high calcium permeability (see refs. 1 and 2); if this were coupled with slow desensitization, then cells would be more vulnerable to excessive calcium influx.
Seven P2X receptor subunits have been cloned and expressed (refs. 9 and 10 and references therein). They belong to a structural class of channels distinct from the nicotinic superfamily (gated in vertebrates by acetylcholine, 5-hydroxytryptamine, γ-aminobutyric acid, or glycine) and the glutamate family (gated by glutamic or aspartic acid). Each channel subunit appears to have only two transmembrane segments and a large, extracellular loop (see refs. 1, 11, and 12). In this general respect, although not in primary amino acid sequence, they resemble the sodium-selective channels found in epithelia and their homologs in neurons (degenerins of Caenorhabditis elegans, the mammalian brain sodium channel, and the Phe-Met-Arg-Phe-NH2 (FMRFamide)-gated channel of Helix aspersa; reviewed in ref. 12). For both the P2X receptors and the epithelial sodium channels there is evidence that the ion channel is a multimer, but in neither case is the subunit stoichiometry known (12).
The isolation of P2X receptor cDNAs allows one to compare the properties of expressed receptors with the responses observed in native cells; this is useful both to determine the likely subunit composition of the native receptors and to understand more about the function of the channels in molecular terms. Some of the differences among tissue responses described above are clearly seen in channels expressed from cloned cDNAs. For example, the agonist action of [αβ-CH2]ATP is observed for the expressed P2X1 receptor (cloned from vas deferens smooth muscle; ref. 13) but not for the P2X2 receptor (cloned from PC12 cells; ref. 14). Furthermore, the P2X4 receptor is insensitive to PPADS when expressed in heterologous cells (3, 15, 16), and it is the predominant form expressed by salivary glands (3). The molecular basis of the difference in sensitivity to [αβ-CH2]ATP is not known, but the insensitivity of P2X4 receptors to PPADS results in part from a single amino acid difference with P2X1 and P2X2 (3).
The focus of the present study was the difference in desensitization. Currents evoked by ATP at P2X1 receptors undergo marked desensitization within a few hundred milliseconds, whether expressed in human embryonic kidney cells or Xenopus oocytes (13, 17), and, as mentioned above, similar strong desensitization is observed in the native smooth muscle cells. The P2X3 receptor also shows strong desensitization (18, 19). In contrast, currents evoked at P2X2 receptors, similarly expressed, undergo little or no desensitization on this time scale (14, 17), as do responses of PC12 cells. The 399 amino acids of the P2X1 subunit are 37% identical with the corresponding portion of the P2X2 receptor (which has 472 amino acids due to a longer C terminus). The aim of the experiments was to determine the region(s) of the molecule responsible for this difference in desensitization.
MATERIALS AND METHODS
Mutagenesis and in Vitro Transcription.
P2X receptors were generated by overlap extension polymerase chain reaction (PCR) (20) using the original P2X1 and P2X2 expression plasmids (13, 14) as DNA templates. Mutagenesis primers were pairs of chimeric sense and antisense that were 42-mer long, with the crossover site positioned in the center (i.e., 21 nucleotides of P2X1 or P2X2 to either side). The first set of PCR produced individual DNA segments, which self-assembled to chimeric cDNA in a second PCR with universal flanking primers (see below). The PCR fragments carrying the start codons were primed with 46-mer long primers (5′-GGGAACTAGTGGATCCGACCATGGCTCGGCGGCTGCAAGATGAGCT for P2X1 or 5′-GGGAACTAGTGGATCCGACCATGGTCCGGCGCTTGGCCCGGGGCTG for P2X2) in the first amplification reaction. The 3′ half of those primers is specific for the start of P2X1 or P2X2 coding regions, whereas the first 23 nucleotides are recognized by the universal T3 primer used in the second amplification reaction. The T3 primer (5′-GCACTGAATTAACCCTCACTAAAGGGAACTAGTGGATCCGACCATG) introduces a T3 RNA polymerase promoter followed by a Kozak consensus sequence. Subtype specific 34-mer primers located in the 3′-untranslated region fixed the limits of the 3′-end (5′-TATAGAATTCGTTCAAACTGACACATCTGGCTAG for P2X1 and 5′-TATAGAATTCGCTCATCACTGGTTTATTGAACTC for P2X2). Vent polymerase (Biolabs, Northbrook, IL) was used in 100 μl reaction mix, according to the supplier’s protocol, with 4 mM magnesium. The template for the first amplification was 10 ng of plasmid DNA, and the second amplification combined the first PCRs (2 μl each) in 100 μl. Thirty cycles were executed: 20 s at 96°C, 20 s at 50°C, t s at 72°C (in the first round of amplification, t was 0.5 min for fragments <0.2 kb, 1 min for fragments between 0.2 and 0.5 kb, and 2 min for fragments between 0.5 and 2 kb; in the fusion reaction, t was 2.5 min). The most 5′ fragments of chimeras 7, 9, 12, 15, 16, and 18 were primed within the vector sequence of P2X1 and P2X2 pBKCMV constructs at the unique NheI site (5′-ATAAGCAGAGCTGGTTTAGTGAA). As controls, wild-type DNA templates for in vitro transcription also were generated by two rounds of PCR.
