

Abstract
Disruption of NF-E2–related factor (Nrf2), a redox-sensitive basic leucine zipper transcription factor, causes early-onset and more severe emphysema due to chronic cigarette smoke. Nrf2 determines the susceptibility of lungs to cigarette smoke–induced emphysema in mice through the transcriptional induction of numerous antioxidant genes. The lungs of Nrf2−/− mice have higher oxidative stress as evident from the increased levels of lipid peroxidation (4-hydroxy-2-nonenal) and oxidative DNA damage (7,8-dihydro-8-Oxo-2′deoxyguanosine) in response to cigarette smoke. Glutathione peroxidases (GPX) are the primary antioxidant enzymes that scavenge hydrogen peroxide and organic hydroperoxides. Among the five GPX isoforms, expression of GPX2 was significantly induced at both mRNA and protein levels in the lungs of Nrf2+/+ mice, in response to cigarette smoke. Activation of Nrf2 by specific knock down of the cytosolic inhibitor of Nrf2, Keap1, by small inhibitory RNA (siRNA) upregulated the expression of GPx2, whereas Nrf2 siRNA down-regulated the expression of GPX2 in lung epithelial cells. An ARE sequence located in the 5′ promoter–flanking region of exon 1 that is highly conserved between mouse, rat, and human was identified. Mutation of this ARE core sequence completely abolished the activity of promoter–reporter gene construct. The binding of Nrf2 to the GPX2 antioxidant response element was confirmed by chromatin immunoprecipation, electrophoretic mobility shift assays, and site-directed mutagenesis. This study shows that GPX2 is the major oxidative stress–inducible cellular GPX isoform in the lungs, and that its basal as well as inducible expression is dependent on Nrf2.
Keywords: antioxidant response element, cigarette smoke, emphysema, GPX2, Nrf2
Oxidative stress caused by chronic exposure of the lungs to free radicals and reactive nitrogen species present in cigarette smoke contributes to the pathogenesis of chronic obstructive pulmonary disease (COPD) (1–6). The degree of pulmonary oxidant/antioxidant imbalance correlates well with the severity of COPD (6). The ability of the lungs to respond to this stress could be an important determinant of their relative resistance or susceptibility to chronic obstructive pulmonary disease. A battery of protective genes can be induced in a rapid and highly coordinated response to oxidants through the activation of redox-sensitive transcription factors. The coordinated induction of these genes is fundamental to maintaining antioxidant protection against both free radical and electrophilic species that arise from exposure to cigarette smoke. We recently showed that in response to oxidative stress due to cigarette smoke (CS) exposure, Nrf2, a basic leucine zipper transcription factor, detaches from its cytosolic repressor kelch-like ECH-associated protein 1 (Keap1), and binds to the antioxidant response element (ARE) in the promoter of target antioxidant genes, leading to their transcriptional induction in lungs (7). Glutathione peroxidases (GPX) are the primary antioxidant enzymes that scavenge hydrogen peroxide or organic hydroperoxides and thus protect biomembranes and cellular components against oxidative stress (8). GPX has also been shown to protect against peroxinitrite, a potent oxidant generated from the reaction of superoxide 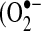 ) and nitrous oxide. The isoforms of GPx differ in their primary structure and localization. The classic cytosolic-mitochondrial GPX1 (cGPX) is a selenium-dependent enzyme and was the first of the GPx family to be discovered. GPX2, which is also known as gastrointestinal GPX, is a cytosolic enzyme expressed predominantly in the epithelium of the gastrointestinal tract. Extracellular plasma GPx (pGPx, or GPx3) is expressed mainly in the kidney, from where it is released into the blood circulation. Phospholipid hydroperoxide (GPx4) is expressed in most tissues, and GPx5 (or eGPx) is epididymis-specific secretory GPx (8, 9).
) and nitrous oxide. The isoforms of GPx differ in their primary structure and localization. The classic cytosolic-mitochondrial GPX1 (cGPX) is a selenium-dependent enzyme and was the first of the GPx family to be discovered. GPX2, which is also known as gastrointestinal GPX, is a cytosolic enzyme expressed predominantly in the epithelium of the gastrointestinal tract. Extracellular plasma GPx (pGPx, or GPx3) is expressed mainly in the kidney, from where it is released into the blood circulation. Phospholipid hydroperoxide (GPx4) is expressed in most tissues, and GPx5 (or eGPx) is epididymis-specific secretory GPx (8, 9).
Nrf2 in response to CS regulates expression of genes involved in two major redox systems, the glutathione (GSH) and thioredoxin systems. Among the various isoforms of GPx, GPx2 showed maximum induction in lungs of mice in response to CS (3). Similarly, in humans GPx2 had the highest transcriptional induction in the airway epithelium of smokers compared with nonsmokers (10, 11). A better understanding of the regulation of important downstream target genes of Nrf2 such as GPX will help elucidate its protective role in lungs in response to oxidative stress caused by environmental pollutants. Here, we show that GPx2 is the only isoform of GPx that is induced in the lungs in response to CS, and that both basal and CS-inducible expression are directly dependent on Nrf2.
MATERIALS AND METHODS
Animals and Care
Nrf2-deficient CD-1 (ICR) mice were generated as described (12). Mice were genotyped for Nrf2 status by PCR amplification of genomic DNA extracted from blood as described previously (3). All experimental protocols conducted in the mice were performed in accordance with the standards established by the U.S. Animal Welfare Acts, set forth in the National Institutes of Health guidelines and the Policy and Procedures Manual of the Johns Hopkins University Animal Care and Use Committee.
Cell Culture
A549 cells, human alveolar type II epithelial cell line, were obtained from American Type Culture Collection (Rockville, MD) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1% penicillin–streptomycin (Invitrogen, Carlsbad, CA).
Exposure to CS
Eight-week-old mice were divided into four groups (n = 3 per group): I, control Nrf2+/+ mice; II, experimental Nrf2+/+ mice; III, control Nrf2−/−mice; and IV, experimental Nrf2−/− mice. Groups I and III were kept in a filtered air environment, and groups II and IV were subjected to CS according to the protocol of Rangasamy and coworkers (3). CS exposure was performed by burning 2R4F reference cigarettes (2.45 mg nicotine per cigarette; purchased from the Tobacco Research Institute, University of Kentucky, Lexington, KY) using a smoking machine (Model TE-10; Teague Enterprises, Woodland, CA) as previously described (3).
Immunohistochemical Staining of 4-Hydroxy-2-Nonenal
Nrf2+/+ and Nrf2−/− mice exposed to CS for 1 mo (7 h/d) were killed and the lungs dissected out for staining with antibody to 4-hydroxy-2-nonenal (4-HNE). Antigen retrieval was performed for 20 min with vector citrate buffer, and the slides were washed in 1× PBS for 5 min. Sections were incubated with normal blocking serum for 20 min, followed by incubation overnight at 4°C with primary 4-hydroxynonenal antibody (1:1,000) (Vector Labs, Burlingame, CA). The slides were then washed in 1× PBS for 5 min and incubated with diluted biotinylated universal secondary antibody solution for 30 min. Next the slides were washed for 5 min in 1× PBS and incubated with VECTASTATIN ABC-AP reagent (Vector Labs) for 30 min. The slides were then washed for 5 min in PBS and incubated with alkaline phosphatase substrate (5 min). The sections were rinsed in distilled water and counterstained with hematoxylin QS for 30 s. Finally, the sections were rinsed in distilled water, dehydrated, and mounted.
Immunohistochemical Localization of 8-Oxo-dG in the Lungs
To detect the oxidative DNA damage marker 7, 8-dihydro-8-Oxo-2′deoxyguanosine (8-Oxo-dG), the mice were exposed to CS for 1 mo (5 h/d). Lungs were inflated with 10% buffered formalin and embedded in paraffin. The lung sections were incubated with anti–8-oxo-dG antibody and stained using the Iso-IHC DAB kit (InnoGenex, San Ramon, CA) using mouse antibodies. Normal mouse-IgG1 antibody was used as a negative control (3).
