

Abstract
Cystic fibrosis is a fatal genetic disorder involving dysfunction of the cystic fibrosis transmembrane regulator protein (CFTR) resulting in progressive respiratory failure. Previous studies indicate that CFTR regulates cellular glutathione (GSH) transport and that dysfunctional CFTR is associated with chronic pulmonary oxidative stress. The cause and the source of this oxidative stress remain unknown. The current study examines the role of the mitochondria in CFTR-mediated pulmonary oxidative stress. Mitochondrial GSH levels and markers of DNA and protein oxidation were assessed in the lung mitochondria from CFTR-knockout mice. In addition, in vitro models using human CFTR-sufficient and -deficient lung epithelial cells were also employed. Mitochondrial GSH levels were found to be decreased up to 85% in CFTR-knockout mice, and 43% in human lung epithelial cells deficient in CFTR. A concomitant 29% increase in the oxidation of mitochondrial DNA, and a 30% loss of aconitase activity confirmed the existence of a mitochondrial oxidative stress. Flow cytometry revealed significantly elevated levels of cellular reactive oxygen species (ROS) in CFTR-deficient human lung cells. These studies suggest that dysfunctional CFTR leads to an increase in the level of ROS and mitochondrial oxidative stress. This oxidative stress, however, appears to be a consequence of lower mitochondrial GSH levels and not increased oxidation of GSH. Further studies are needed to determine how CFTR deficiency contributes to mitochondrial oxidative stress and the role this plays in CFTR-mediated lung pathophysiology.
Keywords: aconitase, CFTR, glutathione, HPLC, mitochondrial DNA
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane regulator protein (CFTR). CFTR is a 168-kD integral membrane protein located on the apical cell membrane of secretory epithelia. On the apical membrane, CFTR couples ATP hydrolysis to the extracellular transport of chloride and other organic anions (1). CF is characterized by pancreatic insufficiency, intestinal obstructions, impaired nutrient absorption, male sterility, and most importantly, recurrent episodes of bronchitis and pneumonia. While the gastrointestinal complications can be managed through diet and enzyme replacement therapy, treatment of the pulmonary complications is more challenging. In fact, mortality in CF is primarily due to respiratory failure (2).
While CFTR has long been recognized as an anion channel, clinical and experimental observations over the last decade indicate that CFTR has an important role in the extracellular transport of glutathione (GSH) (3–6). In the extracellular environment, GSH serves as an antioxidant protecting cells from the damaging effects of oxidants, and as a substrate for extracellular glutathione peroxidase involved in the detoxification of hydrogen peroxide (7). In an early clinical study investigators found significantly lower concentrations of GSH in the epithelial lining fluid (ELF) and serum of patients with CF compared with control subjects without CF (8). ELF is a thin, biochemically complex layer of fluid that covers the entire airspace surface of the lung. ELF was initially recognized for its surface tension–reducing properties that facilitate alveolar compliance, but now it is appreciated as a first line of defense against inhaled chemicals and pathogens (9, 10). In vitro studies demonstrated that functional CFTR was indeed necessary for a significant portion of apical GSH secretion (3, 4, 6). In vivo studies with CFTR knockout (KO) mice revealed ∼ 50% lower ELF GSH concentration, and strongly supported an important role of CFTR in ELF GSH homeostasis (5) and an adaptive GSH response to Pseudomonas lung infection (11).
In addition to the diminished ELF GSH levels, the lungs of CFTR KO mice also exhibit a mild but significant increase in markers of oxidative stress. Increases in both a lipid oxidation marker (thiobarbituric acid reactive substances) and a DNA oxidation marker (8-hydroxy-2-deoxyguanonsine [8OHdG]) were detected in the lung tissue of CFTR KO mice (5). There is an important debate in the literature as to whether a low-level inflammatory response exists even before the onset of infection in newborns with CF (10, 12, 13). An equally plausible explanation for the previously observed increases in oxidative stress markers is mitochondrial generation of oxidants. Mitochondria are often the primary source of intracellular oxidative stress and can produce intramitochondrial reactive oxygen species (ROS) such as superoxide 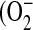 ) and hydrogen peroxide (H2O2) even under basal conditions (14). A small number of studies have postulated that mitochondrial dysfunction may play a role in CF (15–18). These studies have noted changes in mitochondrial morphology, elevated oxygen consumption, increased activity of the mitochondrial electron transport chain with higher rates of energy utilization, and altered mitochondrial calcium metabolism in the CF phenotype.
) and hydrogen peroxide (H2O2) even under basal conditions (14). A small number of studies have postulated that mitochondrial dysfunction may play a role in CF (15–18). These studies have noted changes in mitochondrial morphology, elevated oxygen consumption, increased activity of the mitochondrial electron transport chain with higher rates of energy utilization, and altered mitochondrial calcium metabolism in the CF phenotype.
Based on these previous observations of mitochondrial dysfunction, we sought to determine whether mitochondria were a source of oxidative stress under basal conditions when CFTR is deficient. Oxidative stress is defined as an imbalance between oxidant production and antioxidant defense resulting in an increase in the steady-state levels of oxidized cellular macromolecules. The results of our investigations show that compared with the control mouse, lung mitochondria from CFTR KO mice have significantly lower levels of GSH, decreased aconitase activity, and increased levels of 8OHdG in mitochondrial DNA. Additional studies with human CFTR-sufficient and CFTR-deficient cells corroborated the presence of a deficiency in mitochondrial GSH steady-state levels and revealed elevated levels of ROS in CFTR-deficient cells.
MATERIALS AND METHODS
Materials
Unless noted otherwise, all reagents were obtained from commercial suppliers such as Sigma Chemical Co. (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
Animal Care and Use
This study compared the mitochondrial consequences of the CFTR mutation from two congenic CFTR KO strains (S489X and FABP breeding stock were kind gifts from Dr. Anna van Heeckeren, Case Western Reserve University, Cleveland, OH) with C57B6 control mice. In the S489X congenic C57B6 strain, a mutation creates a stop codon and produces a truncated CFTR protein (19). Without functional CFTR in the gastrointestinal tract, these mice can develop bowel obstructions when maintained on a regular solid diet. To avoid this problem, S489X mice are maintained on a liquid diet (Peptamen; Nestle, Glendale, CA). In the FABP congenic C57B6 strain, human CFTR is expressed in gastrointestinal tissue via the fatty acid–binding protein promoter (FABP) that allows the animals to be maintained on the same solid diet as the control mice (20). With the exception of the liquid diet for the S489X mice, all mice were provided solid mouse chow and autoclaved tap water ad libitum. These animal studies were approved by the IACUC committee at National Jewish Medical and Research Center.