Chimeric PCR products (the average yield was 10–20 μg from three 100 μl reactions) were purified by phenol-chloroform extractions, ethanol precipitated, and resuspended in 20 μl diethyl-pyrocarbonate-treated water. Aliquots of the material were analyzed with the DNA sequencing system from Applied Biosystems. The nucleotide sequences of the chimeras reported were as follows (GenBank accession nos. X80477X80477 for P2X1 = X1; U14414U14414 for P2X2 = X2; X91167X91167 for P2X3 = X3). 1: X2 37–183, X1 357-1775; 2: X1 210-1193, X2 1018–1831; 3: X2 37–183, X1 357-1193, X2 1018–1831; 4: X1 210–356, X2 184-1831; 5: X2 37–1017, X1 1194–1775; 6: X1 210–356, X2 184-1017, X1 1194–1775; 7: X2 1–75, X1 249-1775; 8: X2 37–123, X1 297-1775; 9: X1 1–296, X2 124–183, X1 357-1775; 10: X1 1–248, X2 76–183, X1 357-1775; 11: X1 210-1193, X2 1018–1134, X1 1311–1775; 12: X2 1–123, X1 297–356, X2 184-1017, X1 1194–1775; 13: X1, 210–338, X2 166-1017, X1 1194–1795; 14: X1 210–296, X2 124-1017, X1 1194–1775; 15: X2 1–75, X1 249–356, X2 184-1017, X1 1194–1775; 16: X2 1–75, X1 249–356, X2 184-1831; 17: X2 37–1017, X2 1194–1310, X1 1135–1831; 18: X2 1–75, X1 249–356, X2 184-1017, X1 1194–1310, X2 1135–1831; 19: X1 1–356, X2 184-1041, X1 1218–1775; 20: X1 1–356, X2 184-1059, X1 1236–1775; 21: X1 210–356, X2 184-1077, X1 1254–1775; 22: X2 37–75, X3 193–300, X2 184-1017, X3 1126–1242, X2 1135–1831; 23: X2 1–75, X1 249–356, X2 184-1041, X1 1218–1280, X2 1105–1831; 24: X2 1–75, X1 249–356, X2 184-1041, X1 1218–1280, X2 1105–1831; 25: X1 210-1253, X2 1078–1831; 26: X1 210-1310, X2 1135–1831. Ten microliters of the purified PCR products was transcribed in vitro in 100 μl Promega reaction mix containing cap analog m7G(5′)ppp(5′)G. cRNA was purified for injection into oocytes as described above for the PCR products. Chimeras 2, 23, 24, 25, and 26 did not express functional channels.
Recording from Oocytes.
Stage V oocytes were isolated as described (13, 17) and injected 24 h later with 50 ng of cRNA encoding wild-type or chimeric P2X receptors. Oocytes were maintained at 18°C in a solution containing 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 5 mM sodium pyruvate, and 5 mM Hepes (pH 7.5) supplemented with penicillin and streptomycin (10 units/ml) and gentamycin (1 mg/ml). Two-electrode voltage-clamp recordings were made 2–5 days later using electrodes (0.5–1 MΩ) containing 3 M potassium chloride and a Geneclamp amplifier (Axon Instruments, Foster City, CA). The usual holding potential was −60 mV. Oocytes were perfused continuously (5 ml/min) with a solution (96 mM NaCl/2 mM KCl/1 mM MgCl2/0.1 mM BaCl2/5 mM Hepes, pH 7.5). Agonists were applied using a fast-flow U-tube delivery system; the position and flow rate of the U-tube was adjusted to ensure that agonist solution completely surrounded the exposed surface of the oocyte. Reproducible peak responses were obtained by applying agonist for 10 s. For oocytes expressing P2X1, P2X3, and chimeric receptors, agonists were applied at 5-min intervals to ensure reproducible responses (see below). With oocytes expressing P2X2 receptors, 2-min intervals were sufficient. Currents were sampled at 200 Hz, filtered at 20–50 Hz, digitized, stored, and analyzed using pclamp and axograph software (Axon Instruments). All experiments were performed at room temperature (≈20°C).
Except where stated, desensitization was measured during 10 s applications of 100 μM ATP (sodium salt); this concentration evokes maximal currents for both P2X1 and P2X2 receptors (17). This measure of desensitization was used because the time course of the decay of the current varied considerably among oocytes, requiring from one to three exponential components. Both the peak current amplitude (p) and the current at the end of 10 s (mean value of current from 9.6 to 10 s, d) were measured; desensitization is expressed as 100(1 − d/p). Numerical values are presented ± SEM. At least five oocytes were tested for each receptor.