Immunohistochemical Staining of GPx2
Nrf2+/+ and Nrf2−/− mice were exposed to CS for 5 h. After 24 h, mice were killed and the lungs dissected out for staining with antibody to GPX2. Six-micron-thick lung sections were heated at 95°C for 10 min in 10 mM citrate buffer (pH 6) and allowed to cool for 10 min. The sections were blocked in 10% donkey serum for 30 min; endogenous biotin was blocked using an avidin–biotin blocking kit (Zymed, South San Francisco, CA). Sections were then incubated with 1.0 μg/ml goat anti GPX antibody for 18 h at 4°C. Nonimmune goat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) was used as a negative control. Antibody binding was detected by incubation with biotinylated donkey anti-goat antibody diluted 1:1,000 (Jackson ImmunoResearch Laboratories). Antibody binding was visualized by incubation with avidin–peroxidase conjugate (Vector Laboratories, Burlingame, CA) for 30 min and DAB (Zymed) for 5 min.
GPX Enzyme Activity Assay
To measure the enzyme activity of GPX, mice were exposed to CS for 5 h and killed after 24 h. The lungs were excised (n = 5 per group) and processed as described (3). GPx activity was measured according to the procedure of Flohé and Gunzler using t-butyl hydroperoxide as the substrate (13).
Quantitation of GPx mRNA Expression
To measure the enzyme activity of GPX, mice were exposed to CS for 5 h. Total RNA was extracted from lung tissues and or cells with TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed using Superscript First Strand Synthesis system (Invitrogen) as per the manufacturer's instructions. Quantitative real-time RT-PCR analyses of murine GPx1, GPx2, GPx3, and GPx4 and human GPx2 were performed by using assay on demand primers and probe sets from Applied Biosystems (Foster City, CA). Assays were performed by using the ABI 7000 Taqman system (Applied Biosystems). GAPDH was used for normalization.
Western Blot
Western blot analysis of GPX2 was performed by following the protocol described by Banning and colleagues (14). Briefly, 50 μg of total protein were separated by 15% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and transferred to PVDF membrane by semi-dry blotting. The PVDF membrane was blocked and incubated with polyclonal rabbit anti-GPX2 antibody (1:500) or actin (1:1,000) followed by incubation with horseradish peroxidase–conjugated secondary antibody and developed using an ECL chemilumineiscence detection system (Amersham Biosciences, Piscataway, NJ).
Identification of ARE(s) in the Promoter of GPx2
To identify the presence and location of ARE(s) in the GPx2 promoter, the 2-kb upstream region from the translation start site was downloaded from the NCBI database (Human Genome resources, www.ncbi.nlm.nih.gov/genome/guide/human). The 2-kb sequence was used to search for ARE(s) with the help of Genamics Expression 1.1 software (Hamilton, New Zealand) using the primary core sequence of ARE (RTGAYNNNGCR) as the probe. The location of Transcription start site for human GPX2 gene has already been experimentally determined (15). The promoter sequence of mouse and rat homologs of GPx2 were downloaded from the NCBI database and scanned for the presence of ARE. The Position of the transcription start site for mouse GPx2 was determined using Mouse Genome Build 33, version 1 of NCBI database. The transcription start site for rat GPx2 gene was determined using the Rat Genome Build 2.
Plasmids and Mutagenesis
The 5′ flanking region of human GPX2 promoter region (−2,030 to +539) was PCR amplified from genomic DNA isolated from human blood with high-fidelity Taq polymerase (Applied Biosystems). The isolated PCR product was ligated into pCR2.1 (Invitrogen), and a KpnI–XhoI fragment from this construct was cloned into pGL3 basic vector (Promega, Madison, WI). Two deletion constructs (−1,029 to +539 and −140 to +539) were generated from the full-length promoter construct. The primers used for amplification were forward 1 (−2,030), CAGAGAAAGACCCTGTCTCAAT; forward 2 (−1,029), GCTAG GTCAA-GCCCCATTGCTAG; and forward 3 (−140), GCTCTGGTTT CCTGTAAGCAGTGAATTAT. The reverse primer (+539) was the same for all three deletion constructs: TATGTCACATGC-ACCTA AAGACCTAGCTTA. A 201–base pair (bp) fragment containing the ARE (−140/+60) was amplified by PCR from the full-length promoter vector and ligated into the pTAL luciferase reporter vector (BD Biosciences, San Jose, CA). Mutated ARE sequences were generated by using a site-directed mutagenesis kit from Stratagene (La Jolla, CA). Primers containing the mutated ARE sequences GACCTGTTTTTGCTAAGGCCTCCTGGGGATGCTCAAAG (ARE-Mu1) and CAGGTGGG-GACCTGTTTTTTATAAGTCATCCTGGGGATGC (ARE-Mu2) were used for PCR amplification of the mutated GPx2 promoter, and PCR products were digested with DpnI for 1 h to cleave the wild-type promoter. The mutation of each promoter was verified by sequencing. The NQO1 ARE reporter construct contains a 41-bp rQR-ARE/EpRE inserted into a TATA-Inr minimal promoter vector (16).
Screening of Nrf2 and Keap1 siRNA Duplexes
Nrf2 siRNA duplexes and Keap1 siRNA duplexes were purchased from Dharmacon Research, Inc. (Lafayette, CO) (Table 1). We first identified siRNA sequences that permitted potent and selective silencing of the human Nrf2 or Keap1 coding sequence. To confirm the specificity of the inhibition, the siCONTROL nontargeting siRNA 1 (SS siRNA) with microarray-defined signature was used as a negative control. Cells in the exponential growth phase were plated in 6-well plates at a density of 5 × 105 cells/well, grown for 12 h, and then transfected with 20 pmol of siRNA duplexes by using Lipofectamine 2000 and OPTI-MEMI reduced serum medium (Invitrogen) according to the manufacturer's protocol. Concentrations of siRNAs were chosen on the basis of dose–response studies. Knockdown of the target gene was quantified by real-time RT-PCR at 24, 48, and 72 h after transfection using assay on demand primers and probe sets from Applied Biosystems. Assays were performed using the ABI 7000 Taqman system (Applied Biosystems). GAPDH was used for normalization.
TABLE 1.
siRNA SEQUENCES
| Gene Name | siRNA Duplex No. | siRNA Sequence |
|---|---|---|
| Keap1 | Duplex-1 | Sense: 5′ GGACAAACCGCCUUAAUUCUU 3′ |
| Antisense: 3′ UUCCUGUUUGGCGGAAUUAAG 5′ | ||
| Duplex-2 | Sense: 5′ CAGCAGAACUGUACCUGUUUU 3′ | |
| Antisense: 3′ UUGUCGUCUUGACAUGGACAA 5′ | ||
| Duplex-3 | Sense: 5′ GGGCGUGGCUGUCCUCAAUUU 3′ | |
| Antisense: 3′ UUCCCGCACCGACAGGAGUUA 5′ | ||
| Duplex-4 | Sense: 5′ CGAAUGAUCACAGCAAUGAUU 3′ | |
| Anrisense: 3′ UUGCUUACUAGUGUCGUUACU 5′ | ||
| Nrf2 | Duplex-1 | Sense: 5′ GAGAAAGAAUUGCCUGUAAUU 3′ |
| Antisense: 3′ UUCUCUUUCUUAACGGACAUU 5′ | ||
| Duplex-2 | Sense: 5′ CCAAAGAGCAGUUCAAUGAUU 3′ | |
| Antisense: 3′ UUGGUUUCUCGUCAAGUUACU 5′ | ||
| Duplex-3 | Sense: 5′ GAAGCCAGAUGUUAAGAAAUU 3′ | |
| Antisense: 3′ UUCUUCGGUCUACAAUUCUUU 5′ | ||
| Duplex-4 | Sense: 5′ UAAAGUGGCUGCUCAGAAUUU 3′ | |
| Antisense: 3′ UUAUUUCACCGACGAGUCUUA 5′ | ||
| siCONTROL Nontargeting | Sense: 5′ UAGCGACUAAACACAUCAAUU 3′ | |
| Antisense: 3′ UUAUCGCUGAUUUGUGUAGUU 5′ | ||
| GPX2 | Sense: 5′ CCCUCUGGUUGGUGAUUCATT 3′ | |
| Antisense: 3′ TTGGGAGACCAACCACUAAGU 5′ |
Definition of abbreviation: siRNA, small inhibitory RNA.
