

Abstract
Endotoxin (LPS), a Gram-negative cell wall component, has potent proinflammatory properties. Acute LPS exposure causes airway inflammation; chronic exposure causes airway hyperreactivity and remodeling. IL-10 is an important antiinflammatory cytokine, which is decreased in patients with airway disease, such as asthma and cystic fibrosis. To examine the physiologic and therapeutic role of IL-10 in acute and chronic LPS-induced airway disease. Mice were exposed to aerosolized LPS once or daily for 4 wk. Endpoints were airway inflammation, airway reactivity to methacholine, extracellular matrix protein expression, and histologic analysis. IL-10–deficient mice developed significantly enhanced airway cellularity and remodeling when compared with C57BL/6 mice after chronic LPS inhalation. However they demonstrated less airway hyperreactivity associated with higher inducible nitric oxide synthase (iNOS), endothelial NOS (eNOS), and lung lavage fluid nitrite levels. In a bone marrow transplantation model, the IL-10 antiinflammatory effect was dependent on the hematopoietic but not on the parenchymal IL-10 expression. Induced epithelial human IL-10 expression protected from the LPS effects and led to decreased collagen production. IL-10 attenuates chronic LPS-induced airway inflammation and remodeling. Physiologically, the antiinflammatory effect of IL-10 is mediated by hematopoietic cells. Therapeutically, adenovirus-driven expression of human IL-10 in airway epithelia is sufficient for its protective effect on inflammation and remodeling. The role of IL-10 on airway hyperreactivity is complex: IL-10 deficiency protects against LPS-induced hyperreactivity, and is associated with higher eNOS, iNOS, and airway nitrate levels.
Keywords: airway hyperreactivity, airway remodeling, endotoxin, IL-10
CLINICAL RELEVANCE
This research elucidates the mechanism of chronic LPS-induced airway inflammation and remodeling and provides insights on the effect of IL-10. IL-10 is identified as a potential therapeutic agent for chronic LPS-induced airway inflammation and remodeling.
Endotoxin or LPS, a component of the wall of Gram-negative bacteria with significant proinflammatory properties, is present in high concentrations in organic dusts (1) and air pollution (2). Chronic occupational LPS exposure leads to airway inflammation and persistent obstruction (3), whereas LPS concentration in urban homes has been correlated with the clinical severity of allergic asthma (4). In animal models, acute LPS inhalation leads to neutrophilic airway inflammation, release of proinflammatory cytokines, and airway hyperreactivity (5, 6), whereas chronic LPS inhalation causes airway inflammation, persistent airway hyperreactivity, and airway remodeling with thickening of the subepithelial space (7). Regulation of the biological response to inhaled LPS is a complex process and requires the interaction of many cell types, including alveolar macrophages (8), airway epithelia (9), and smooth muscle (10). The specific cellular interactions that regulate this process and ultimately lead to airways remodeling remain to be elucidated.
IL-10 is a potent antiinflammatory cytokine with direct antiinflammatory activity on macrophages and neutrophils (11, 12). IL-10 substantially reduces the inflammatory response to intravenous LPS by downregulating the release of cytokines, CC chemokines, free radicals, and coagulation factors (13–15). IL-10 may also play a role as an endogenous antiinflammatory agent in asthma (16, 17) and cystic fibrosis (18, 19). Overexpression of IL-10 in the airways inhibited airway inflammation in an ovalbumin model of allergic asthma (20), and attenuated acute lung injury to inhaled LPS (21) and grain dust exposure (22). However, the role of IL-10 expression in the context of chronic inflammation, a clinically more relevant situation, has not been examined.
Elucidating the role of IL-10 in chronic LPS-induced airway disease will lead to a better understanding of the nature and mechanisms of chronic inflammatory airway diseases, such as asthma and cystic fibrosis. Furthermore, because airway remodeling is thought to be a sequela of chronic inflammation and may be amenable to antiinflammatory treatment, IL-10 may represent a useful therapeutic option in these diseases. We hypothesized that IL-10 would minimize the effects of chronic LPS inhalation exposure in mice, and that IL-10 can be used therapeutically to decrease chronic LPS effects on the airways. To test this hypothesis, we compared the biological and physiologic response in IL-10–deficient (C57BL/6-IL10tm1Cgn) and wild-type (WT; C57BL/6) mice after acute and chronic inhalation of LPS. We also induced expression of human IL-10 (hIL-10) in murine airways by using an adenoviral expression vector, and investigated the same endpoints. Finally, we used bone marrow transplant to determine whether hematopoietic cell-derived IL-10 or IL-10 derived from the lung parenchyma was involved in modulating the response to inhaled LPS.