For the desensitizing receptors (P2X1, P2X3, and some chimeras), the amplitude of the response to the second application of ATP (p2) was always less than that to the first (p1) (see Fig. 1B). This loss of response is expressed as the ratio (%) of the two amplitudes when the applications were 5 min apart (100 p2/p1). Applications following the second one evoked responses of similar amplitude (pn) so long as the interval between them (Δt) was 5 min. The time constant of recovery from desensitization (τ) was estimated from the plot of pn+1/pn = (1 − exp(Δt/τ)) for Δt in the range 5–600 s. Comparison of the time course of the currents has been facilitated in Figs. 2, 3, 4, 5 by scaling the peak amplitudes; the actual currents are noted in the legends. In each figure, the horizontal bar indicates the 10-s application of 100 μM ATP.
Figure 1.
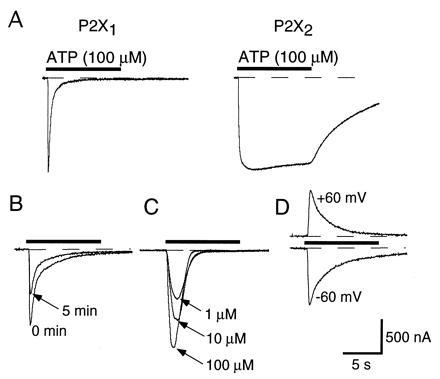
P2X1 receptor desensitization. (A) ATP induces a rapidly desensitizing current when applied to oocytes expressing P2X1 receptors (Left) but a weakly desensitizing current in an oocyte expressing P2X2 receptors (Right). In this and subsequent figures, the ATP is applied for 10 s (indicated by horizontal bar above trace). Current traces have been normalized for comparison of time courses; the mean peak amplitude of currents at P2X1 receptors was 1600 ± 160 nA (n = 38) and at P2X2 receptors 3150 ± 260 nA (n = 47). (B) Desensitization at P2X1 receptors is unaffected by run-down of the response. The superimposed responses were evoked by 100 μM ATP, and applied for the first (0 min) and second times (5 min) to the same oocyte. The decline in the response is not associated with a change in the rate of desensitization. (C) Desensitization at P2X1 receptors is not strongly dependent on concentration. The superimposed traces were evoked by the concentrations indicated (10 s) applied at intervals of 20 min. (D) Desensitization at P2X1 receptors is not voltage-dependent. Two applications of 100 μM ATP for 10 s at an interval of 5 min are shown.
Figure 2.
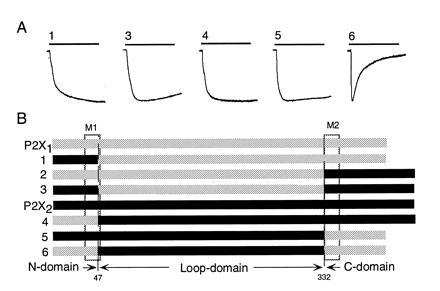
Desensitization of P2X1/P2X2 chimeras distinguishes three domains. (A) Representative currents evoked by 10-s applications of ATP (100 μM) to oocytes expressing five different chimeric receptors. (B) Schematic representation of the three domain chimeras: grey, P2X1; black, P2X2. Rectangles indicate positions of putative membrane-spanning regions M1 and M2. Top four rows show P2X1 and substitutions of P2X2 segments into P2X1: N-domain (chimera 1), C-domain (chimera 2), and N- and C-domains (chimera 3). Bottom four rows show P2X2 receptor and substitutions of P2X1 segments into P2X2: N-domain (chimera 4), C-domain (chimera 5), and N- and C-domains (chimera 6). Mean currents in P2X1, chimeras 1-3, P2X2, and chimeras 4-6 were (measured in nA, number of oocytes in parentheses) 1600 ± 160 (38), 545 ± 82 (19), zero, 824 ± 250 (7), 3150 ± 260 (47), 1340 ± 260 (6), 1390 ± 239 (12), and 2310 ± 210 (14), respectively.
Figure 3.
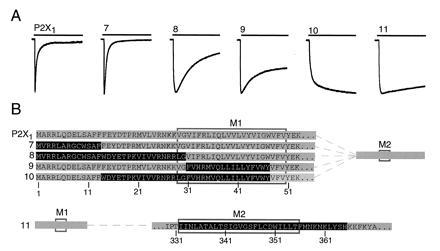
Determination of P2X2 domains that prevent desensitization in the P2X1 receptor. (A) Representative currents evoked by 10-s applications of ATP (100 μM) to oocytes expressing six different chimeric receptors. (B) Schematic representation of chimeras: grey, P2X1; black, P2X2. Rectangles indicate positions of putative membrane-spanning regions M1 and M2. Upper five rows show P2X1 receptor and four chimeras in which N-terminal segments of P2X2 were inserted into P2X1. Lower row shows P2X1 receptor in which an extended second transmembrane domain of P2X2 receptor was inserted. Mean currents in chimeras 7-11 were (measured in nA, number of oocytes in parentheses) 440 ± 58 (3), 1130 ± 260 (14), 1190 ± 420 (4), 788 ± 487 (3), and 319 ± 56 (14), respectively.