Cell Culture
Two human lung epithelial cell lines that only differed in the presence or absence of a functional CFTR were used in this study. The IB3 cell line was derived from bronchial epithelial cells from a patient with CF and was immortalized by viral transformation (16). These cells possess heterozygous mutations (ΔF508 and W1282X) for the CFTR gene. The C38 cell line was generated by stable transfection of the IB3 line with a cDNA encoding a wild-type human CFTR. Thus, the C38 and IB3 cells provide CFTR-sufficient and -deficient cell lines, respectively, for these studies. Cell lines were cultured in LHC-8 medium with 5% fetal bovine serum and maintained at 37°C with humidified air containing 5% CO2. For these experiments, both cell types were grown to near-confluence (∼ 90%) in T-75 flasks or 24-well plates. To obtain mitochondria, cell monolayers in T-75 flasks were rinsed once with PBS, scraped, transferred to 15-ml conical centrifuge tubes, and pelleted at 2,000 × g for 10 min at 4°C. Cells from three separate T-75 flasks were combined in a single 15-ml conical to produce a single sample. Cell mitochondria were then isolated for subsequent analysis of mitochondrial GSH, aconitase activity, and fumarase activity.
Mitochondrial Isolation
Isolation of lung mitochondria for analysis of oxidative stress markers was achieved using a procedure based on differential centrifugation (21). Mice were killed with a pentobarbital overdose and the lungs excised. Lungs were rinsed in cold PBS and homogenized in 2.0 ml of ice-cold isotonic mitochondrial isolation buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HCl, 1 mM EDTA, pH 7.5) with a Kontes-Duall glass homogenizer. The homogenate was then centrifuged (1,300 × g for 10 min at 4°C) to pellet tissue debris and nuclei, yielding a supernatant enriched in mitochondria. This centrifugation was repeated on the supernatant until no visible pellet was obtained (usually two to four centrifugations). Mitochondria from the supernatant were isolated by centrifugation at 17,000 × g for 10 min at 4°C. To further reduce the presence of any cytosolic contamination, the mitochondrial pellet was resuspended in the mitochondrial isolation buffer and again pelleted by centrifugation.
Isolation of C38 and IB3 mitochondria from cell pellets was achieved with a slight modification of the tissue procedure outlined above. The pelleted C38 and IB3 cells were resuspended in 250 μl of mitochondrial isolation buffer and homogenized with 15 strokes in a Dounce homogenizer (Fisher Scientific, Pittsburgh, PA) on ice. The homogenate was transferred to a 1.5-ml microcentrifuge tube. The homogenizer was then washed with 1.0 ml of mitochondrial isolation buffer that was then added to the microcentrifuge tube. To obtain mitochondria, the homogenate then was subjected to the same differential centrifugation procedure described above for lung tissue. Mitochondrial purity and isolation efficiency were assessed by determination of lactate dehydrogenase (LDH) and glutamate dehydrogenase (GDH) activities.
GDH and LDH Activities
Assessing the contamination of the mitochondrial preparation for cytosolic components is necessary to ensure that our oxidative stress markers are not overtly influenced by cytosolic or nuclear constituents. In these studies, the effectiveness of our mitochondria isolation procedures were assessed by determining LDH and GDH activities in the mitochondrial and cytosolic fractions (22). LDH and GDH are enzymes exclusively found in the cytosol and mitochondria, respectively. The proportion of LDH activity in the mitochondrial fraction provides a measure of cytosolic contamination, while the proportion of GDH activity indicates the efficiency of the isolation procedure.
Before LDH and GDH analyses, the mitochondrial pellets were resuspended in PBS and lysed by the addition of lauryl dimethylamine N-oxide to a final concentration of 0.3% (vol/vol). An aliquot was removed for determination of protein concentration. An equal volume of the cytosolic fraction was prepared in a similar fashion for analysis.
LDH activity in both fractions was determined kinetically by monitoring the loss of NADH at 340 nm. Briefly, 5 μl of sample was combined with NADH in Tris-HCl using a 96-well plate. The reaction was initiated by the addition of pyruvate and the loss of NADH was monitored at 340 nm for 10 min. LDH activity in the samples was calculated based on an extinction coefficient of 6.2 mM-1.
GDH activity was determined using an assay developed for a 96-well plate based on previously published methods (23). Briefly, a solution containing 10 mM NADH and 50 mM ADP were mixed with 3.3 M ammonium acetate as a source of ammonium ions. An aliquot of the sample was added and followed by the addition of 1 unit of LDH to eliminate pyruvate. The assay quantified GDH activity based on the consumption of NADH in the transamination of α-ketoglutarate (oxoglutarate). To initiate the reaction, 233 mM α-ketoglutarate was added and absorbance at 340 nm monitored. GDH activity in the samples was calculated based on an extinction coefficient of 6.2 mM-1.
Ultimately, the total activity of LDH and GDH were determined for the entire sample volumes. Purity and isolation efficiency were expressed as percent of the total (i.e., combined) activity in the samples.
Analysis of Mitochondrial GSH Concentrations
Mitochondrial pellets from tissue samples were initially resuspended in 175 μl of deionized-distilled water, and a 25-μl aliquot was retained for subsequent determination of protein concentration. To the remaining 150 μl, 10 μl of 17.5% (wt/vol) metaphosphoric acid was added and cooled on ice for ∼ 15 min. To remove precipitated proteins, the acidified preparation was centrifuged (20,000 × g for 10 min at 4°C) and the supernatant retained for mitochondrial GSH analysis. GSH in the lung mitochondria was analyzed by HPLC coupled with coulometric electrochemical detection (CoulArray Model 5600; ESA Inc., Chelmsford, MA). Sample analysis was done using a 7- × 53-mm C-18 reverse phase (Platinum EPS C18 100A 3 μm; Alltech Associates Inc., Deerfield, IL) and a mobile phase of 125 mM potassium acetate in 1% acetonitrile at pH 3.0. The electrode potentials in channels one through four in the array were set at 100, 215, 485, and 650 mV, respectively. Under these conditions GSH exhibited a retention time of 3.4 min. The GSH signal was distributed across electrodes 2–4, with the dominant signal on channel 3. GSH concentrations were determined from a 5-μl injection and quantified from a five-point calibration curve generated from standards that were prepared fresh daily and analyzed in duplicate.