MATERIALS AND METHODS
Experimental Animals
C57BL/6J and C57BL/6-IL10tm1Cgn mice were purchased from Jackson Laboratories (Bar Harbor, ME). Experimental protocols were approved by the Duke Institutional Animal Care and Use Committee, and performed in accordance with the U.S. Animal Welfare Acts standards.
Intratracheal Administration of Ad5RSVhIL-10
Ad5RSVhIL-10 (adenovirus carrying RSV promoter and hIL-10 gene) was purchased from the University of Iowa Vector Core Facility. A total of 5 × 108 plaque-forming units of the virus were given one time intratracheally in 50 μl of 3% Sucrose/PBS. Adenoviral vector-mediated expression after intratracheal infection usually abates after 2–3 wk, and would be insufficient for chronic LPS exposure, which lasts 4 wk. For chronic LPS exposures, mice were therefore treated intraperitoneally with 100 μg of anti-murine CD40L on days −3, 0, +3, and +6 of intratracheal instillation. This treatment prolongs vector-mediated expression to over 42 d (Ref. 23 and our observations). Control mice received adenovirus carrying the β-galactosidase gene (Ad5RSVLacZ). Acute LPS exposure was performed 7 d after adenoviral instillation (no CD40L used). Chronic LPS exposure was initiated 14 d after adenoviral instillation to avoid effects of anti-CD40L on the immune response. Immunohistochemical staining revealed the presence of hIL-10 in bronchial and alveolar epithelial cells, but not alveolar macrophages, at the end of both acute and chronic exposures.
Bone Marrow Transplantation
Chimeric animals were created by lethal irradiation (1,050 rad) of 6- to 8-wk recipient mice, which then received 4 × 106 donor bone marrow cells intravenously. The chimeric groups were: C57BL/6-IL10tm1Cgn bone marrow into C57BL/6-IL10tm1Cgn recipients, C57BL/6-IL10tm1Cgn into C57BL/6, C57BL/6 into C57BL/6-IL10tm1Cgn, and C57BL/6 into C57BL/6. CD45.1-positive C57BL/6 mice were used to differentiate from CD45.2-positive C57BL/6-IL10tm1Cgn. Engraftment was verified 6 wk after transplantation. Peripheral engraftment of B-lymphocytes was > 99%, and that of T-lymphocytes was 83%, by flow cytometry. LPS exposure started 8 wk after transplantation.
LPS Exposure
Lyophilized, reconstituted LPS (Escherichia coli serotype 0111:B4; Sigma, St. Louis, MO) was used. LPS aerosol was generated as previously described (7). Briefly, we used a six-jet atomizer (Model 9306; TSI, Inc., Shoreview, MN) at a constant pressure of 35 psi. Mice were exposed for 2.5 h (acute exposure), or for 2.5 h/d, 5 d/wk, for 4 wk (chronic exposure). LPS concentrations were determined by sampling the total chamber outflow using the quantitative chromogenic Limulus amebocyte lysate assay (QCL-1000; Whittaker Bioproducts, Walkersville, MD). The concentrations of LPS aerosol (Limulus amebocyte lysate assay) in these experiments were 6–8 μg/m3.
Airway pressure–time index (APTI) was measured as previously described (24). Mice were anesthetized, paralyzed, tracheally intubated, and received intravenous methacholine in doses of 25 μg/ml, 100 μg/ml, and 250 μg/ml.
Whole-Lung Lavage
Whole-lung lavage was performed, and lavage cells were spun and stained as previously described (7). In acute LPS exposure, mice were killed 4 h after the exposure. In chronic LPS exposures, mice were killed 3 d after the last exposure to avoid confounding acute LPS effects. Supernatants were stored at −80°C. Levels of murine TNF-α and transforming growth factor (TGF)-β in lavage fluid were measured by commercially available ELISA (R&D Systems, Minneapolis, MN).
Nitrite Level Determination
Nitrite in the whole-lung lavage fluid was measured as described previously (25) using a NOA 280 (Sievers, Boulder, CO).