Figure 4.
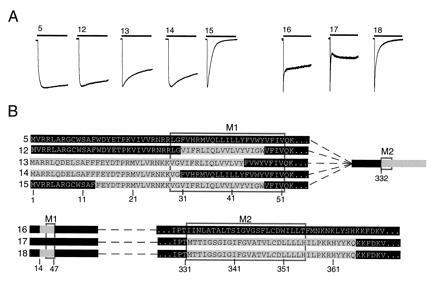
Determination of P2X1 domains that cause desensitization in the P2X2 receptor. (A) Representative currents evoked by 10-s applications of ATP (100 μM) to oocytes expressing eight different chimeric receptors. (B) Schematic representation of chimeras: grey, P2X1; black, P2X2. Rectangles indicated postions of putative membrane-spanning regions M1 and M2. Top row shows the P2X2 receptor with C terminus of P2X1 receptor (chimera 5, see Fig. 2). Next four rows show substitution of N-terminal segments of P2X1 receptor into chimera 2. Lower three rows show P2X2 receptor in which one or both of the minimal segments of P2X1 receptor was inserted. Mean currents in chimeras 5 and 12-18 were (measured in nA, number of oocytes in parentheses) 1390 ± 239 (12), 1460 ± 165 (6), 3110 ± 190 (5), 3000 ± 368 (8), 993 ± 236 (11), 660 ± 82 (11), 424 ± 136 (6), and 200 ± 46 (4), respectively.
Figure 5.
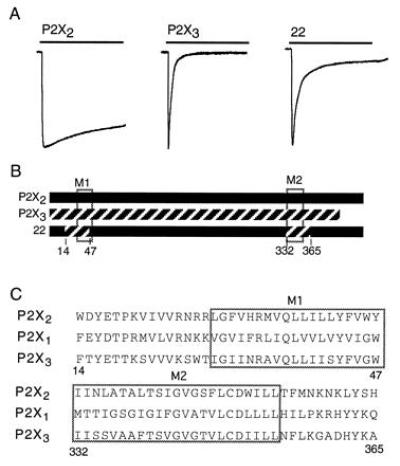
The domains required for desensitization are the same in P2X3 and P2X1 receptors. (A) Representative currents evoked by 10-s applications of ATP (100 μM) to oocytes expressing P2X2 receptors, P2X3 receptors, and a chimeric P2X2/P2X3 receptor. (B) Schematic representation of the portions of the P2X3 receptor (hatched line) that were introduced into the P2X2 receptor to produce desensitization. Mean currents in P2X3 and chimera 22 were (measured in μA, number of oocytes in parentheses) 3.4 ± 1.2 (2) and 2.5 ± 0.7 (3), respectively. (C) Domains required to transfer desensitization include the hydrophobic, presumed membrane-spanning domains (M1, M2, and boxed) and 11–15 residues in the cytoplasmic region.
RESULTS
The basic features of the responses to ATP were as described previously (13, 14, 17). At the P2X1 receptor, ATP evoked concentration-dependent inward currents; the half-maximal current was produced by 3.2 ± 0.9 μM (n = 16). [αβ-CH2]ATP (30 μM) produced 44 ± 4% (n = 11) of the current evoked by 100 μM ATP. For oocytes expressing the P2X2 receptor, the concentration of ATP giving half-maximal current (EC50) was 5.7 ± 0.2 μM (n = 17); these oocytes were insensitive to [αβ-CH2]ATP (30 μM gave 3 ± 1% of the current evoked by 100 μM ATP, n = 32).
Desensitization of P2X1 Receptors.
P2X1 and P2X2 receptors were readily distinguished by their different rates of desensitization (Fig. 1A) (2, 17). In oocytes expressing P2X1 receptors, the current evoked by 100 μM ATP desensitized by 90 ± 1% (n = 37) during a 10-s application of ATP. For oocytes expressing P2X2 receptors, this value was 14 ± 1% (n = 130).
For P2X1 receptors, the peak amplitude of the current evoked by a second 10-s application beginning 5 min after the first was reduced by 60 ± 3% (n = 45) (Fig. 1B). Third and subsequent responses had peak amplitudes similar to that of the second, so long as the interval between them was at least 5 min. For shorter intervals, the peak response was reduced; the recovery of the peak amplitude was related to the time interval between applications by an exponential with time constant 108 ± 7.4 s (n = 5). Responses to ATP (100 μM) of oocytes expressing P2X2 receptors were essentially unchanged during repeated applications at 5-min intervals (amplitude of second response declined by 4 ± 2% relative to that of the first, n = 32).