In an effort to also obtain oxidized mitochondrial GSH (GSSG) concentrations with the reduced GSH concentrations, the C38 and IB3 mitochondria were analyzed by HPLC coupled with fluorescence detection. To accomplish this, C38 and IB3 mitochondrial preparations were treated with glutathione reductase and subsequently derivatized with monobromobinane (mBBr) (24). Mitochondrial pellets from C38 and IB3 cells were resuspended in 90 μl of water and frozen at −80 C for ∼ 30 min. Mitochondrial pellets were then thawed quickly at 37°C and an equal volume of KPBS (50 mM potassium phosphate buffer, 17.5 mM EDTA, 50 mM serine, 50 mM boric acid; pH 7.4) buffer was added. To each sample, 10 μl of reduced des-Gly-glutathione (0.1 mM stock solution) was added as an internal standard. Samples were then treated with 10 μl of mBBr (5 mM in acetonitrile) and incubated at room temperature in the dark for 30 min. The derivatization reaction was terminated by the addition of 10 μl of 70% perchloric acid. The samples were centrifuged at 16,000 × g for 10 min and 180 μl of supernatant was transferred to HPLC vials for analysis. Samples were analyzed on a Hitachi HPLC (Model Elite LaChrom; San Jose, CA) equipped with a fluorometric detector (Model L-2480). Samples were eluted with a Synergi 4 μ Hydro-RP 80A C18 column (150 × 4.6 mm; Phenomenex, Torrance, CA) using a mobile phase consisting of 1% acetic acid, 7% acetonitrile, and the pH adjusted to 4.25 with NH4OH. GSH analysis was performed with 1-μl injections and a constant flow rate of 1.0 ml/min. Detector excitation and emission wavelengths were set at 390 and 480 nm, respectively. To account for differences in the number of isolated mitochondria among the tissue and the cell culture samples, mitochondrial reduced GSH and GSSG concentrations were normalized to the sample protein concentration.
Aconitase and Fumarase Activity Assays
Immediately before aconitase activities were determined, freshly isolated mitochondria were suspended in 0.5 ml of buffer containing 50 mM Tris-HCl (pH 7.4) and 0.6 mM MnCl2 and sonicated for 2 s. Aconitase activity was measured spectrophotometrically by monitoring the formation of cis-aconitate from added iso-citrate (20 mM) at 240 nm and 25°C. One unit was defined as the amount of enzyme necessary to produce 1 μmol cis-aconitate per minute (ε240 = 3.6 mM−1).
Fumarase activity was determined by measuring the increase in absorbance at 240 nm at 25°C in the reaction mixture to which 30 mM potassium phosphate (pH 7.4), and 0.1 mM L-malate were added. One unit (U) was defined as the amount of enzyme necessary to produce 1 μmol fumarate per minute (ε240 = 3.6 mM−1 cm−1).
Isolation of Mitochondrial DNA
As a marker of mitochondrial oxidative stress, levels of 8OHdG in mitochondrial DNA (mtDNA) from control and CFTR KO mouse lungs were determined. Isolation of mtDNA from mouse lungs for analysis was performed using the Wako mtDNA Extractor Kit (Wako Chemicals USA, Inc.; Richmond, VA) in accordance with the manufacturer's protocol.
Analysis of Mitochondrial DNA for 8-Hydroxy-2-Deoxyguanosine
For 8OHdG analysis, the mouse lung mtDNA was hydrolyzed to nucleosides using nuclease P1 and alkaline phosphatase as described previously (5). Briefly, samples were analyzed for 8OHdG and 2-deoxyguanosine (2 dG) by HPLC coupled with coulometric electrochemical and ultraviolet detection (CoulArray Model 5600; ESA Inc.) for 8OHdG and 2 dG, respectively. Nucleoside concentrations were calculated from multipoint standard curves generated daily with freshly prepared standards at concentrations that encompassed those observed in the samples. The levels of 8OHdG in mtDNA were expressed as a ratio to 105 2 dG bases.
Detection of Reactive Oxygen Species by Flow Cytometry
The dyes MitoSOX and carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Molecular Probes, Inc., Eugene, OR) were used to assess mitochondrial levels of 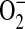 and intracellular H2O2, respectively (25, 26). Briefly, confluent C38 and IB3 cells (∼ 2 × 105 cells) were exposed to 5 μM MitoSOX or 1 μM DCFH-DA for 30 min. Cells were rinsed and resuspended in 0.5 ml of PBS. Cells were immediately analyzed by flow cytometry (FACSCalibur; Becton-Dickinson Biosciences, San Jose, CA) with the FL2 (485 ± 42 nm) and FL1 (530 ± 30 nm) channels set for detection of the oxidation products of MitoSOX and DCFH-DA, respectively. A total of 10,000 cells were counted for each sample.
and intracellular H2O2, respectively (25, 26). Briefly, confluent C38 and IB3 cells (∼ 2 × 105 cells) were exposed to 5 μM MitoSOX or 1 μM DCFH-DA for 30 min. Cells were rinsed and resuspended in 0.5 ml of PBS. Cells were immediately analyzed by flow cytometry (FACSCalibur; Becton-Dickinson Biosciences, San Jose, CA) with the FL2 (485 ± 42 nm) and FL1 (530 ± 30 nm) channels set for detection of the oxidation products of MitoSOX and DCFH-DA, respectively. A total of 10,000 cells were counted for each sample.
Statistical Analysis
Data are presented as means ± SE. Unless noted otherwise, each experimental result was derived from two or more experiments wherein each experimental group consisted of an n ⩾ 3. Data were subsequently analyzed for significant differences using either a Student's t test or one-way ANOVA with a Tukey's range test (Prizm v.3; GraphPad, San Diego, CA). The criterion for statistical significance was P ⩽ 0.05.