DNA Transfection and Luciferase Activity
Cells were transfected at 85% confluence using Lipofectamine 2000 (Invitrogen). Briefly, cells were seeded in 24-well plates at a density of 2 × 105 cells/ml and grown overnight. Next day, cells were transfected with 5 pmol of siRNA duplexes using Lipofectamine 2000 and OPTI-MEMI reduced serum medium (Invitrogen) according to the manufacturer's protocol. Medium containing 30% serum was added 5 h after the transfection was begun, and cells were incubated for another 7 h. After ∼ 12 h, the media containing siRNA complex was removed and fresh transfection complex containing 200 ng of plasmid DNA, 1 ng of pRL-TK plasmid (Promega), and transfection reagent were added to each well in the presence of fetal bovine serum. Cells were incubated for another 36 h. After 48 h, cells were lysed and Renilla and firefly luciferase activities measured using the dual luciferase assay kit (Promega) with a luminometer (EG&G Wallac, Gaithersberg, MD). Luciferase activity was normalized relative to Renilla luciferase activity, the internal control.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed according to our previously described procedure (3). GPX2 wild-type ARE was annealed and end labeled with [γ-32P]. For gel shift analysis, 10 μg of nuclear proteins isolated from the lungs of air- and CS-exposed (5 h) Nrf2+/+ and Nrf2−/− mice were incubated with the labeled human GPX2 ARE and the mixtures analyzed on a 5% nondenaturing polyacrylamide gel. To determine the specificity of the proteins binding to the ARE sequence, 100-fold excess of unlabeled competitor oligo (ARE consensus sequence) was incubated with the nuclear extract for 10 min before the addition of radiolabeled probe. The 32P-labeled consensus sequence for the octamer transcription factor 1 was used as a control for gel loading. Similarly, 10 μg of nuclear proteins isolated from A549 cells transfected with Nrf2, Keap1, or SS siRNA were incubated with radiolabeled wild-type ARE. The competition assays were performed with 50-fold and 100-fold excess of unlabeled wild-type ARE, unlabeled mutant ARE-Mu1, and unlabeled mutant ARE-Mu2. The wild-type ARE oligo sequence used for EMSA was ACCTGTTTTTGCTAAGT CATCC-TG, the ARE-Mu1 oligo sequence was ACCTGTTTTTGC TAAGGCCTCCTG, and the mutant ARE-Mu2 oligo sequence was CTGTTTTTTATAAGTCATCCTGGG. For supershift analysis, the labeled wild-type ARE was first incubated for 30 min with 10 μg of nuclear protein and then with 4 μg of anti-Nrf2 antibody (Nrf2 sc-722X; Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h. Normal rabbit IgG (4 μg) was used as a control for the supershift assay.
Chromatin Immunoprecipiation Assay
Cells (∼ 107) were transfected with Keap1, Nrf2, and nonspecific siRNA. After 72 h, the cells were harvested and the chromatin immunoprecipitation (CHIP) assay was performed by using a commercially available kit (Upstate Biotechnology, Lake Placid, NY). Immunoprecipitates and total chromatin input were reverse cross-linked, DNA was isolated and 1 μl of DNA was used for PCR (35 cycles) with primers specific for the GPX2 promoter. The GPX2 promoter primer sequence is: forward, CATAGATATCAATTGGCCCTTCC; reverse, CAGGATGACTTAGCAAAAACAGGT.
High-Performance Liquid Chromatography Analysis of Lipid Peroxidation Products
The thiobarbutyric acid reactive substances (TBARS) in the cells were quantified using the high-performance liquid chromatography (HPLC) method of Agarwal and Chase (17). Briefly, the cell pellets were sonicated on ice with PBS (containing 0.02% BHT). Fifty microliters of the homogenate was incubated with a solution containing 1 μl of BHT (250 mM BHT), 50 μl of TCA (20%), and 200 μl of thiobarbutyric acid (0.67% [wt/vol] in 100 ml/liter glacial acetic acid [pH 3.5]) in a boiling water bath for 45 min. The samples were allowed to cool for 10 min at room temperature. Three hundred microliters of n-butanol was added to the sample, vortexed, and centrifuged for 5 min at 13,000 × g. To quantify the TBARS in the sample, 50 μl of the organic layer was injected on a Supelcosil LC-18 HPLC column (4.6 × 250 mm; Supelco, Bellefonte, PA) with a mobile phase consisting of 50 mM potassium phosphate pH 6.4, 52.5% methanol at a flow rate of 1 ml/min using a Hewlett Packard 1050 chromatography system (Agilent, Palo Alto, CA). TBARS were detected by fluorescence (ex. 515 nm, em. 553) using a Hewlett Packard fluorescence detector (Agilent) and quantified using a standard curve of 1,1,2,2-tetraethoxypropane (10–0.625 μM). The values are represented as nmoles of TBARS/mg of protein.
MTT Cell Viability Assay
Cells were transfected with 50 nM siRNAs two times with a 48-h interval and, 24 h after the second transfection, cells were plated at a density of 7,500 cells/well in 180 μl of growth media in 96-well plates. After 12 h, cells were treated with various concentrations of CS condensate (Murthy Pharmaceuticals, Lexington, KY) for 48 h. Cell viability was evaluated by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma, St. Louis, MO) reduction conversion assay (18). Cell survival was expressed as absorbance relative to that of untreated controls. Results are presented as means ± SD.
Statistical Analysis
Results are presented as the means ± SE. Statistical comparisons were performed by paired Student's t tests. A value of P < 0.05 was considered statistically significant.
RESULTS
Increased Oxidative Stress Due to CS Exposure in Nrf2-Deficient Mice
Immunohistochemical staining with anti-4HNE and anti–8-Oxo-dG antibodies were used to assess oxidative damage in the lungs of CS exposed Nrf2+/+ and Nrf2−/− mice. In response to 1 mo CS exposure, formation of the aldehyde 4-HNE was higher in lungs from Nrf2−/− mice than in lungs from wild-type mice (Figure 1A). Levels of 8-Oxo-dG, which is a sensitive biomarker of oxidative DNA damage, were greatly elevated in the alveolar epithelial cells of the lungs from CS exposed Nrf2−/− mice compared with the CS exposed Nrf2+/+ mice, as well as the respective air-exposed control mice (Figure 1B). These observations further document the occurrence of excessive oxidative stress in the lungs of Nrf2-deficient mice exposed to chronic CS.
Figure 1.

Increased sensitivity of Nrf2−/− mice to CS-induced oxidative stress. (A) Immunohistochemical staining for 4HNE in lung sections from Nrf2+/+ and Nrf2−/− mice exposed to air or CS for 1 mo. Lung sections from the CS-exposed Nrf2−/− mice show greater staining for 4HNE than the CS-exposed Nrf2+/+ mice and the respective air-exposed control mice. The image is representative of results from three animals in each group. Magnification: ×40. (B) Immunohistochemical staining clearly shows the presence of an increased number of 8-Oxo-dG–positive alveolar epithelial cells in the airways (indicated by arrow in the inset and middle bottom panel) of CS-exposed Nrf2-deficient mice compared with wild-type mice (middle upper panel). Airways of room air–exposed mice shows few or no 8-oxo-dG–positive cells (left panels). Magnification: ×20 (inset magnification: ×40) (n = 3).
CS Induces GPx2 Gene Expression in Lungs
We exposed Nrf2+/+ and Nrf2−/− mice to CS for 5 h and examined the expression of all four GPx isoforms by real-time reverse transcriptase-polymerase chain reaction (RT-PCR). The basal transcript level of GPx2 mRNA was 50% higher in Nrf2+/+ mice than in Nrf2−/− mice. The GPx2 mRNA level was further up-regulated by 2-fold in response to CS in Nrf2+/+ mice. Overall, there was a 3-fold increase in CS-inducible GPx2 mRNA between genotypes, as no induction was observed in the lungs of CS-exposed Nrf2−/− mice. Transcript levels of GPx1, GPx3, and GPx4 did not change significantly in Nrf2+/+ mice in response to CS, and expression GPx1 and GPx4 was down-regulated in response to CS in Nrf2−/− mice (Figure 2A).
Figure 2.