Tissue Preparation
Lungs were perfused with 0.9% saline, the right lung removed, snap-frozen in liquid nitrogen, and stored at −80°C, and the left lung instilled and fixed with 4% paraformaldehyde in PBS. Tissue was embedded in paraffin, and 5 μm sections were stained as previously described (7).
Real-Time RT-PCR
Total lung RNA was isolated using the Trizol method. RT-PCR for hIL-10, murine IL-10, murine collagen I, III, and IV, fibronectin, eNOS, iNOS, neuronal NOS, and S-nitrosoglutathione reductase was performed using SYBR-Green assay and the 7900HT sequence detection system (Applied Biosystems, Foster City, CA). Fluorescence values for each gene were normalized to those of housekeeping genes, and expressed as fold-change over control groups.
Morphometric Analysis
Measurements of cross-sectional areas were performed on transverse bronchial sections (Masson-Trichrome stained) as previously described (7). Briefly, based on airway lumen diameter, all airway profiles were divided into small (⩽ 90 μm), medium (> 90–129 μm), and large (> 129 μm). For every profile, the subepithelial area was normalized to the length of adjacent basal membrane. Measurements were averaged per animal. Data are reported as group mean ± SEM of these average values for each airway size.
Immunohistochemical Analysis
Staining for human and murine IL-10 was performed using respective goat antibodies (R&D Systems) and Vectastain ABC (Vector Laboratories, Burlingame, CA).
Statistical Analyses
All data were expressed as means ± SEM. Individual comparisons between groups were tested by the two-tailed Student's t test. Statistical calculations were performed using SPSS software (SPSS, Inc; Chicago, IL). Probability values of P < 0.05 (two-tailed) were considered statistically significant.
RESULTS
hIL-10 Attenuates Airway Inflammation after Acute LPS Exposure in Mice
First, we sought to confirm the efficacy of hIL-10 in ameliorating murine LPS–induced inflammation. After a single acute LPS inhalation challenge, IL-10–deficient mice had significantly increased cellularity and TNF-α protein in whole-lung lavage fluid compared with C57BL/6 control animals (Table 1). In contrast, expression of hIL-10 in the airways of C57BL/6 mice led to a decreased concentration of cells and TNF-α. Expression of hIL-10 in the airways of IL-10–deficient mice reconstituted the WT phenotype both in terms of cells and TNF-α (no statistical difference from C57BL/6 mice).
TABLE 1.
IL-10 REDUCES CELLULAR INFLAMMATION AND PROINFLAMMATORY CYTOKINE PRODUCTION IN WHOLE-LUNG LAVAGE FLUID AFTER ACUTE LPS EXPOSURE
| IL-10–Deficient | C57BL/6 | IL-10–Deficient Expressing hIL-10 | C57BL/6 Expressing hIL-10 | |
|---|---|---|---|---|
| Total cells /ml lavage fluid | 1,134.0 ± 169.1 | 469.5 ± 45.8* | †594.4 ± 61.5 | 245.4 ± 43.5‡ |
| Neutrophils/ml lavage fluid | 1,082.6 ± 162.6 | 446.9 ± 46* | 563 ± 59.8* | 232.6 ± 43.5‡ |
| TNF-α (pg/ml lavage fluid) | 4,284.8 ± 545.9 | 2,864.6 ± 297.8† | 1,404.3 ± 419.4* | 564.9 ± 105.6‡ |
Definition of abbreviation: hIL = human IL.
Data are expressed as mean ± SEM.
P < 0.05 compared to IL-10–deficient mice.
P < 0.01 compared to IL-10–deficient mice.
P < 0.001 compared to IL-10–deficient mice.