RESULTS
Lung Mitochondrial Purity
To successfully perform these studies, it was critical that the mitochondrial isolation procedures provide relatively pure mitochondria in sufficient quantities to reliably measure the selected endpoints. In these studies, 82.6 ± 3.1% of the total GDH activity was present in the mitochondrial preparation. The remaining 17.4% was present in the cytosolic fraction and represents lost or damaged mitochondria. More importantly, only 6.5 ± 1.5% of the total LDH activity was present in the mitochondrial preparation. Based on the LDH and GDH assay, the mitochondrial preparation used in these studies provided sufficient numbers of mitochondria that were relatively free of cytosolic contamination.
Lung Mitochondrial GSH Levels
The concentrations of GSH in lung mitochondria from the CFTR KO mouse lines were significantly lower than those in control mice (Figure 1). Mitochondrial GSH concentrations in the lung of control mice were 7.07 ± 1.05 nmol/mg protein. In the S489X CFTR KO mice on a liquid diet, mitochondrial GSH concentrations were decreased 43% compared with controls. The FABP CFTR KO mice on a solid diet revealed a more profound 85% decrease in mitochondrial GSH concentrations compared with controls. These results indicate that the absence of functional CFTR can alter the lung concentration of mitochondrial GSH. It is unclear why there is such a profound difference between the two CFTR KO mice strains since they are both congenic on the C57B6 mouse background and the only major differences are the transgene and diet.
Figure 1.

Levels of GSH in lung mitochondria from control and CFTR KO mice. Mitochondria isolated from the lungs of S489X and FABP CFTR KO mice displayed significantly lower levels of GSH than mitochondria from control mice. Data represent n ⩾ 5 mice in each group. Statistical significance (P < 0.05) was determined by one-way ANOVA coupled with a Tukey's range test. Bars with different letters are statistically significant from one another.
Mitochondrial GSH Levels in Human Lung Epithelial Cells
The concentration of GSH in mitochondria isolated from C38 and IB3 cells were determined (Figure 2A). Analogous to the mouse experiments, the human lung epithelial C38 and IB3 cell lines present a CFTR-sufficient and CFTR-deficient model. These cell lines serve to bridge the results of this study to humans. In these experiments, the CFTR-deficient IB3 cells displayed a 32% lower concentration of mitochondrial GSH compared with the CFTR-sufficient C38 cells. In addition, the mitochondrial GSSG concentrations between C38 and IB3 cells were compared (Figure 2B). For mitochondrial GSSG, the C38 and IB3 cells displayed no significant differences in concentrations. These data suggest the depletion in steady-state mitochondrial GSH levels was not due to oxidation of GSH to GSSG.
Figure 2.

Levels of mitochondrial GSH and GSSG in human lung epithelial cells sufficient (C38) and deficient (IB3) for CFTR. (A) CFTR-deficient (IB3) cells displayed significantly lower levels of reduced mitochondrial GSH than CFTR-sufficient (C38) cells. (B) CFTR-deficient (IB3) cells displayed similar levels of oxidized GSSG as CFTR-sufficient (C38) cells. Statistical significance was determined by a Student's t test and is defined as P < 0.05 (*).
In Vivo and In Vitro Markers of Mitochondrial Oxidative Stress
The activities of the enzymes aconitase and fumarase are often used to assess the presence or absence of oxidative stress in the mitochondria. Aconitase contains an iron-sulfur cluster in its active site that is sensitive to oxidative inactivation (27). Fumarase, in contrast, is not inactivated by oxidants, and determination of its activity can indicate whether other processes could be affecting aconitase activity (e.g., mitochondria number) (28). In these studies, the activities of aconitase and fumarase from control and FABP CFTR KO lung mitochondria were compared. Lung mitochondria from FABP CFTR KO mice displayed 30% lower aconitase activity than control mice (Figure 3A). Mitochondrial fumarase activities between the FABP CFTR KO and control mice, however, were comparable (Figure 3B). These results indicate that the mitochondria from lungs devoid of functional CFTR are encountering oxidative stress. The absence of any difference in fumarase activity between the FABP CFTR KO and control mice further supports the presence of an oxidative stress by ruling out differences in mitochondria abundance or isolation artifacts.
Figure 3.

Comparison of aconitase and fumarase activities in lung mitochondria between control and CFTR KO mice. (A) Mitochondria isolated from lungs of FABP CFTR KO mice displayed significantly lower aconitase activity than mitochondria from control mice. (B) Mitochondria isolated from the lungs of FABP CFTR KO mice displayed a similar level of fumarase activity as mitochondria isolated from control mice. Aconitase and fumarase activities were determined from n ⩾ 12 from at least three separate experiments. Statistical significance was determined by a Student's t test and is defined as P < 0.05 (*).
As a more direct measure of oxidative stress, levels of 8OHdG in mtDNA from control and FABP CFTR KO lungs were compared (Figure 4). Levels of 8OHdG in mtDNA from FABP CFTR KO mouse lungs were elevated 29% compared with control mice. The significant increase in lung mtDNA 8OHdG from FABP CFTR KO mice further supports the evidence of an underlying mitochondrial oxidative stress associated with CFTR deficiency.
Figure 4.

Comparison of the levels of lung mtDNA oxidation between control and CFTR KO mice. Levels of mtDNA 8-hydroxy-2-deoxyguanosine (8OHdG) in CFTR KO mouse lungs were significantly increased compared with control mice. Statistical significance was determined by a Student's t test and is defined as P < 0.05 (*).
Analogous to the in vivo studies, the activities of the enzymes aconitase and fumarase from C38 and IB3 mitochondria were compared. The CFTR-deficient IB3 lung cells had a small 13% decrease in aconitase activity that was not significantly different from the CFTR-sufficient C38 lung cells (97.3 ± 8.1 and 84.5 ± 7.9 U/mg protein, respectively). Fumarase activities between the cell lines were likewise not significantly different and indicate that the measured activities were not affected by differences in protein expression or isolation artifacts (85.2 ± 7.9 and 83.3 ± 15.9 U/mg protein, respectively).