(Continued)
Western blot analysis of lung proteins using GPX2 antibody showed that GPX2 protein was present constitutively and that its level increased significantly on exposure to CS in Nrf2+/+ mice. By contrast, GPX2 protein was remarkably low or undetectable in Nrf2−/− mice and did not change significantly with CS exposure (Figures 2B and 2C). Total GPX enzyme activity was increased in the lungs of CS-exposed Nrf2+/+ mice in response to CS (Figure 2D). The measured increase in total GPX activity probably represents the cytosolic GPX2 isoform, which would be compatible with the increased mRNA and protein levels of GPX2 seen in response to CS exposure in Nrf2+/+ mice. However, interpretation of such activity measurements is confounded by the fact that the specific activities of individual enzymes (GPX1, GPX2, and GPX3) are largely unknown and only PH-GPX activity can be determined individually by using phosphatidylcholine hydroperoxide as a substrate.
Immunohistochemical staining with anti-GPX2 antibody revealed an increased number of GPX2-stained airway epithelial cells in the lungs of CS-exposed Nrf2+/+ mice when compared with its knockout counterpart and the age-matched control mice (Figure 2E). Nrf2−/− mice showed reduced level of staining with GPX2 antibody, further suggesting that Nrf2−/− mice have reduced levels of GPX2 protein.
siRNA-Mediated Knockdown of Nrf2 Results in Down-Regulation of GPX2 Expression
To further elucidate the role of Nrf2 in regulating transcription of GPX2, we used Nrf2 siRNA to inhibit Nrf2 function. We first identified siRNA sequences that permit potent and selective silencing of human Nrf2 gene. siRNAs were designed to target four different regions spanning the Nrf2 cDNA. The siRNA duplexes were transfected into A549 cells, and 48 h later cells were harvested and Nrf2 message was quantified by real-time RT-PCR. The time points for measuring the effects of siRNA in these cells were chosen after preliminary optimization experiments (data not shown). To confirm the specificity of the inhibition, the siCONTROL nontargeting siRNA 1 (SS siRNA) with microarray defined signature was used as a negative control. All the siRNA duplexes targeting Nrf2 displayed significant reductions in Nrf2 message (> 60%). Duplex-2 showed maximum knockdown of the Nrf2 message (> 85%) and was chosen for further experiments (Figure 3A). The cells transfected with nontargeting siRNA showed no evidence of silencing and contained Nrf2 levels comparable with the vehicle control. To further confirm the specificity of the inhibitory effect of duplex-2 on Nrf2 gene expression, we measured the transcript level of GCLc, a well-characterized target of Nrf2 (19). Decreases in Nrf2 transcript by duplex-2 resulted in parallel decreases in GCLc transcript (Figure 3B), which suggests that the decrease in Nrf2 protein resulted in down-regulation of transcription of ARE-dependent genes. In addition, we transfected the NQO1-ARE reporter vector into A549 cells already transfected with Nrf2 siRNA or SS siRNA. Nrf2 is the only protein known to strongly activate the ARE, and hence the measurement of an ARE reporter is a reliable measure of the efficiency of Nrf2 silencing by siRNA. As shown in Figure 3C, cells transfected with Nrf2 siRNA displayed significant reduction in the amount of ARE luciferase activity as compared with the cells transfected with SS siRNA.
Figure 3.

Down-regulation of Nrf2 gene expression by Nrf2 siRNA decreased the expression of GPX2 in A549 cells. (A) Screening of Nrf2 siRNA duplexes. Cells transfected with nontargeting SS siRNA– and vehicle-treated cells were used as controls. (B) Nrf2 siRNA duplex-2 (showing maximum knockdown) was transfected into A549 cells. Total RNA was isolated 72 h after transfection and transcript level of Nrf2 its target gene GCLc were quantified by real-time RT-PCR. (C) Transfection of A549 cells with Nrf2 siRNA inhibited NQO1-ARE luciferase activity. (D) Transient transfection of A549 cells with Nrf2 siRNA inhibited the expression of Nrf2 in a time-dependent manner. (E) Down-regulation of GPX2 expression in cells transfected with Nrf2 siRNA. The inhibition of Nrf2 and GPX2 mRNA expression was progressive from 24 to 72 h after Nrf2 siRNA transfection (n = 3 independent experiments). * Significant when compared with the respective SS siRNAs at P ⩽ 0.05.
To test whether time-dependent degradation of Nrf2 message results in a parallel decrease in GPX2 transcription, we isolated total RNA 24, 48, and 72 h after transfection of Nrf2 siRNA and quantified Nrf2 and GPX2 transcripts by real-time RT-PCR. The Nrf2 message level decreased with time, and the maximum knockdown (∼ 75%) was observed at 72 h (Figure 3D). GPX2 message followed a similar trend, with the maximum inhibition (∼ 75%) of GPX2 transcription at 72 h after transfection (Figure 3E). Western blot analysis of proteins isolated from A549 cells after 72 h of transfection with Nrf2 siRNA showed that the decrease in Nrf2 message led to a parallel decrease in GPX2 protein at 72 h (Figure 4F), indicating that Nrf2 is essential for basal expression of GPX2 in lungs.
Figure 4.

Nrf2 induces GPX2 expression in A549 cells. (A) Screening of Keap1 siRNA duplexes. Cells transfected with nontargeting SS siRNA– and vehicle-treated cells were used as controls. (B) Transient transfection of A549 cells with duplex-3 of keap1 siRNA inhibited the expression of keap1 and upregulated the expression of NQO1 mRNA. * Significant when compared with SS siRNA transfected cells. (C) Transfection of A549 cells with keap1 siRNA significantly increased NQO1-ARE luciferase activity. † Significant when compared with vector control transfected with the respective siRNA; * significant when compared with SS siRNA. (D) Transient transfection of A549 cells with keap1 siRNA significantly inhibited the expression of Keap1. * Significant when compared with SS siRNA. (E) Up-regulation of GPX2 expression in cells transfected with Keap1 siRNA. * Significant when compared with SS siRNA. (F) Western blot analysis of A549 cells transfected with Nrf2, keap1, or SS siRNA. Total protein was isolated 72 h after transfection, and GPX2 protein levels were determined. β-Actin was used as the loading control. (G) Quantification of GPX2 protein in the Western blots of A549 cells transfected with Nrf2, Keap1, and SS siRNA (mean ± SE of the GPX2/actin protein ratio in three independent samples). * Significant when compared with SS siRNA; † significant when compared with Nrf2 siRNA-transfected A549 cells. (H) Induction of GPX2 expression in response to multiple oxidative stress inducing agents. Cells transfected with Nrf2 siRNA or SS siRNA were exposed to tBHP (400 μM), tBHQ (400 μM), and CS condensate (100 μg/ml), and expression of GPX2 was measured by real-time RT-PCR. *Significant when compared with vehicle-treated sample. Data are mean ± SEM of three independent experiments (P ⩽ 0.05).
Nrf2 Activation Induces GPX2 Expression in Lung Epithelial Cells
Next we examined whether activation of Nrf2 induces GPX2 expression in A549 cells. We used Keap1 siRNA to activate the expression of Nrf2 dependent genes. siRNA duplexes targeting Keap1 mRNA were screened, and duplex-3 was selected for further experiments (Figure 4A). To further confirm the specificity of the inhibitory effect of duplex-3 on Keap1 gene expression and activation of Nrf2, we measured the transcript level of NQO1, another well-characterized target of Nrf2 (20, 21). Decreases in Keap1 transcript levels by duplex-3 resulted in parallel increase in NQO1 transcript levels (Figure 4B). To establish that degradation of the Keap1 transcript results in activation of Nrf2, we transfected A549 cells with Keap1 siRNA or SS siRNA followed by second transfection with NQO1-ARE reporter vector and measured luciferase activity. As shown in Figure 4C, cells transfected with Keap1 siRNA displayed a significant increase in the amount of ARE luciferase activity as compared with the cells transfected with SS siRNA. To demonstrate that activation of Nrf2 results in upregulation of GPX2 transcription, we isolated total RNA after 24, 48, and 72 h of transfection and quantified Keap1 and GPX2 transcripts by real-time RT-PCR. Keap1 transcript levels decreased with time and maximum knockdown of Keap1 mRNA was achieved at 72 h (> 85%) (Figure 4D). However, the initial decrease in Keap1 message did not alter the GPX2 message level, which remained unchanged until 48 h and increased by 3-fold at 72 h (Figure 4E). The above observation suggests that near complete knockdown (> 80%) of Keap1 is essential for nuclear accumulation of Nrf2 and consequent upregulation of the transcription of GPX2. Increased GPX2 mRNA was reflected in increased levels of GPX2 protein in A549 cells in response to down-regulation of Keap1 expression by siRNA (Figures 4F and 4G).