IL-10 Attenuates Airway Inflammation and Remodeling after Chronic LPS Exposure
IL-10 had a significant effect on cellular airway inflammation and remodeling after chronic LPS exposure (Table 2 and Figure 2). There was a statistically significant increase in inflammatory cells in the airways of IL-10–deficient mice, whereas the hIL-10–expressing mice had a significant decrease in cells. Additionally, real-time RT-PCR showed that murine IL-10 expression was increased by 350% in C57BL/6 mice after the end of chronic LPS exposure (Table 3). The subepithelial space in airways of IL-10–deficient mice was significantly thickened compared with that of LPS-exposed C57BL/6 mice (Figures 2 and 3A). In a separate experiment, hIL-10–expressing mice had significantly decreased thickness of the airway subepithelial area compared with mice treated with a control virus (Figures 2 and 3B). After a 4-wk recovery period, there was no difference in subepithelial thickness between IL-10–deficient and C57BL/6 mice (not shown). Expression of mRNA for collagen I, III, IV, and fibronectin was not significantly different between IL-10–deficient and control animals 3 d after chronic LPS exposure or 4 wk later (Table 3). Total levels of TGF-β1 in the lavage fluid were significantly higher in IL-10–deficient mice (616.3 ± 67.6 pg/ml versus 399 ± 43.9 pg/ml; P = 0.02); however, active TGF-β1 levels were similar (113.2 ± 11.6 pg/ml versus 94.6 ± 23.7 pg/ml). Mice that expressed hIL-10 in their airways had significantly decreased expression of collagen I and III in their lungs compared with control mice (Table 3).
TABLE 2.
IL-10 REDUCES CELLULAR INFLAMMATION AFTER CHRONIC LPS EXPOSURE
| IL-10–Deficient | C57BL/6 | C57BL/6 Expressing hIL-10 | |
|---|---|---|---|
| Total cells/ml lavage fluid | 813,234 ± 124,449* | 479,285 ± 79,350† | 263,100 ± 16,486 |
| Macrophages/ml lavage fluid | 692,222 ± 118,654 | 382,764 ± 72,194 | 230,876 ± 25,292 |
| Percent macrophages | 83.8 ± 3.2 | 78.8 ± 3.5 | 87.2 ± 6.3 |
For definition of abbreviation see Table 1.
Data are expressed as mean ± SEM.
P < 0.05 compared to C57BL/6 mice and P < 0.01 compared to C57BL/6 hIL-10 mice.
P < 0.05 compared to mice expressing hIL-10.
Figure 2.
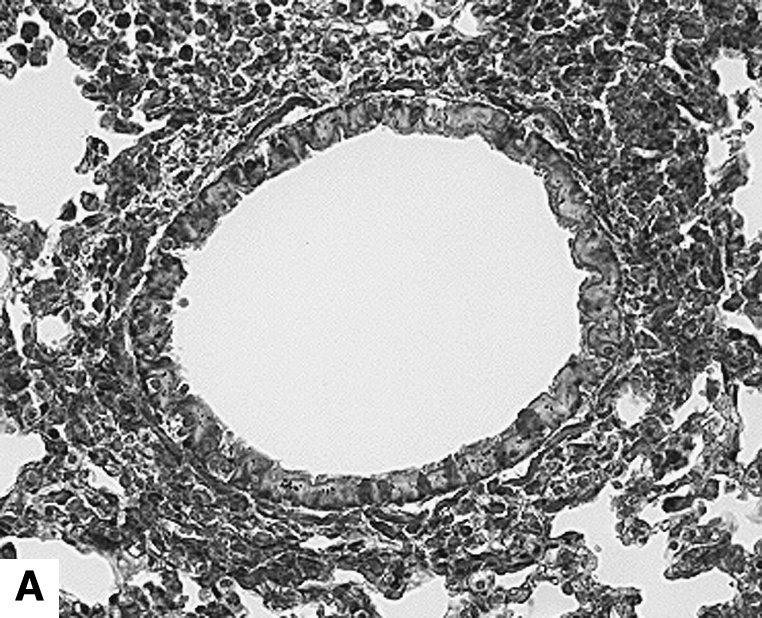
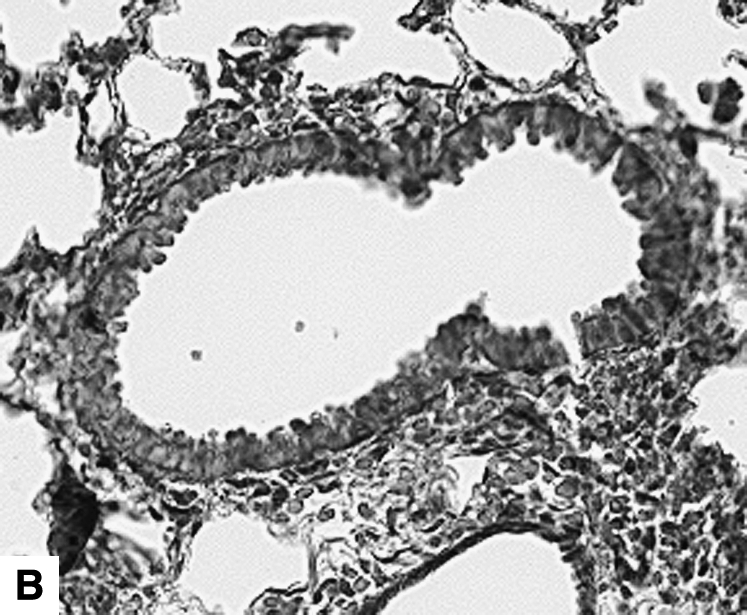
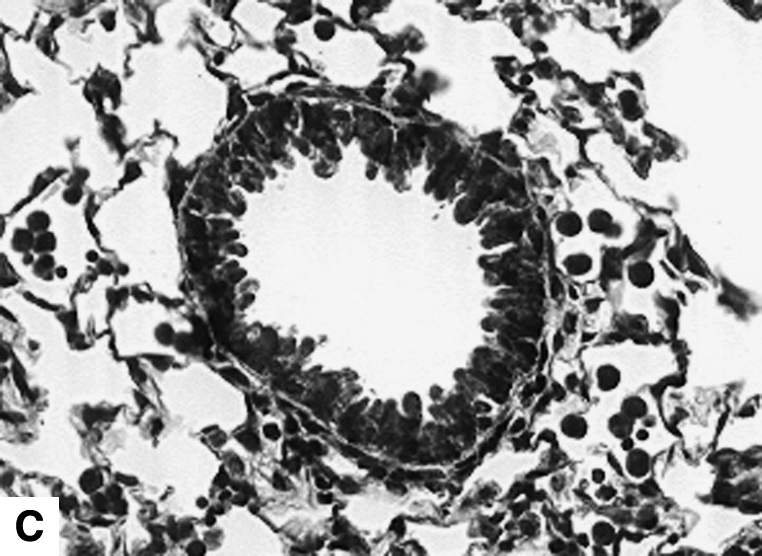
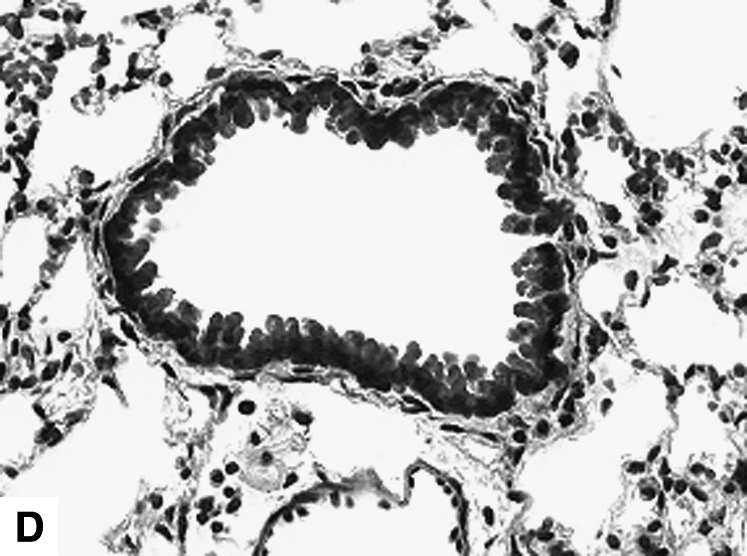
Airway changes after chronic LPS exposure. (A) IL-10–deficient mice. (B) C57BL/6 mice. (C) Mice expressing hIL-10. (D) C57BL/6 mice exposed to saline for same amount of time (control) (Masson-Trichrome).
TABLE 3.