Detection of ROS in CFTR-Deficient and -Sufficient Human Lung Epithelial Cells
Accumulation of MitoSOX in mitochondria and its fluorescence upon reaction with ROS such as 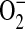 was used to assess mitochondrial ROS formation in C38 and IB3 cells (25). DCFH-DA is recognized as a marker of intracellular hydrogen peroxide (H2O2) formation (26). However, intracellular localization of the H2O2 production is limited due to its diffusivity and the widespread intracellular distribution of DCFH-DA. In this study, basal
was used to assess mitochondrial ROS formation in C38 and IB3 cells (25). DCFH-DA is recognized as a marker of intracellular hydrogen peroxide (H2O2) formation (26). However, intracellular localization of the H2O2 production is limited due to its diffusivity and the widespread intracellular distribution of DCFH-DA. In this study, basal 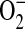 - and H2O2-like levels in C38 and IB3 cells were compared with MitoSOX and DCFH-DA, respectively (Figure 5). CFTR-deficient IB3 cells revealed a statistically significant increase in mitochondrial ROS levels compared with the CFTR-sufficient C38 cells (Figures 5A and 5B). Similarly, IB3 cells demonstrated significantly increased H2O2 levels compared with the C38 cells (Figures 5C and 5D). The observation of increased levels of ROS in the CFTR-deficient cells strongly suggests that defects in CFTR can lead to an endogenous oxidative stress. Moreover, this oxidative stress appears to arise from mitochondrial processes and is a likely cause of the macromolecular oxidations observed in this study.
- and H2O2-like levels in C38 and IB3 cells were compared with MitoSOX and DCFH-DA, respectively (Figure 5). CFTR-deficient IB3 cells revealed a statistically significant increase in mitochondrial ROS levels compared with the CFTR-sufficient C38 cells (Figures 5A and 5B). Similarly, IB3 cells demonstrated significantly increased H2O2 levels compared with the C38 cells (Figures 5C and 5D). The observation of increased levels of ROS in the CFTR-deficient cells strongly suggests that defects in CFTR can lead to an endogenous oxidative stress. Moreover, this oxidative stress appears to arise from mitochondrial processes and is a likely cause of the macromolecular oxidations observed in this study.
Figure 5.

Flow cytometry of CFTR(+) (C38) and CFTR(−) (IB3) cells using MitoSOX (A and B) and DCFH-DA (C and D) as dyes using the FL2 and FL1 channels for the detection of mitochondrial ROS 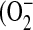 ) and intracellelular ROS (H2O2), respectively. The shifts of fluorescence using MitoSOX and DCFH-DA were statistically significant (B and D, respectively) (n = 6), suggesting that the lack of CFTR induces higher levels of ROS. Statistical significance was determined by a Student's t test and is defined as P < 0.05 (*).
) and intracellelular ROS (H2O2), respectively. The shifts of fluorescence using MitoSOX and DCFH-DA were statistically significant (B and D, respectively) (n = 6), suggesting that the lack of CFTR induces higher levels of ROS. Statistical significance was determined by a Student's t test and is defined as P < 0.05 (*).
DISCUSSION
Previous studies with human bronchoalveolar lavage fluid (BALF) and cultured cells indicate that CFTR modulates apical transport of GSH from pulmonary epithelial cells (4). Subsequent studies in our lab with CFTR KO mice provided additional evidence that CFTR modulates ELF transport of GSH in vivo (5). This previous study demonstrated a 50% decrease in the steady-state levels of ELF GSH in CFTR KO mice compared with wild-type mice. In addition to the deficit of ELF GSH, the study also revealed increases in general markers of oxidative stress in lung tissue. Specifically, CFTR KO mice demonstrated an increase in lung lipid and DNA oxidation. Similar findings have been noted in patients with CF; however, there exists considerable debate over the contribution from chronic infectious processes (29). Our current study suggests that the mitochondria may be an important contributor to the previously observed lung oxidative stress (5, 30, 31).
It is well accepted that mitochondrial processes (i.e., oxidative phosphorylation) is a major endogenous source of oxidative stress (32). The current study sought to determine whether a mitochondrial-derived oxidative stress was present in CFTR KO mouse lungs. The results of these experiments strongly suggest that mitochondrial-derived ROS contribute to the oxidative stress previously observed in the CFTR KO mouse lung. This oxidative stress appears to be a result of a decreased availability of GSH to neutralize endogenous ROS generation. The current study revealed that mitochondrial GSH is significantly lower in lungs from mice lacking a functional CFTR protein. Both S489X and FABP CFTR KO mice had significantly lower mitochondrial GSH concentrations than control mice. The mitochondrial GSH levels reported in this study are consistent with concentrations reported in other studies (33–35).
Between the two CFTR KO mouse strains, however, the FABP GSH levels were considerably lower than the levels in the S489X mice. The S489X mutation generates a stop codon and produces a truncated protein (19). The S489X CFTR KO mouse must be maintained on a liquid diet to prevent fatal bowel obstructions. The FABP CFTR KO mouse contains a transgene consisting of a functional human CFTR driven by the FABP. Thus, the FABP CFTR KO mice express the functional hCFTR only in the GI tract and can be maintained on the same solid diet as control mice. Therefore, the finding of lower mitochondrial GSH concentrations in the “gut-corrected” mice was unexpected. One explanation for this observation is that the hCFTR expression in the gut is occurring in a nontypical cell type. Normally, CFTR expression in the GI tract is localized to the cells of the crypts and not those of the villi (36). A previous study has suggested that hCFTR expression in the FABP CFTR KO mice is primarily found on the villi and not the crypt cells (19, 20). While the decrease in mitochondrial GSH could be attributed to increased oxidative stress, alterations in other cellular processes cannot be ruled out by this observation alone (e.g., GSH transport).
Measurement of the mitochondrial aconitase and fumarase activities is a useful approach to identifying mitochondrial oxidative stress (28). Aconitase contains an iron-sulfur center that is easily oxidized and renders the enzyme inactive. In contrast, fumarase contains no such oxidant-sensitive moieties and is resistant to oxidative stress. Thus, a decrease in aconitase activity without a concomitant decrease in fumarase suggests the presence of an oxidative stress. In the current study, mitochondria from CFTR KO mice demonstrated lower levels of aconitase activity than control mice in the absence of any fumarase activity differences. While the source of any ROS cannot be identified by this assay, ROS derived from the electron transport system in GSH-deficient mitochondria is the most likely source.