Next we treated the A549 cells transfected with Nrf2 siRNA or SS siRNA with multiple oxidative stress–inducing agents. The cells were exposed to 400 μM tert-butyl hydroperoxide (tBHP), 400 μM tert-butyl hydroquinone (tBHQ) for 4 h, and cigarette smoke condensate (100 μg/ml) for 12 h, and the GPX2 induction in response to these treatments was quantified by real-time RT-PCR. SS siRNA–transfected cells up-regulated the expression of GPX2 in response to all three oxidative stress inducing agents, whereas cells transfected with Nrf2 siRNA did not show any induction when exposed to these agents, suggesting that Nrf2 is essential for the inducible expression of GPX2 (Figure 4H).
ARE Mediates the Transcriptional Regulation of GPX2
To decipher the underlying mechanism of GPX2 induction by Nrf2, we examined the human GPX2 genomic locus in detail for the presence of ARE consensus sequences. Functional AREs, such as the one in HO-1 or the NQO1 gene, are often represented by an extended ARE consensus sequence (5′-TMAnnRTGAYnnnGCR-wwww-3′), and detailed in silico analysis of the human GPX2 genomic locus revealed a potential ARE sequence in the 5′ region of the first exon (+23), which correlates with ARE sequence identified by Banning and coworkers (14). The ARE sequence matches the long ARE consensus sequence and is similar to the ARE identified in the human HO-1, NQO1, and GCLm regulatory elements. NCBI database searches led to the identification of a putative ARE sequence in the mouse (+12) and rat GPx2 promoters (−140). The ARE consensus sequence is highly conserved between mouse, rat, and human promoter sequences (Figure 5A).
Figure 5.

ARE mediates the transcriptional regulation of the GPX2 gene. (A) Comparison of the first exon and the 5′ upstream regulatory regions of the human, mouse, and rat GPX2 genes. The short ARE core, long ARE consensus, GPX2-ARE, ARE from the human HO-1 gene (StREb), human NQO1, and human GCLm (EpRE) are aligned. The vertical bar indicates nucleotides that are identical between the human, mouse, and rat ARE as well as long ARE, HO-1 ARE. The abbreviations follow the standard IUPAC nomenclature (M = A or C, R = A or G, Y = C or T, W = A or T, S = G or C). (B) Deletion analysis of GPX2 promoter by transient transfection in A549 cells. A 2.569-kb portion of the GPX2 promoter was cloned into pGL3 basic luciferase reporter vector (−2,030/+539) to monitor the activity of this promoter. Two deletion constructs (−1,029/+539 and −140/+539) were prepared by PCR. All three constructs were transfected into A549 cells transiently transfected with SS, Nrf2, or Keap1 siRNA, and luciferase activities were measured after 48 h. The three constructs had similar reporter activity. The −140/+539 deletion construct displayed the maximum reporter activity. * Significant when compared with vector control transfected with respective siRNA; † significant when compared with SS siRNA; ‡ significant when compared with Nrf2 siRNA. (C) Reporter constructs (ARE-Mu1 and ARE-Mu2) bearing mutations in the ARE (−140/+539) were generated by using the −140/+539 deletion construct. These constructs were transiently transfected into A549 cells already transfected with siRNA, and luciferase reporter activity was monitored. Mutation of the ARE core sequence completely abolished the reporter activity of the deletion construct. * Significant when compared with ARE-WT. (D) 201 bp (−140/+61) ARE enhancer sequence was amplified from the full-length promoter and cloned in pTAL vector. Mutations were introduced in the ARE core sequence (ARE-Mu1 and ARE-Mu2) by site-directed mutagenesis. These constructs were transfected into A549 cell lines transiently transfected with siRNA and luciferase activity was measured. Luciferase activities were normalized by measuring the Renilla luciferase activity from a cotransfected reporter vector. * Significant when compared with ARE-WT (n = 3 independent experiments). Values are mean ± SE from three different experiments (P < 0.05).
The promoter activity of GPX2 was measured using a luciferase reporter system to further test the hypothesis that nuclear translocation of Nrf2 may enhance transcriptional activation of GPX2. Three deletion constructs (−2,030/+539, −1,029/+539 and −140/+539) had similar luciferase activities, with the deletion construct containing the region between −140 and +539 displaying maximal reporter activity, suggesting that this region likely contains the cis-acting enhancer elements (Figure 5B). The basal activity of the −140 to +539 deletion construct was 8-fold higher than that of the pGL3 basic vector. The luciferase activity of the deletion construct was further increased to 15-fold in the presence of Keap1 siRNA, whereas cells transfected with Nrf2 siRNA showed only 4-fold increase in luciferase activity (Figure 5B). These results suggest that the region between −140 and +539 contains the necessary cis-elements for directing minimal, basal, and Nrf2-inducible GPX2 expression.
Sequences containing the ARE (−130/+69) were amplified from the full-length promoter construct and cloned into pTAL luciferase vector containing a TATA-like promoter (PTAL) region from the Herpes simplex virus thymidine kinase (HSV-TK) promoter for enhancer analysis. The ARE enhancer construct in pTAL had ∼ 17-fold higher basal activity than the pTAL vector alone. The ARE enhancer had ∼ 26-fold higher luciferase activity in cells transfected with Keap1 siRNA, whereas luciferase activity was reduced to ∼ 8-fold in Nrf2 siRNA transfected cells (Figure 5D). To assess the contribution of these motifs (AREs) to Nrf2-dependent GPX2 promoter activity, we introduced mutations across the 20-bp region of the ARE. These alterations not only abolished the luciferase activity in the cells transfected with Keap1 siRNA, but also completely eliminated the constitutive luciferase activity in SS siRNA–transfected cells when compared with the wild-type ARE (Figures 5C and 5D). The luciferase reporter activity observed with ARE-Mu1 and ARE-Mu2 was same with all three siRNA and was comparable with the background luciferase activity obtained with the pGL3 Basic vector or the pTAL vector. Together, these transient transfection studies establish the role of Nrf2 in the transcriptional activation of GPX2 gene expression.
Activation of Nrf2 Leads to Increased Binding of Nuclear Proteins to GPX2 ARE
EMSA were performed to determine the activation and DNA-binding activity of Nrf2 in the lungs after acute exposure to CS (5 h). In response to CS, there was an increased binding of nuclear proteins isolated from the lungs of Nrf2+/+ mice to an oligonucleotide probe containing the GPX2 ARE sequence as compared with the binding of nuclear proteins isolated from CS-exposed Nrf2−/− mice or air-exposed control mice (Figure 6A). These results suggest that Nrf2 binds to the ARE and likely mediates the GPX2 ARE response to CS. Minimal binding observed in Nrf2+/+ and Nrf2−/− air-exposed samples and Nrf2−/− CS-exposed samples represents the binding of other nuclear proteins to antioxidant response element.
Figure 6.

EMSA was used to determine the DNA-binding activity of Nrf2 to GPX2 ARE. (A) Nuclear proteins isolated from the lungs of CS-exposed Nrf2+/+ mice showed increased binding to GPX2 ARE than the nuclear proteins from the lungs of CS-exposed Nrf2−/− mice or air-exposed control mice. A 100-fold molar excess of cold WT-ARE was used as a competitor oligo. Octamer transcription factor 1 consensus sequence was used as the endogenous control. (B) EMSA analysis with the nuclear proteins isolated from the A549 cells transfected with Nrf2, SS, or Keap1 siRNA. Incubation with 50- and 100-fold molar excess unlabeled cold GPX2 WT-ARE significantly abolished the binding suggesting the specificity of the binding of nuclear proteins to GPX2 ARE. However, incubation with mutant AREs did not abolish the binding of nuclear proteins to GPX2 ARE. (C) Supershift assay with anti-Nrf2 antibody revealed the binding of Nrf2 to GPX2 ARE. Normal rabbit IgG1 was used as the control. (D) CHIP assay was performed with A549 cells transfected with Nrf2 siRNA, Keap1 siRNA, or SS siRNA. No DNA, PCR without DNA. (E) Quantification of CHIP assay results (GPX2 promoter binding-antibody/input ratio). Filled bars, Keap siRNA; open bars, SS siRNA; shaded bars, Nrf2 siRNA.