Desensitization of the P2X1 receptor was the same during the second 10-s application of ATP as during the first (94 ± 1%, n = 35; Fig. 1B), even though the peak amplitude of the second responses was less than that of the first. In a sample of 26 oocytes expressing the P2X1 receptor, the peak currents evoked by 100 μM ATP ranged from 142 to 3467 nA; the degree of decline of the current during a 10-s application showed no correlation with the initial amplitude (r = 0.255, P > 0.05).
Different concentrations of ATP (Fig. 1C) or different agonists caused similar desensitization. The peak current evoked by 10 μM ATP was 80 ± 3% (n = 9) of that produced in the same oocytes by 100 μM; in 30 oocytes tested with 10 μM ATP, currents desensitized by 95 ± 1% during a 10-s application. For 1 μM ATP, the peak current was 31 ± 8% (n = 7) of that evoked by 100 μM ATP in the same oocytes; currents evoked by 1 μM ATP declined by 92 ± 3% (n = 9) during a 10-s application. Similar applications of [αβ-CH2]ATP (30 μM) and ADP (100 μM) evoked currents that desensitized by 94 ± 1% (n = 20) and 94 ± 4% (n = 4), respectively.
Membrane potential had little or no effect on desensitization. Thus, hyperpolarizing oocytes from −60 mV to −90 mV increased the mean amplitude of the inward current from 919 ± 120 nA to 1333 ± 175 nA (n = 9) but had no significant effect on the time course of desensitization (546 ± 34 ms and 598 ± 32 ms to reach 50% of peak amplitude, respectively). Even when oocytes were depolarized to +60 mV (Fig. 1D), ATP evoked outward currents (661 ± 120 nA, n = 7) that declined with a similar time course (585 ± 72 ms, n = 7). Other treatments that did not affect desensitization were phorbol 12-myristate 13-acetate (100 nM, 5–40 min, n = 7), dibutyryl cAMP (1 mM), together with isobutylmethylxanthine (500 μM, 40–60 min, n = 3), cytochalasin B (10 and 50 μM, 30–60 min, n = 6), and cyclothiazide (100 μM, 5 min, n = 4).
In the P2X1 Receptor, Introduction of Either of Two P2X2 Receptor Segments Removes Desensitization.
The P2X receptors were initially divided into three large domains, corresponding roughly to what are thought to comprise (i) the intracellular N terminus and first transmembrane region (N-domain; residues 1–47), (ii) the large, presumed extracellular, region (loop domain; residues 48–331), and (iii) the second transmembrane region and the intracellular C terminus (C-domain; residues 332–399) (Fig. 2). All numbers refer to the P2X1 receptor.
Substitution of the P2X2 N-domain into the P2X1 receptor completely abolished desensitization (chimera 1) (Fig. 2); in this construct, ATP (100 μM) evoked a current that declined by only 2 ± 0.3% (n = 55) in 10 s. Substitution of the loop-domain of P2X2 into the P2X1 receptor (chimera 6) did not affect desensitization (90 ± 1%, n = 49). Introduction of the P2X2 C-domain into the P2X1 receptor (chimera 2) gave a nonfunctional channel; however, function was restored when both the N-domain and C-domains from P2X2 were substituted together (chimera 3). Chimera 3 showed the minimally desensitizing currents (13 ± 2%, n = 12) typical of the wild-type P2X2 receptor.
These experiments indicate that exchange of the first 47 amino acid residues fully eliminates desensitization from the P2X1 receptor. The region responsible within this N-domain was mapped with further chimeras (Fig. 3). Substitution of the first 13 amino acid residues of P2X2 into the P2X1 receptor was not sufficient; this chimera (7) had properties similar to the wild-type P2X1 receptor (desensitization 96 ± 1%, n = 14). Introduction of the first 30 residues of P2X2 (i.e., the N terminus, but not the first transmembrane domain; chimera 8) only slightly reduced desensitization (76 ± 3%, n = 41), and substitution of residues 31–47 (approximately equivalent to the first transmembrane domain; chimera 9) was also partly effective to remove desensitization (38 ± 2%; n = 15) (Fig. 3). On the other hand, substitution of only residues 14–47 (chimera 10) completely removed desensitization (4 ± 2%, n = 9), indicating that the minimum portion of the P2X2 receptor that completely removes desensitization from the P2X1 receptor is residues 14–47. The EC50 for ATP was not different from control (5.4 ± 0.8 μM, n = 3).
A 34 amino acid segment within the C-domain was also effective to relieve desensitization. Thus, substitution of the corresponding P2X2 segment into position 332–365 of the P2X1 receptor (chimera 11) provided a channel in which the current declined by only 10 ± 1%, n = 25) during a 10-s application of ATP (Fig. 3).