Measurement of oxidized deoxyguanosine in the mitochondrial genome served as a second and more specific marker for mitochondrial oxidative stress (37). In this study, mtDNA from the lungs of CFTR KO mice demonstrated significantly elevated levels of 8OHdG. The presence of oxidative stress suggested by the aconitase data is further supported by the detection of elevated levels of 8OHdG in the mtDNA. There is considerable debate in the literature regarding the baseline level of 8OHdG in mitochondrial and nuclear DNA (38). To minimize artifactual oxidation of DNA in these studies, care was taken to minimize sample exposure to elevated temperatures, oxidizing conditions (e.g., sonication, phenol), and light throughout mtDNA isolation and hydrolysis. While the 8OHdG/105 2 dG ratios reported herein are not as low as reported by others, the ratios are consistent with data reported in other studies (5). Taken together, the decreased aconitase activity and increased mtDNA 8OHdG level in CFTR KO mice strongly support the possibility of a mitochondrial-derived oxidative stress.
The C38 and IB3 cell lines provide a useful model system in which to compare the biological effect of defective CFTR. Similar to the mouse lung studies above, the mitochondrial GSH in the CFTR-defective IB3 cells was significantly lower than the CFTR-sufficient C38 cells. The results from the flow cytometry experiments clearly demonstrate an oxidative stress in both the mitochondria and the cell. MitoSOX is taken up by cells and transported to the mitochondria (25). Once it undergoes reaction with 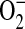 to yield its fluorescent product, it remains in the mitochondria through an association with mtDNA. In these experiments, CFTR-deficient IB3 cells consistently demonstrated higher levels of MitoSOX fluorescence. Due to the ability of H2O2 to diffuse across membranes, DCFH-DA cannot be relied upon to localize a source of H2O2. Instead, DCFH-DA fluorescence can be used to differentiate between cellular levels of H2O2. In these experiments, the IB3 cells consistently demonstrated elevated levels of DCFH-DA fluorescence indicative of higher levels of cellular H2O2. These results clearly show that cells deficient in CFTR are under oxidative stress that is comprised of mitochondrial
to yield its fluorescent product, it remains in the mitochondria through an association with mtDNA. In these experiments, CFTR-deficient IB3 cells consistently demonstrated higher levels of MitoSOX fluorescence. Due to the ability of H2O2 to diffuse across membranes, DCFH-DA cannot be relied upon to localize a source of H2O2. Instead, DCFH-DA fluorescence can be used to differentiate between cellular levels of H2O2. In these experiments, the IB3 cells consistently demonstrated elevated levels of DCFH-DA fluorescence indicative of higher levels of cellular H2O2. These results clearly show that cells deficient in CFTR are under oxidative stress that is comprised of mitochondrial 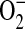 and H2O2. While it is very likely that the H2O2 stems from dismutation of mitochondrial
and H2O2. While it is very likely that the H2O2 stems from dismutation of mitochondrial 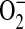 , other cellular sources cannot be ruled out by these experiments.
, other cellular sources cannot be ruled out by these experiments.
The lack of a difference in GSSG levels suggests that the reduced GSH levels are more likely a result of impaired transport of GSH into the mitochondria from the cytosol. Unlike the mouse model, aconitase and fumarase activities between the human lung epithelial C38 and IB3 cells were not significantly different. While the IB3 cells did display decreased aconitase activity, this difference never attained statistical significance. This discontinuity, however, may be explained if a threshold mitochondrial GSH concentration is not reached. Given that the mitochondrial GSH concentration is generally maintained at a high level, it has been suggested that measurable oxidative damage does not occur until GSH is depleted to a threshold level (39, 40). Compared with these earlier studies, the decrease in mitochondrial GSH between C38 and IB3 cells is much less marked and not accompanied by a significant decrease in aconitase activity. Thus, despite the lower mitochondrial GSH levels in IB3 cells, a critical threshold concentration that would give rise to detectable changes in aconitase activity may not have been reached.
The observation of mitochondrial dysfunction in CF is not new. Previous studies from patients with CF have reported differences in mitochondrial Ca+2 concentrations, O2 uptake, energy use, and activity of enzymes of the electron transport system (ETS) (41–45). One explanation put forth to explain the increased activity of ETS enzymes was that alterations in the composition of the inner mitochondrial membrane could affect these activities. While there is no direct evidence to support this hypothesis in CF, it is widely accepted that the altered composition and fluidity of the mitochondrial membrane in chronic alcohol exposure contributes to decreases in mitochondrial GSH concentrations in the heart, liver, and lung (46–48). Furthermore, a cell membrane lipid imbalance has been identified in the organs most affected by CF: small intestine, pancreas, and lung in CFTR KO mice (49). Although these studies focused on the effect of this membrane lipid imbalance on the inflammatory response, it raises the possibility that lipid imbalances may exist in organelles such as the mitochondria.
GSH is synthesized in the cytosol from its constituent amino acids by two successive ATP-dependent enzymatic steps. Mitochondria do not possess the enzymatic machinery to perform de novo synthesis of GSH (50). Cytosolic GSH is then distributed among the intracellular organelles including the mitochondria, endoplasmic reticulum (ER), and nucleus. Except for the ER, GSH in the cytosol, mitochondria, and nucleus exists predominantly in its reduced form. Mitochondrial GSH represents ∼ 10% of the total cellular GSH pool (51). Based on the volume of the mitochondrial matrix, the GSH concentration is estimated to be ∼ 10–14 mM (52–54). The transport of GSH across the inner mitochondrial membrane has been attributed to transporters of inorganic phosphate (Pi) and metabolites of the tricarboxylic acid cycle. Studies with kidney mitochondria demonstrate that the mitochondrial dicarboxylate and 2-oxoglutarate carriers are involved in the mitochondrial uptake of GSH (54, 55). At physiologic pH, a small proportion of the GSH pool exists as its thiolate anion, GS−. For both the dicarboxylate (DC) and 2-oxoglutarate (OG) carriers, import of GS− into the matrix is coupled to the export of dicarboxylic acids (e.g., malonate, 2-oxoglutarate) and Pi into the intermembrane space (34). Thus, since GSH import is critical to maintaining mitochondrial GSH levels, it appears likely that the decreased GSH levels observed in this study may be due to transport issues.