Similarly, nuclear extracts were prepared from A549 cells transfected with Keap1 siRNA, Nrf2 siRNA, and SS siRNA and incubated with radiolabeled GPX2-ARE. In A549 cells transfected with Keap1 siRNA, there was an increase in binding of the nuclear proteins to the GPX2 ARE compared with cells transfected with SS siRNA. A549 cells transfected with Nrf2 siRNA showed diminished protein binding to the ARE. The binding involves a specific complex of GPX2 ARE and nuclear proteins as demonstrated by the competition assays in which cold GPX2 WT-ARE effectively competed away the complex. By contrast, the two unlabeled mutant AREs with mutations in the core ARE sequence competed poorly with the complex. As shown in Figure 6B, excess amounts of unlabeled WT-ARE inhibited the binding of nuclear proteins to these sequences. To examine whether the protein binding to the GPX2 ARE is Nrf2, we performed supershift experiment using an antibody against Nrf2. As shown in Figure 6C, the complex was supershifted in the presence of an antibody to Nrf2. Collectively, the results from both lungs and A549 cells conclusively show that Nrf2 binds to the ARE (+23) present on the human GPX2 gene.
Nrf2 Binds to the GPX2 Promoter In Vivo
We next used a CHIP assay to determine whether Nrf2 is bound to the endogenous GPX2 promoter constitutively and after activation by Keap1 siRNA. As shown in Figure 6D, Nrf2 was present in the basal transcription complex formed with GPX2 promoter in cells transfected with SS siRNA. Activation of Nrf2 by silencing Keap1 expression enhanced the recruitment of Nrf2 to the GPX2 promoter as demonstrated by increased amplification of the GPX2 promoter region. Nrf2 was present at very low or undetectable levels in cells transfected with Nrf2 siRNA, and no amplification was detected using GPX2 promoter primers. Immunoprecipitations with IgG or mock failed to select the GPX2 promoter (Figure 6D), which suggests that the sites were not enriched in nonspecific fashion. To gain insight into the pathway of GPX2 gene activation, we also performed a CHIP assay with an antibody to RNA-pol II. Recruitment of RNA polymerase II to the promoter of the GPX2 was observed in both Keap1 and SS siRNA transfected A549 cells. However, no amplification was detected in cells transfected with Nrf2 siRNA, which is consistent with the expectation that Nrf2 regulates transcription of GPx2.
Silencing of GPX2 by siRNA Sensitizes the Lung Epithelial Cells to Oxidative Damage
We downregulated the expression of GPX2 in A549 cells by using siRNA targeting GPX2 mRNA (Figure 7A) (22). To determine the contribution of GPX2 in protecting the lung epithelial cells against oxidative stress, we first transfected the cells with SS siRNA and GPX2 siRNA and then treated them with tBHP (400 μM). The lipid peroxidation products were measured using HPLC with fluorescence detector. Treatment of GPX2 siRNA–transfected cells with tBHP significantly enhanced the formation of Thiobarbutyric acid reactive (TBARS) compared with SS siRNA–transfected cells, suggesting the occurrence of excessive oxidative stress in the absence of GPX2 in lungs (Figure 7B). TBARS were significantly higher in A549 cells treated with tBHP compared with vehicle-treated cells. To further validate the significance of GPX2 in conferring protection against oxidative stress, we exposed the cells transfected with GPX2 siRNA, Nrf2 siRNA, and SS siRNA to CS condensate and measured the cell viability after 48 h. Down-regulation of GPX2 or Nrf2 significantly enhanced the cytotoxicity induced by CS condensate and increased cell death (Figure 7C). Thus, GPX2 plays a crucial role in conferring cytoprotection against oxidative damage in lungs.
Figure 7.

GPX2 confers protection against oxidative damage in lung epithelial cells. (A) A549 cells were transfected with GPX2 siRNA and non targeting SS siRNA. Cells were harvested after 72 h and GPX2 message was quantified by real-time RT-PCR. (B) Inhibition of GPX2 enhances the production of lipid hydroperoxides in lung epithelial A549 cells. Knockdown of GPX2 by siRNA significantly increased the production of thiobarbutyric acid substances in tBHP-treated A549 cells. Data are mean ± SEM of three independent experiments. * Significant when compared with vehicle-treated cells; † significant when compared with SS siRNA–transfected cells. †P ⩽ 0.05. (C) Enhanced sensitivity of Nrf2 siRNA– and GPX2 siRNA–transfected A549 cells to CS condensate. Cells were exposed to CS for 48 h and viable cells were determined by MTT assay. Data are represented as percentage of viable cells relative to the vehicle-treated control. Data are means of six independent replicates, combined to generate the mean ± SD for each concentration. *P < 0.01 relative to SS siRNA.
DISCUSSION
The oxidative burden in the lungs of smokers has been estimated to be very high—on the order of 1014 free radicals per puff (23). The potent oxidants in cigarette smoke include superoxide anion  ), nitric oxide (NO•), and, through their interaction, the even more reactive peroxynitrite (ONOO−) (25). Semiquinones and benzoquinones present in the cigarette tar along with superoxide radicals lead to production of reactive hydroxyl radicals (•OH) and hydrogen peroxide (H2O2) (24). Iron in the fluid of the epithelial lining and in tar results in the production of •OH radicals through the Fenton reaction. Some of these reactive oxygen species cause lipid peroxidation, leading to destruction of membrane lipids (4, 25). Lipid peroxidation, a well-established mechanism of cellular injury, is used as an indicator of oxidative stress. Markers of oxidative stress (e.g., hydrogen peroxide and the end-products of lipid peroxidation, such as ethane, pentane, and 8-isoprostane) are elevated in the breath and serum of patients with chronic obstructive pulmonary disease (6, 26). Recently, it has been reported that the degree of oxidative stress increases greatly with the severe exacerbations in COPD (6). The levels of pulmonary antioxidants determine the degree of lung destruction as a result of inflammation and oxidative stress. Nrf2, a redox-sensitive basic leucine zipper transcription factor, is a critical determinant of susceptibility to lung inflammation and oxidative stress and the subsequent emphysema caused by chronic exposure to CS (3). Consistent with the central role of Nrf2 in protection against CS-induced stress, there was an enhanced formation of 4HNE, a marker of lipid peroxidation, and 8-Oxo-dG, a marker of oxidative DNA damage in the lungs of Nrf2−/− mice exposed to CS relative to wild-type mice.
), nitric oxide (NO•), and, through their interaction, the even more reactive peroxynitrite (ONOO−) (25). Semiquinones and benzoquinones present in the cigarette tar along with superoxide radicals lead to production of reactive hydroxyl radicals (•OH) and hydrogen peroxide (H2O2) (24). Iron in the fluid of the epithelial lining and in tar results in the production of •OH radicals through the Fenton reaction. Some of these reactive oxygen species cause lipid peroxidation, leading to destruction of membrane lipids (4, 25). Lipid peroxidation, a well-established mechanism of cellular injury, is used as an indicator of oxidative stress. Markers of oxidative stress (e.g., hydrogen peroxide and the end-products of lipid peroxidation, such as ethane, pentane, and 8-isoprostane) are elevated in the breath and serum of patients with chronic obstructive pulmonary disease (6, 26). Recently, it has been reported that the degree of oxidative stress increases greatly with the severe exacerbations in COPD (6). The levels of pulmonary antioxidants determine the degree of lung destruction as a result of inflammation and oxidative stress. Nrf2, a redox-sensitive basic leucine zipper transcription factor, is a critical determinant of susceptibility to lung inflammation and oxidative stress and the subsequent emphysema caused by chronic exposure to CS (3). Consistent with the central role of Nrf2 in protection against CS-induced stress, there was an enhanced formation of 4HNE, a marker of lipid peroxidation, and 8-Oxo-dG, a marker of oxidative DNA damage in the lungs of Nrf2−/− mice exposed to CS relative to wild-type mice.