In those chimeras in which the minimal N-region or the minimal C-region of P2X2 were introduced into the P2X1 receptor, the run-down of response became less as did the desensitization: thus, in chimeras 7-11 (Fig. 3) run-down was 28 ± 4% (n = 3), 42 ± 9% (n = 10), 0 ± 3% (n = 5), 4 ± 3% (n = 2), and 9 ± 2% (n = 9), respectively.
In the P2X2 Receptor, Both of Two P2X1 Receptor Segments Are Needed to Confer Desensitization.
The simple reciprocal to chimera 1 has the N-domain of the P2X1 receptor substituted into P2X2 (chimera 4) (Fig. 2); this construct showed nondesensitizing currents (2 ± 0.3, n = 24). Likewise, introducing the C-domain of P2X1 into P2X2 did not cause any desensitization (chimera 5; 4 ± 1%, n = 12) (Fig. 2). However, as described above, the P2X2 receptor with both the C-domain and the N-domain of P2X1 (chimera 6) desensitized similarly to wild-type P2X1 receptors, implying that portions of both N-domain and C-domain are required for full desensitization.
In the P2X2 receptor with the P2X1 C-domain (chimera 5), the minimum region of the P2X1 N-domain that was required to confer desensitization was residues 14–47 (chimera 15); in this construct, the current declined by 99 ± 0.2% (n = 40) (Fig. 4). Residues 1–30 (chimera 14; 31 ± 2%, n = 28), 1–43 (chimera 13; 34 ± 1, n = 12), or 31–47 (chimera 12; 12 ± 1%, n = 16) were only slightly effective (Fig. 4).
In the P2X2 receptor with the P2X1 N-domain (residues 1–47; chimera 4), desensitization was complete when the entire C-domain of P2X1 was also provided (chimera 6; Fig. 2); beginning at M332) (see above). It was also complete (99.6 ± 0.4%, n = 15) when the P2X1 C-domain was shortened by five residues (beginning at S337; chimera 19), but became very much less with further shortening of the C domain (beginning at A345, chimera 20, 43 ± 1%, n = 14; beginning at L351, chimera 21, 25 ± 2%, n = 33). This indicates that the desensitization requires almost all of the second transmembrane domain of the P2X1 receptor, although it does not require the highly conserved region that immediately precedes M2. In the P2X2 receptor with the P2X1 N-domain minimal region (residues 14–47), desensitization was complete when the entire C-domain of P2X1 was also provided (beginning at M332; chimera 15) (see above). The shortest segment of this P2X1 C-domain that conferred desensitization was 332–365 (99.6 ± 0.4%, n = 11; chimera 18; Fig. 4). The EC50 for ATP was approximately the same as for wild-type receptors (4.8 ± 0.2 μM, n = 4). Constructs in which this domain was further shortened (337–365 in chimera 23 and 332–357 in chimera 24) did not express measurable currents.
Whereas full desensitization was obtained with both 34-residue segments of the P2X1 receptor (chimera 18), introduction of either segment alone produced a intermediate and more variable phenotype in which a very rapidly desensitizing current was followed by a sustained component (Fig. 4). Chimera 16 (first segment only) declined by 32 ± 4% (n = 36), and chimera 17 (second segment only) declined by 50 ± 8% (n = 10) (Fig. 4).
P2X2 receptors containing the N and C domains of P2X1 receptors (chimeras 15, 18, and 19) showed strong desensitization but little or no rundown [4 ± 4% (n = 10), 3 ± 4% (n = 4), and 1 ± 11% (n = 4), respectively]. Recovery from desensitization occurred quickly; in the case of chimera 18, recovery occurred with a time constant of 27 ± 4.9 s (n = 3), in comparison to 108 s for the P2X1 receptor.
The P2X3 receptor also exhibits strong desensitization (18, 19). Introduction of the two 34-residue segments of the P2X3 that are analogous to the minimal N- and C-terminal domains of the P2X1 receptor described above also led to a receptor that showed profound desensitization (Fig. 5). Chimera 22 desensitized by 90 ± 2% (n = 7), and wild-type P2X3 by 98 ± 0.4% (n = 5). Recovery from desensitization occurred much more rapidly (time constant 7.3 ± 1.1 s, n = 4) than for the wild-type P2X3 receptor (τ about 170 s). Thus, the transfer of desensitization with these minimal domains can be generalized to two donor P2X receptors, despite only limited homology of amino acid sequences (Fig. 5C).
DISCUSSION
Fig. 6 summarizes the main results of the present experiments; it also indicates the presumed topology of the receptor in the membrane (see refs. 11 and 12), which provides the context for the present discussion. Both gain or loss of desensitization can be attributed to a symmetrical pair of segments. Desensitization can be removed from the P2X1 receptor by providing either one of the two segments from the P2X2 receptor. Desensitization can be introduced into the P2X2 receptor only by providing both segments, and these can come either from P2X1 or P2X3 receptors. There are several implications of these findings.