EVALUATION OF mRNA EXPRESSION BY REAL-TIME RT-PCR AFTER CHRONIC LPS EXPOSURE, EXPRESSED AS PERCENT OF CONTROL
| 3 d after Chronic Exposure
|
4 wk after Chronic Exposure
|
|||||||
|---|---|---|---|---|---|---|---|---|
| mIL-10 | Collagen I | Collagen III | Fibronectin | Collagen III | Collagen IV | eNOS | iNOS | |
| Control | 100 ± 4.5 | 100 ± 1.5 | 100 ± 3.7 | 100 ± 3 | 100 ± 0.8 | 100 ± 1.2 | 100 ± 3.7 | 100 ± 3.7 |
| IL-10–deficient | ND | 90.5 ± 8.7 | 421.6 ± 88.3* | 1,576 ± 217‡ | 170.0 ± 38.8* | 282.0 ± 22.5‡ | 133.9 ± 11.1* | 153.1 ± 8.6* |
| C57BL/6 | 345.2 ± 94* | 105.6 ± 10.7 | 329.1 ± 129.1* | 1,375 ± 395‡ | 183.9 ± 57.9* | 276.9 ± 46.3‡ | 92.9 ± 9.2 | 111.1 ± 11.2 |
| C57BL/6 expressing hIL-10 | NM | 144.4 ± 26.7 | 265.0 ± 24.8* | 686 ± 133‡ | NA | NA | NA | NA |
| C57BL/6 expressing LacZ | NM | 348.5 ± 72.5* | 464.2 ± 113.8† | 1,421 ± 410† | NA | NA | NA | NA |
Definition of abbreviations: eNOS, endothelial nitric oxide synthase; hIL, human IL; iNOS, inducible nitric oxide synthase; mIL, murine IL; NA, not applicable; ND, not detected; NM, not measured.
Data expressed as mean ± SEM.
P < 0.05 compared to control mice.
P < 0.05 compared to hIL-10–expressing mice.
P < 0.01 compared to control mice.
Figure 3.



Stereometric measurement of subepithelial space thickness after chronic LPS exposure. (A) At 3 d after cessation of exposure, IL-10–deficient mice have dramatically increased subepithelial space thickness (*P < 0.001 compared with all other groups). C57BL/6 mice have moderate thickening of the subepithelial space (†P < 0.001 compared with control C57BL/6 mice; P < 0.05 compared with control IL-10–deficient mice). Filled bars, IL-10 KO PBS; open bars, C57BL/6 PBS; shaded bars, IL-10 KO PBS; striped bars, C57BL/6 LPS. (B) C57BL/6 mice expressing hIL-10 have decreased epithelial thickness compared with control mice expressing β-galactosidase (*P < 0.001 compared with all other groups; †P < 0.05 compared with control animals). Filled bars, AdhIL-10 PBS; open bars, AdLacZ PBS; shaded bars, AdhIL-10 LPS; striped bars, AdLacZ LPS. (C) At 3 d after cessation of exposure in the bone marrow transplantation model. Mice receiving IL-10–deficient bone marrow had significantly increased thickness of the subepithelial space of medium and large airways regardless of the recipient strain (*P < 0.05). Open bars, IL-10 KO bone marrow to IL-10 KO mice; light gray bars, IL-10 KO bone marrow to C57BL/6 mice; dark gray bars, C57BL/6 bone marrow to IL-10 KO mice; solid bars, C57BL/6 bone marrow to C57BL/6 mice.
We have previously demonstrated that chronic LPS exposure results in increased airway reactivity to methacholine even after a 4-wk recovery (7). Interestingly, APTI at that point demonstrated that LPS-treated IL-10–deficient mice had decreased airway hyperreactivity compared with C57BL/6 control animals (Figure 1). This was associated with statistically higher nitrite levels in the bronchoalveolar lavage fluid (154.4 ± 18.3 nM versus 79.4 ± 7.8 nM, P < 0.01), as well as increased mRNA expression of eNOS and iNOS (Table 3). Expression levels of neuronal NOS and S-nitrosoglutathione reductase did not differ between groups (data not shown).
Figure 1.

Airway hyperreactivity (measured as APTI) after chronic inhaled LPS exposure (*P < 0.01). Open bars, IL-10–deficient 3 d after chronic LPS; shaded bars, C57BL/6 3 d after chronic LPS; striped bars, IL-10–deficient 4 wk after chronic LPS; solid bars, C57BL/6 4 wk after chronic LPS.
Hematopoietic but Not Structural Cell-Derived IL-10 Is Important for the Attenuation of Airway Remodeling after LPS Exposure
To examine whether the origin of IL-10 is important for the effects on airway remodeling, we created bone marrow chimeric mice expressing IL-10 in their leukocytes but not in structural cells (WT > knockout [KO]) by engrafting bone marrow from C57BL/6 mice into IL-10–deficient mice (Figure 4F). Conversely, mice that expressed IL-10 in structural cells but not leukocytes (Figure 4C) were created by engrafting bone marrow from IL-10–deficient mice into C57BL/6 mice (KO > WT). Control groups were created by engrafting same-strain bone marrow to mice (WT > WT and KO > KO). We found that leukocyte-derived IL-10 was essential for the attenuation of airway remodeling after chronic LPS exposure. Mice with IL-10 expressing leukocytes had substantially less subepithelial thickening compared with mice with IL-10–deficient leukocytes, regardless of IL-10 expression in their pulmonary parenchyma (Figure 4). In morphometric analysis, mice expressing IL-10 in leukocytes (WT > KO and WT > WT) had significantly reduced subepithelial space thickness when compared with mice expressing IL-10 in structural cells only (Figure 3C).