Glutathione peroxidases decompose H2O2 and organic hydroperoxides produced during oxidative insults and prevent peroxide-induced DNA damage, lipid peroxidation, and protein degradation (27). Mice with targeted disruption of only GPx1 remain largely asymptomatic; however, a double knockout of GPx1 and GPx2 results in inflammatory bowel disease and increased cancer incidence (28), making a role for GPx2 in preventing inflammation and carcinogenesis likely. The regulation of pulmonary GPX in response to CS has not been defined. To date, only GPx1 has been widely investigated as the representative isoform of GPx in lungs in animal models. However, a study using a mouse model with targeted disruption of GPx1 showed that the mice develop normally and do not show increased sensitivity to hyperoxia (29). Accumulating evidence has suggested that GPx2 is highly inducible in lungs as a protective mechanism against lung injury inflicted by hyperoxia (3, 10, 30), lipopolysaccharide (31), and so on. GPx2 antagonizes oxidative stress–induced apoptosis in a p53-dependent manner (32). In response to hyperoxia, GPx2 expression increases significantly followed by GPx3, whereas expression of the cellular isoform GPx1 does not change (29, 33). In humans, among different isoforms of glutathione peroxidase, GPX2 showed the highest transcriptional induction in the airway epithelium of smokers compared with nonsmokers (10, 11). Chronic CS exposure induces GPX3 expression in human airway epithelial cells and alveolar macrophages (34). The results of the present study show that GPx2 is the only glutathione peroxidase isoform selectively upregulated in the lungs of Nrf2+/+ mice in response to CS exposure. CS did not significantly induce the expression of GPx1, GPx3, or GPx4. Both basal and CS-inducible expressions of GPx2 are Nrf2-dependent in murine lungs. Consistent with the Nrf2-dependent gene expression patterns, GPX2 protein and enzyme activity were significantly higher in the lungs of Nrf2+/+ mice in response to acute CS exposure. More recently, it has been shown that even a single functional allele of GPx2 can protect against inflammation in the colon (35).
GPX2 is a cytosolic protein that efficiently reduces H2O2 and fatty acid hydroperoxides, but not phospholipid hydroperoxides or cholesterol peroxides (36, 37). The human GPX2 gene consists of two exons, codes for a 22-kD protein, and maps to chromosome 14 (38). Mouse GPx2 maps to chromosome 12 and is 89% identical to the human gene (39). A c/EBP element overlaps with the ARE sequence. Ubiquitous transcription element sites of the SP1 and AP series are present throughout the region. There are liver-specific sites (HNF-1, -3, and -4), several GATA sites, and a potential retinoic acid responsive element (40). Although GPX2 expression is classically associated with the intestine, the mammalian sites that are commonly considered to be intestine-specific (NF-LPH1, SIF series, cdx series, etc.) are not present (15, 41).
A common feature of the promoter regions of genes induced by oxidative and electrophilic insult in an Nrf2-dependent fashion is the presence of a cis-acting enhancer sequence, the ARE (7). Enhancers are DNA sequences that increase transcription in a manner that is independent of their orientation and distance relative to the RNA start site (42). To our knowledge, all AREs have been reported in the sequences upstream of the transcription start site. However, active enhancer sequences that bind to other transcription factors are known to be present in the downstream of the transcription start site. For example, the murine immunoglobulin Hu-core enhancer lies in the second intron of the transcription unit, and the mouse Nrf2 gene contains two xenobiotic response element (XRE)-like elements located at +755 (XREL2) and +850 (XREL3) (43). AREs have been identified in several antioxidant and xenobiotic genes, including mouse GSTA1 (44), HO-1 (45), and Ferritin H (46) and rat GSTA2 (47), GSTA3 (48), NQO1 (20), and GCLc (19). We searched for AREs in the human GPX2 locus and identified a putative ARE in the proximal promoter region of exon 1 at +23. Very recently, Banning and colleagues identified the same ARE in human GPX2 promoter and characterized it in Human Colon carcinoma (Caco) cells (14). The GPX2 ARE is similar to the ARE found in the HO-1 or NQO1 regulatory region. Detailed sequence analysis revealed that the sequence of the GPX2 ARE is highly conserved in different species; an identical core sequence is found in human, rat, and mouse. NQO1-ARE is the only other ARE shown to be highly conserved in its sequence in human, mouse, and rat genomes (21). The nucleotide sequences immediately adjacent to the core ARE in the human, mouse, and rat GPX2 genes are also highly conserved. We suspect that these AREs are evolutionarily conserved for GPX2 regulation.
Silencing a transcription factor post-transcriptionally by RNA interference provides a direct approach to understanding the regulation of its downstream target genes. Chemical activators, however, suffer from the limitation of having nonspecific effects on other pathways. In the present study, down-regulation of Nrf2 by siRNA in human type II A549 cells caused decreased expression of well-established target genes, such as GCLc (19). Targeting the cytosolic repressor of Nrf2, Keap1, resulted in upregulation of Nrf2 target genes. Cells transfected with siRNA directed against Nrf2 down-regulated the expression of GPX2 mRNA and protein, whereas activation of Nrf2 by Keap1 siRNA up-regulated the expression of GPX2. These results clearly show that basal and inducible expression of GPX2 mRNA is dependent upon cellular levels of Nrf2.
The activity of the luciferase reporter construct (−2,000 to +539) harboring the GPX2-ARE in pGL3 Basic vector was doubled in the A549 cells transfected with Keap1 siRNA, whereas basal reporter activity was reduced to half in the presence of Nrf2 siRNA. Studies with nested deletion constructs differing in their 5′ ends indicated that the ARE is located between −140 and +539. The reporter construct containing the putative ARE ligated to pTAL luciferase vector (contains TATA-like promoter region) was significantly activated in the presence of Keap1 siRNA, whereas reporter activity was reduced substantially in the presence of Nrf2 siRNA. Mutation of the core sequence of ARE in the pGL3 reporter vector or pTAL vector completely abolished the GPX2 promoter or luciferase reporter activity. Collectively, these results suggest that Nrf2 binds to the GPX2 ARE and regulates its expression.
In response to CS exposure, there was an increased binding of nuclear proteins from the lungs of Nrf2+/+ mice to the GPX2 ARE sequence. This binding is presumably due to the interaction of nuclear proteins with the ARE. EMSA conducted on nuclear proteins isolated from A549 cells transfected with Nrf2, Keap1, or SS siRNA showed maximum binding to ARE in the cells transfected with Keap1 siRNA, followed by A549 cells transfected with SS siRNA. The binding of nuclear proteins with the GPX2 ARE, particularly of Nrf2 in the A549 cells, was validated by the supershift analysis with antibody to Nrf2. Binding of Nrf2 to the ARE sequence in the GPX2 promoter was further confirmed by the CHIP assay with Nrf2 antibody. Both the EMSA and the CHIP experiments demonstrated convincingly that Nrf2 is recruited to the GPX2 ARE after Nrf2 activation.
To determine the relative contribution of GPX2 in protecting the lung epithelial cells against oxidative stress, we down-regulated the expression of GPX2 by siRNA and then treated them with tBHP. Treatment of A549 cells having low basal levels of GPX2 with tBHP significantly enhanced the production lipid peroxidation byproducts when compared with the cells treated SS RNA, suggesting that GPX2 is essential in protecting the cells against oxidative stress. Silencing of GPX2 or Nrf2 in A549 cells enhanced the sensitivity of these cells to oxidative stress–induced by CS condensate, further highlighting the role of Nrf2 and GPX2 in protection against oxidative damage and apoptosis (32).
In conclusion, we found that the so-called “gastrointestinal-specific” GPX2 is the major oxidative stress–inducible isoform of GPX in the lungs in response to CS that is regulated by Nrf2. Furthermore, this isoform is dependent on Nrf2 for its basal expression in lungs. The Nrf2-dependent transcriptional regulation of GPX2 may play an important role in counteracting CS-induced oxidative stress. Basal and inducible expression of GPX2, along with other Nrf2-regulated antioxidative enzymes, may help in maintaining the pulmonary antioxidant defense to protect against chronic obstructive pulmonary disorders.
Acknowledgments
The authors thank Brian Schofield and Judith Coram for their help in immunohistochemical staining of GPX2.