Figure 6.
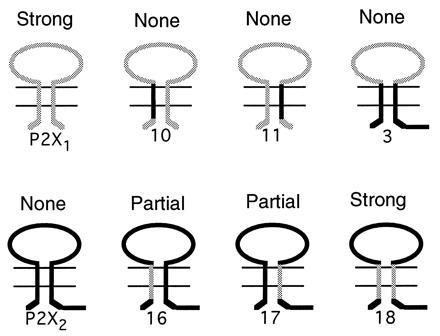
Schematic representation of domains needed to change desensitization. P2X1 receptor (grey lines) shows “full” desensitization; introduction of one or other domains of the P2X2 receptor (black lines) prevents desensitization. P2X2 receptor shows no desensitization; introduction of both minimal domains of the P2X1 receptor is needed to produce full desensitization.
First, the results indicate that the large extracellular loop is not involved in desensitization and, by implication, probably does not contribute directly to the pore-forming portion of the molecule.
Second, the findings provide evidence that certain domains of the molecule operate as functional units. Thus, the overall function of the molecule as a ligand-gated channel was not compromised by the exchange of the minimal domains (Fig. 5C) among P2X1, P2X2, and P2X3 receptors. The exchange of larger domains (e.g., chimera 2, Fig. 2B) or smaller domains (e.g., the second domain of P2X1 shortened by five N-terminal or seven C-terminal amino acids) often led to nonfunctional channels. Efforts to exchange domains between P2X1 and P2X2 receptors within the large extracellular loop also usually led to nonfunctional channels (unpublished observation). Third, some of the results imply interactions between domains of the protein. Thus, simple replacement of the entire C-domain of the P2X1 receptor with that of the P2X2 receptor produced a null phenotype (chimera 2), but function was fully restored by substituting in addition the first 47 residues of the P2X1 receptor (chimera 3).
The domains that could be exchanged among the P2X receptors to alter the desensitization phenotype include the most hydrophobic segments of the molecules, which are thought to be membrane-spanning segments. However, they additionally include segments of 11–15 amino acids that would be located at the immediate cytoplasmic aspect; these segments have an abundance of positively charged residues, as is commonly found in such a location (21). Although we have not systematically shortened or mutated these regions, it appears from the results with chimeras 9 and 10 that the first charged region (pre-M1) of P2X2 is needed to remove desensitization from P2X1, and the results with variants of chimera 6 and 15 suggest that the second charged region (post-M2) of P2X1 is needed (along with pre-M1, M1, and M2) to bring desensitization to the P2X2 receptor. The amino acid sequences of these domains of the three receptors are compared in Fig. 5C; they are 26–48% pairwise identical, with the closest being P2X2 and P2X3 receptors for both the first and second domains. This seems to reflect overall identity between subunits (9) rather than revealing a specific sequence motif for desensitization. A comparison with the sequences of P2X4, P2X5, and P2X6 receptors, in which currents also desensitize much more slowly than with P2X1 and P2X3 (18), does not provide any indication that a particular residue in a given position correlates with desensitization. This is not to say that single amino acid substitutions might not have effects on desensitization, but it makes it unlikely that a difference in one or a few amino acids could account for the large differences between P2X1 and P2X2. In homomeric α7 nicotinic channels and 5-HT3 receptors, manmade changes at a single position in the amphipathic M2 (pore-lining) domain can considerably alter the rate of desensitization (22, 23). This seems true also for a naturally occurring receptor variant in C. elegans (24). On the other hand, glutamate receptors can fine-tune their kinetic properties with the help of alternative splicing (25) or RNA editing (26), although in this case the crucial sites do not reside in the pore.
We have concentrated on a relatively crude measure of desensitization, namely the fraction of the current remaining at the end of a 10-s application of a fixed concentration of ATP. This was chosen because it was obvious from early experiments that in some chimeras desensitization could not readily be fitted by one or two exponentials. Clearly, this simple approach fails to detect more subtle intermediate desensitizing states that are entered by some of the chimeric constructs, such as were apparent for P2X2 receptors carrying only one or other 34-residue segment of P2X1 (Fig. 4). On the one hand, with substitution of relatively small sections of the entire receptor molecule, we were able to effect a complete transition of phenotype. The desensitizing P2X2 receptor variants (chimeras 18 and 22, in which desensitization was introduced by providing both minimal domains of P2X1 or P2X3) were fully equivalent to the wild-type P2X1 or P2X3 receptors as measured by the current at 10 s. On the other hand, it is evident that the desensitized state(s) entered by these chimeric receptors is very much less stable than for the wild-type P2X1 or P2X3 receptors, because recovery from desensitization occurred some 3–10 times more rapidly.