The ABC “super-family” of proteins is characterized by the presence of an ATP-binding moiety called the ATP-binding cassette (ABC) and transmembrane domains (TMDs). CFTR is a member of the ABCC subfamily, which also includes other transporters and receptors. In addition to CFTR, several members of the ABCC subfamily have been associated with GSH transport. These include multidrug resistance protein (MDR-1, MDR-2, and MRP-5) (56–58). GSH transport, however, is not unique to the ABCC subfamily and has been associated with members of ABCB (58). The ABC proteins found on mitochondria are members of the ABCB subfamily and have been generally associated with iron homeostasis in the mitochondria (59). An immunohistochemical study of rat and human proximal small intestine found high levels of intracellular CFTR expression (36). While still speculative, it is possible that an ABC protein such as CFTR could be present on the mitochondrial membrane and contribute to the maintenance of mitochondrial GSH.
This study suggests that CFTR may play a role in modulating mitochondrial GSH levels in lung epithelium and is a contributing factor of the observed lung oxidative stress. Importantly, evidence of elevated levels of mitochondrial and cellular ROS were associated with a CFTR-deficient state. These data suggest that impaired CFTR function is associated with decreased mitochondrial GSH which contributes to increased steady-state ROS levels in the mitochondria. These alterations in mitochondrial ROS are likely the cause of potentially deleterious oxidations of cellular components. Further studies are needed to determine how CFTR deficiency contributes to altered GSH steady-state levels and the role this plays in CFTR-mediated lung pathophysiology.
Acknowledgments
The authors thank Mark Goldstein, M.S., Elysia Min, M.S., Pablo R. Castello, Ph.D., and Jie Huang, Ph.D. for their excellent technical assistance in this study.
This work is supported by a grant from NIH RO1 HL075523.
Originally Published in Press as DOI: 10.1165/rcmb.2005-0473OC on June 8, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev 1999;79:S23–S45. [DOI] [PubMed] [Google Scholar]
- 2.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1996;154:1229–1256. [DOI] [PubMed] [Google Scholar]
- 3.Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am J Physiol 1998;275:C323–C326. [DOI] [PubMed] [Google Scholar]
- 4.Gao L, Kim KJ, Yankaskas JR, Forman HJ. Abnormal glutathione transport in cystic fibrosis airway epithelia. Am J Physiol 1999;277:L113–L118. [DOI] [PubMed] [Google Scholar]
- 5.Velsor LW, van Heeckeren A, Day BJ. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Physiol Lung Cell Mol Physiol 2001;281:L31–L38. [DOI] [PubMed] [Google Scholar]
- 6.Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J 2003;22:1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comhair SA, Lewis MJ, Bhathena PR, Hammel JP, Erzurum SC. Increased glutathione and glutathione peroxidase in lungs of individuals with chronic beryllium disease. Am J Respir Crit Care Med 1999;159:1824–1829. [DOI] [PubMed] [Google Scholar]
- 8.Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol 1993;75:2419–2424. [DOI] [PubMed] [Google Scholar]
- 9.Verkman AS, Song Y, Thiagarajah JR. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am J Physiol Cell Physiol 2003;284:C2–15. [DOI] [PubMed] [Google Scholar]
- 10.Tirouvanziam R, Khazaal I, Peault B. Primary inflammation in human cystic fibrosis small airways. Am J Physiol Lung Cell Mol Physiol 2002;283:L445–L451. [DOI] [PubMed] [Google Scholar]
- 11.Day BJ, van Heeckeren AM, Min E, Velsor LW. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infect Immun 2004;72:2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995;151:1075–1082. [DOI] [PubMed] [Google Scholar]
- 13.Heeckeren A, Walenga R, Konstan MW, Bonfield T, Davis PB, Ferkol T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J Clin Invest 1997; 100:2810–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 1980;191:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro BL. Evidence for a mitochondrial lesion in cystic fibrosis. Life Sci 1989;44:1327–1334. [DOI] [PubMed] [Google Scholar]
- 16.Flotte TR, Afione SA, Solow R, Drumm ML, Markakis D, Guggino WB, Zeitlin PL, Carter BJ. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J Biol Chem 1993;268:3781–3790. [PubMed] [Google Scholar]
- 17.Shapiro BL. Mitochondrial dysfunction, energy expenditure, and cystic fibrosis. Lancet 1988;2:289. [DOI] [PubMed] [Google Scholar]
- 18.Gardner PR. Superoxide production by the mycobacterial and pseudomonad quinoid pigments phthiocol and pyocyanine in human lung cells. Arch Biochem Biophys 1996;333:267–274. [DOI] [PubMed] [Google Scholar]
- 19.Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev 1999;79:S193–S214. [DOI] [PubMed] [Google Scholar]
- 20.Zhou L, Dey CR, Wert SE, DuVall MD, Frizzell RA, Whitsett JA. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 1994;266:1705–1708. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Vizarra E, Lopez-Perez MJ, Enriquez JA. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods 2002;26:292–297. [DOI] [PubMed] [Google Scholar]
- 22.Andersson BS, Jones DP. Use of digitonin fractionation to determine mitochondrial transmembrane ion distribution in cells during anoxia. Anal Biochem 1985;146:164–172. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt E, Schmidt FW. Methods and value of determination of glutamic acid dehydrogenase activity in the serum: a contribution to the importance of examination of enzyme relations in the serum [in German]. Klin Wochenschr 1962;40:962–969. [DOI] [PubMed] [Google Scholar]
- 24.Gupta S, Rogers LK, Taylor SK, Smith CV. Inhibition of carbamyl phosphate synthetase-I and glutamine synthetase by hepatotoxic doses of acetaminophen in mice. Toxicol Appl Pharmacol 1997;146:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Julian D, April KL, Patel S, Stein JR, Wohlgemuth SE. Mitochondrial depolarization following hydrogen sulfide exposure in erythrocytes from a sulfide-tolerant marine invertebrate. J Exp Biol 2005;208:4109–4122. [DOI] [PubMed] [Google Scholar]