This work was supported by NIH grants HL081205 (S.B.), P50 CA058184, and CA94076; NIEHS center grant P30 ES 038819; and Flight Attendant Research Institute (S.B.).
Originally Published in Press as DOI: 10.1165/rcmb.2005-0325OC on June 22, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.MacNee W. Oxidants/antioxidants and COPD. Chest 2000;117:303S–317S. [DOI] [PubMed] [Google Scholar]
- 2.MacNee W, Rahman I. Is oxidative stress central to the pathogenesis of chronic obstructive pulmonary disease? Trends Mol Med 2001;7:55–62. [DOI] [PubMed] [Google Scholar]
- 3.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 2004;114:1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996;154:1055–1060. [DOI] [PubMed] [Google Scholar]
- 5.MacNee W, Morrison D, Rahman I, Li XY, Donaldson K. Cigarette smoke and ozone-induced epithelial perturbation in vivo and in vitro: the role of glutathione. Chest 1996;109:39S. [DOI] [PubMed] [Google Scholar]
- 6.Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, MacNee W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005;60:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 2003;43:233–260. [DOI] [PubMed] [Google Scholar]
- 8.Brigelius-Flohe R. Tissue-specific functions of individual glutathione peroxidases. Free Radic Biol Med 1999;27:951–965. [DOI] [PubMed] [Google Scholar]
- 9.Brigelius-Flohe R, Flohe L. Is there a role of glutathione peroxidases in signaling and differentiation? Biofactors 2003;17:93–102. [DOI] [PubMed] [Google Scholar]
- 10.Hackett NR, Heguy A, Harvey BG, O'Connor TP, Luettich K, Flieder DB, Kaplan R, Crystal RG. Variability of antioxidant-related gene expression in the airway epithelium of cigarette smokers. Am J Respir Cell Mol Biol 2003;29:331–343. [DOI] [PubMed] [Google Scholar]
- 11.Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA 2004;101:10143–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997; 236:313–322. [DOI] [PubMed] [Google Scholar]
- 13.Flohé L, Gunzler WA. Assays of glutathione peroxidase. Methods Enzymol 1984;105:114–121. [DOI] [PubMed] [Google Scholar]
- 14.Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohe R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol 2005;25:4914–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelner MJ, Bagnell RD, Montoya MA, Lanham KA. Structural organization of the human gastrointestinal glutathione peroxidase (GPX2) promoter and 3′-nontranscribed region: transcriptional response to exogenous redox agents. Gene 2000;248:109–116. [DOI] [PubMed] [Google Scholar]
- 16.Tirumalai R, Rajesh Kumar T, Mai KH, Biswal S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol Lett 2002;132:27–36. [DOI] [PubMed] [Google Scholar]
- 17.Agarwal R, Chase SD. Rapid, fluorimetric-liquid chromatographic determination of malondialdehyde in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci 2002;775:121–126. [DOI] [PubMed] [Google Scholar]
- 18.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 1988;48:589–601. [PubMed] [Google Scholar]
- 19.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem 1997;272: 7445–7454. [DOI] [PubMed] [Google Scholar]
- 20.Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem 1991;266:4556–4561. [PubMed] [Google Scholar]
- 21.Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat Res 2004;555:149–171. [DOI] [PubMed] [Google Scholar]
- 22.Morbitzer M, Herget T. Expression of gastrointestinal glutathione peroxidase is inversely correlated to the presence of hepatitis C virus subgenomic RNA in human liver cells. J Biol Chem 2005;280:8831–8841. [DOI] [PubMed] [Google Scholar]
- 23.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 1985;64:111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med 1995;19:161–167. [DOI] [PubMed] [Google Scholar]
- 25.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;166:490–495. [DOI] [PubMed] [Google Scholar]
- 26.Horvath I, MacNee W, Kelly FJ, Dekhuijzen PN, Phillips M, Doring G, Choi AM, Yamaya M, Bach FH, Willis D, et al. “Haemoxygenase-1 induction and exhaled markers of oxidative stress in lung diseases”, summary of the ERS Research Seminar in Budapest, Hungary, September, 1999. Eur Respir J 2001;18:420–430. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radic Biol Med 1990;8:583–599. [DOI] [PubMed] [Google Scholar]
- 28.Chu FF, Esworthy RS, Chu PG, Longmate JA, Huycke MM, Wilczynski S, Doroshow JH. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res 2004;64:962–968. [DOI] [PubMed] [Google Scholar]
- 29.Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem 1997;272:16644–16651. [DOI] [PubMed] [Google Scholar]
- 30.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 2002;26:175–182. [DOI] [PubMed] [Google Scholar]
- 31.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of innate immune response and survival during experimental sepsis. J Clin Invest 2006; 116:984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan W, Chen X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J Biol Chem 2006;281: 7856–7862. [DOI] [PubMed] [Google Scholar]
- 33.Cho HY, Reddy SP, Debiase A, Yamamoto M, Kleeberger SR. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic Biol Med 2005;38:325–343. [DOI] [PubMed] [Google Scholar]
- 34.Comhair SA, Thomassen MJ, Erzurum SC. Differential induction of extracellular glutathione peroxidase and nitric oxide synthase 2 in airways of healthy individuals exposed to 100% O(2) or cigarette smoke. Am J Respir Cell Mol Biol 2000;23:350–354. [DOI] [PubMed] [Google Scholar]
- 35.Esworthy RS, Yang L, Frankel PH, Chu FF. Epithelium-specific glutathione peroxidase, gpx2, is involved in the prevention of intestinal inflammation in selenium-deficient mice. J Nutr 2005;135:740–745. [DOI] [PubMed] [Google Scholar]
- 36.Chu FF, Doroshow JH, Esworthy RS. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J Biol Chem 1993;268:2571–2576. [PubMed] [Google Scholar]
- 37.Esworthy RS, Chu FF, Geiger P, Girotti AW, Doroshow JH. Reactivity of plasma glutathione peroxidase with hydroperoxide substrates and glutathione. Arch Biochem Biophys 1993;307:29–34. [DOI] [PubMed] [Google Scholar]
- 38.Chu FF, Rohan de Silva HA, Esworthy RS, Boteva KK, Walters CE, Roses A, Rao PN, Pettenati MJ. Polymorphism and chromosomal localization of the GI-form of human glutathione peroxidase (GPX2) on 14q24.1 by in situ hybridization. Genomics 1996;32:272–276. [DOI] [PubMed] [Google Scholar]
- 39.Tsuji T, Watanabe Y, Katoh H, Sato K, Kunieda T. Cloning and mapping of the mouse Gpx2 gene encoding gastrointestinal glutathione peroxidase. J Vet Med Sci 1998;60:651–654. [DOI] [PubMed] [Google Scholar]
- 40.Chu FF, Esworthy RS, Lee L, Wilczynski S. Retinoic acid induces Gpx2 gene expression in MCF-7 human breast cancer cells. J Nutr 1999;129:1846–1854. [DOI] [PubMed] [Google Scholar]
- 41.Traber PG, Silberg DG. Intestine-specific gene transcription. Annu Rev Physiol 1996;58:275–297. [DOI] [PubMed] [Google Scholar]
- 42.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science 1998;281:61–63. [DOI] [PubMed] [Google Scholar]
- 43.Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem 2005;280:20340–20348. [DOI] [PubMed] [Google Scholar]
- 44.Friling RS, Bergelson S, Daniel V. Two adjacent AP-1-like binding sites form the electrophile-responsive element of the murine glutathione S-transferase Ya subunit gene. Proc Natl Acad Sci USA 1992;89:668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inamdar NM, Ahn YI, Alam J. The heme-responsive element of the mouse heme oxygenase-1 gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem Biophys Res Commun 1996;221:570–576. [DOI] [PubMed] [Google Scholar]
- 46.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol 2000;20:5818–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rushmore TH, King RG, Paulson KE, Pickett CB. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci USA 1990;87:3826–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jowsey IR, Jiang Q, Itoh K, Yamamoto M, Hayes JD. Expression of the aflatoxin B1–8,9-epoxide-metabolizing murine glutathione S-transferase A3 subunit is regulated by the Nrf2 transcription factor through an antioxidant response element. Mol Pharmacol 2003;64:1018–1028. [DOI] [PubMed] [Google Scholar]