We have used only homomeric channels. There is evidence that native channels can also be heteromultimeric, and heteromultimers formed by coexpression of P2X2 and P2X3 subunits desensitize only slowly; that is, they have the desensitization phenotype of the P2X2 component (19). Those observations are analogous to the present result that the nondesensitizing current is the “dominant” phenotype.
Desensitization was difficult to produce, requiring two interacting segments to be substituted; desensitization was easy to lose, requiring only one segment to be substituted. It is possible that the segments identified in the present experiments on homomeric channels are also involved in the subunit interactions of heteromeric channels.
The failure to alter the desensitization of the wild-type P2X1 receptor by a range of experimental treatments effective at other ligand-gated channels suggests that it may result from intrinsic conformational changes of the receptor protein that do not involve current flow or calcium entry, and might not involve phosphorylation or cytoskeletal interactions. The portions of the molecule that are directly involved in this conformational change appear not to include the large extracellular loop, and the unimportance of the intracellular N and C termini suggests that it does not result from a simple channel block by a tethered “ball.” The results are consistent with the notion that the conducting pore is formed by the two predicted transmembrane domains and adjacent cytoplasmic segments, but further experiments would be required to show this directly.
Acknowledgments
We thank Annmarie Surprenant and Sarah Thomas for carrying out the experiment illustrated in Fig. 1B.
Footnotes
Abbreviation: [αβ-CH2]ATP, α,β-methylene-ATP.
References
- 1.Surprenant A, Buell G, North R A. Trends Neurosci. 1995;18:224–229. doi: 10.1016/0166-2236(95)93907-f. [DOI] [PubMed] [Google Scholar]
- 2.Evans R J, Surprenant A. Semin Neurosci. 1996;8:217–223. [Google Scholar]
- 3.Buell G N, Lewis C, Collo G, North R A, Surprenant A. EMBO J. 1996;15:55–62. [PMC free article] [PubMed] [Google Scholar]
- 4.Jones M V, Westbrook G L. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- 5.Evans R J, Derkach V, Surprenant A. Nature (London) 1992;357:503–505. doi: 10.1038/357503a0. [DOI] [PubMed] [Google Scholar]
- 6.Edwards F A, Gibb A J, Colquhoun D. Nature (London) 1992;359:144–147. doi: 10.1038/359144a0. [DOI] [PubMed] [Google Scholar]
- 7.Bennett M R, Farnell L, Gibson W G, Karunanithi S. Biophys J. 1995;68:925–935. doi: 10.1016/S0006-3495(95)80268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brorson J R, Manzolillo P A, Gibbons S J, Miller R J. J Neurosci. 1995;15:4515–4524. doi: 10.1523/JNEUROSCI.15-06-04515.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collo G, North R A, Kawashima E, Merlo-Pich E, Neidhart S, Surprenant A, Buell G. J Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Surprenant A, Rassendren F, Kawashima E, North R A, Buell G. Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 11.North R A. Semin Neurosci. 1996;8:187–194. [Google Scholar]
- 12.North R A. Curr Opin Cell Biol. 1996;8:474–483. doi: 10.1016/s0955-0674(96)80023-8. [DOI] [PubMed] [Google Scholar]
- 13.Valera S, Hussy N, Evans R J, Adami N, North R A, Surprenant A, Buell G N. Nature (London) 1994;371:516–519. doi: 10.1038/371516a0. [DOI] [PubMed] [Google Scholar]
- 14.Brake A J, Wagenbach M J, Julius D. Nature (London) 1994;371:519–523. doi: 10.1038/371519a0. [DOI] [PubMed] [Google Scholar]
- 15.Bo X, Zhang Y, Nassar M, Burnstock G, Schoepfer R. FEBS Lett. 1995;375:129–133. doi: 10.1016/0014-5793(95)01203-q. [DOI] [PubMed] [Google Scholar]
- 16.Soto F, Garcia-Guzman M, Gomez-Hernandez J M, Hollmann M, Karschin C, Stühmer W. Proc Natl Acad Sci USA. 1996;93:3684–3688. doi: 10.1073/pnas.93.8.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans R J, Lewis C, Buell G, North R A, Surprenant A. Mol Pharmacol. 1995;48:178–183. [PubMed] [Google Scholar]
- 18.Chen C, Akopian A N, Sivilotti L, Colquhoun D, Burnstock G, Wood J N. Nature (London) 1995;377:428–430. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- 19.Lewis C, Neidhart S, Holy C, North R A, Buell G, Surprenant A. Nature (London) 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- 20.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 21.von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 22.Revah F, Bertrand D, Galzi J L, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux J P. Nature (London) 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 23.Yakel J L, Lagrutta A, Adelman J P, North R A. Proc Natl Acad Sci USA. 1993;90:5030–5033. doi: 10.1073/pnas.90.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treinin M, Chalfie M. Neuron. 1995;14:871–877. doi: 10.1016/0896-6273(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 25.Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg P H, Ruppersberg J P. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- 26.Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger J R P, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg P H. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]