- 26.Kobzik L, Godleski JJ, Brain JD. Oxidative metabolism in the alveolar macrophage: analysis by flow cytometry. J Leukoc Biol 1990;47:295–303. [PubMed] [Google Scholar]
- 27.Gardner PR, Nguyen DD, White CW. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci USA 1994;91:12248–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem 2001;276:23357–23361. [DOI] [PubMed] [Google Scholar]
- 29.Brown RK, Wyatt H, Price JF, Kelly FJ. Pulmonary dysfunction in cystic fibrosis is associated with oxidative stress. Eur Respir J 1996;9:334–339. [DOI] [PubMed] [Google Scholar]
- 30.Brown RK, Kelly FJ. Evidence for increased oxidative damage in patients with cystic fibrosis. Pediatr Res 1994;36:487–493. [DOI] [PubMed] [Google Scholar]
- 31.Brown RK, McBurney A, Lunec J, Kelly FJ. Oxidative damage to DNA in patients with cystic fibrosis. Free Radic Biol Med 1995;18:801–806. [DOI] [PubMed] [Google Scholar]
- 32.Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol Pharmacol 2003;64:1136–1144. [DOI] [PubMed] [Google Scholar]
- 33.Lash LH, Putt DA, Matherly LH. Protection of NRK-52E cells, a rat renal proximal tubular cell line, from chemical-induced apoptosis by overexpression of a mitochondrial glutathione transporter. J Pharmacol Exp Ther 2002;303:476–486. [DOI] [PubMed] [Google Scholar]
- 34.Lash LH. Role of glutathione transport processes in kidney function. Toxicol Appl Pharmacol 2005;204:329–342. [DOI] [PubMed] [Google Scholar]
- 35.Rigobello MP, Folda A, Scutari G, Bindoli A. The modulation of thiol redox state affects the production and metabolism of hydrogen peroxide by heart mitochondria. Arch Biochem Biophys 2005;441:112–122. [DOI] [PubMed] [Google Scholar]
- 36.Ameen NA, Ardito T, Kashgarian M, Marino CR. A unique subset of rat and human intestinal villus cells express the cystic fibrosis transmembrane conductance regulator. Gastroenterology 1995;108:1016–1023. [DOI] [PubMed] [Google Scholar]
- 37.Shigenaga MK, Ames BN. Assays for 8-hydroxy-2'-deoxyguanosine: a biomarker of in vivo oxidative DNA damage. Free Radic Biol Med 1991;10:211–216. [DOI] [PubMed] [Google Scholar]
- 38.Anson RM, Hudson E, Bohr VA. Mitochondrial endogenous oxidative damage has been overestimated. FASEB J 2000;14:355–360. [DOI] [PubMed] [Google Scholar]
- 39.Coll O, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003;38:692–702. [DOI] [PubMed] [Google Scholar]
- 40.Boya P, de la Pena A, Beloqui O, Larrea E, Conchillo M, Castelruiz Y, Civeira MP, Prieto J. Antioxidant status and glutathione metabolism in peripheral blood mononuclear cells from patients with chronic hepatitis C. J Hepatol 1999;31:808–814. [DOI] [PubMed] [Google Scholar]
- 41.Feigal RJ, Shapiro BL. Mitochondrial calcium uptake and oxygen consumption in cystic fibrosis. Nature 1979;278:276–277. [DOI] [PubMed] [Google Scholar]
- 42.von Ruecker AA, Bertele R, Harms HK. Calcium metabolism and cystic fibrosis: mitochondrial abnormalities suggest a modification of the mitochondrial membrane. Pediatr Res 1984;18:594–599. [DOI] [PubMed] [Google Scholar]
- 43.Shapiro BL, Feigal RJ, Lam LF. Mitrochondrial NADH dehydrogenase in cystic fibrosis. Proc Natl Acad Sci USA 1979;76:2979–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dechecchi MC, Girella E, Cabrini G, Berton G. The Km of NADH dehydrogenase is decreased in mitochondria of cystic fibrosis cells. Enzyme 1988;40:45–50. [DOI] [PubMed] [Google Scholar]
- 45.Battino M, Rugolo M, Romeo G, Lenaz G. Kinetic alterations of cytochrome-c oxidase in cystic fibrosis. FEBS Lett 1986;199:155–158. [DOI] [PubMed] [Google Scholar]
- 46.Brown LA, Harris FL, Guidot DM. Chronic ethanol ingestion potentiates TNF-alpha-mediated oxidative stress and apoptosis in rat type II cells. Am J Physiol Lung Cell Mol Physiol 2001;281:L377–L386. [DOI] [PubMed] [Google Scholar]
- 47.Vendemiale G, Grattagliano I, Altomare E, Serviddio G, Portincasa P, Prigigallo F, Palasciano G. Mitochondrial oxidative damage and myocardial fibrosis in rats chronically intoxicated with moderate doses of ethanol. Toxicol Lett 2001;123:209–216. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez-Checa JC, Colell A, Garcia-Ruiz C. S-Adenosyl-L-methionine and mitochondrial reduced glutathione depletion in alcoholic liver disease. Alcohol 2002;27:179–183. [DOI] [PubMed] [Google Scholar]
- 49.Freedman SD, Katz MH, Parker EM, Laposata M, Urman MY, Alvarez JG. A membrane lipid imbalance plays a role in the phenotypic expression of cystic fibrosis in cftr(−/−) mice. Proc Natl Acad Sci USA 1999;96:13995–14000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol 2005;204:263–273. [DOI] [PubMed] [Google Scholar]
- 51.Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci USA 1985;82:4668–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatol 2001;34:946–954. [DOI] [PubMed] [Google Scholar]
- 53.Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol Appl Pharmacol 1996;140:1–12. [DOI] [PubMed] [Google Scholar]
- 54.Chen Z, Lash LH. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2- oxoglutarate carriers. J Pharmacol Exp Ther 1998;285:608–618. [PubMed] [Google Scholar]
- 55.McKernan TB, Woods EB, Lash LH. Uptake of glutathione by renal cortical mitochondria. Arch Biochem Biophys 1991;288:653–663. [DOI] [PubMed] [Google Scholar]
- 56.Leslie EM, Deeley RG, Cole SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology 2001;167:3–23. [DOI] [PubMed] [Google Scholar]
- 57.Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst 2000;92:1295–1302. [DOI] [PubMed] [Google Scholar]
- 58.Ballatori N, Hammond CL, Cunningham JB, Krance SM, Marchan R. Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol 2005;204:238–255. [DOI] [PubMed] [Google Scholar]
- 59.Lill R, Kispal G. Mitochondrial ABC transporters. Res Microbiol 2001; 152:331–340. [DOI] [PubMed] [Google Scholar]