Figure 4.
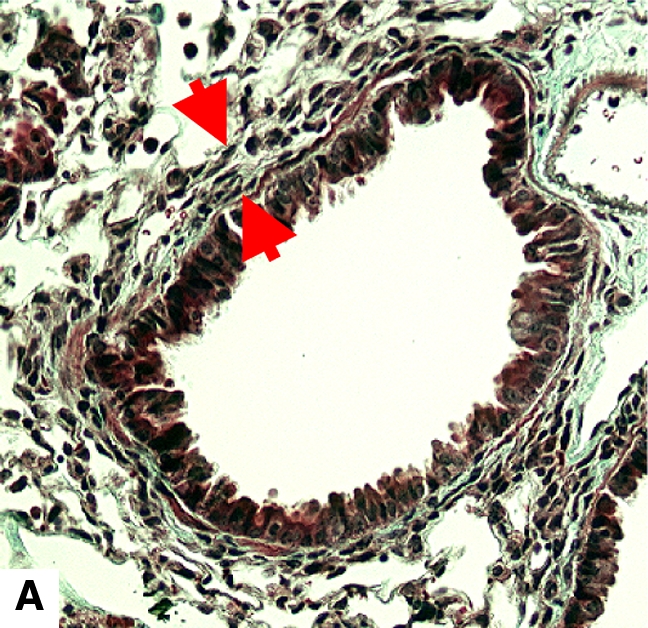
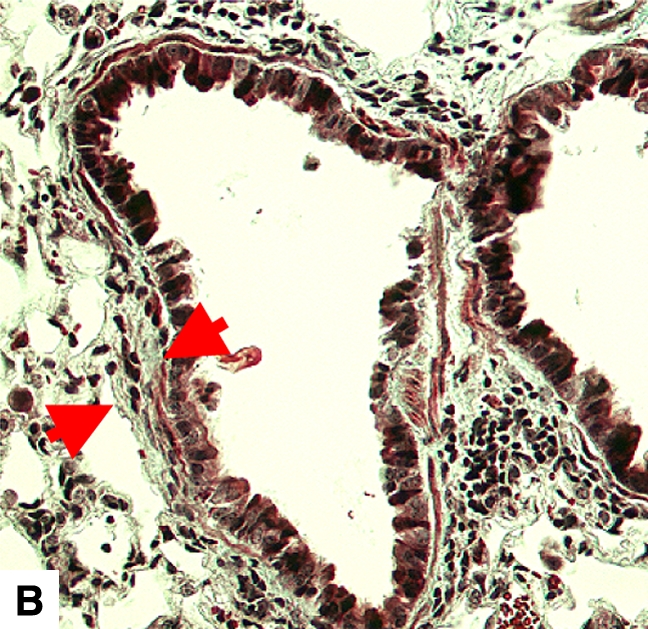
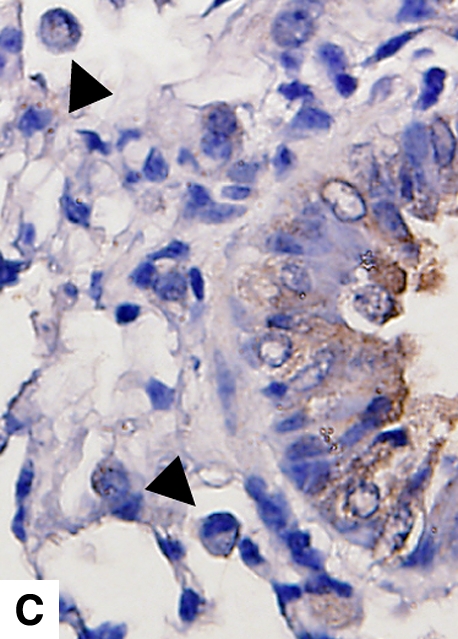
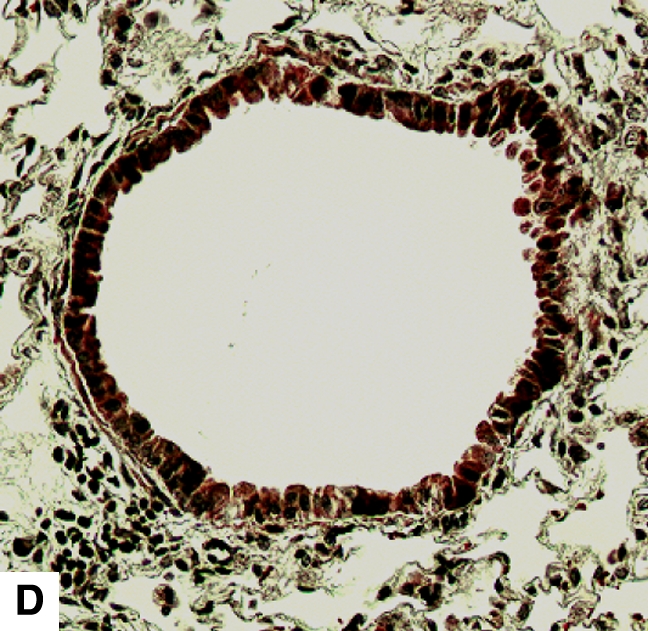
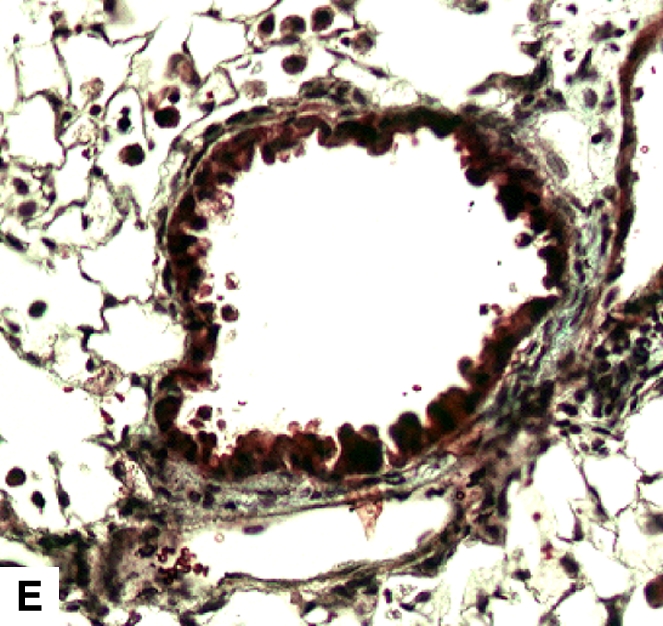
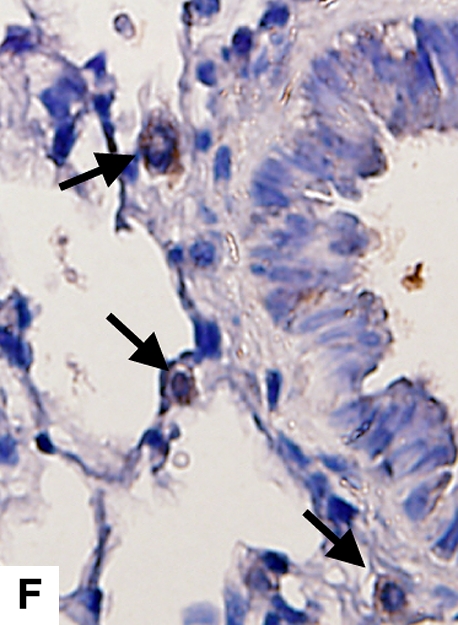
Airway remodeling after chronic LPS exposure, bone marrow transplantation model. (A) IL-10–deficient bone marrow engrafted into IL-10–deficient mice. (B) IL-10–deficient bone marrow engrafted into C57BL/6 mice. (D) C57BL/6 bone marrow engrafted into IL-10–deficient mice. (E) C57BL/6 bone marrow engrafted into C57BL/6 mice. Subepithelial space thickness is depicted between the arrows. (C and F) Representative immunohistochemistry for murine IL-10 in a C57BL/5 mouse receiving IL-10–deficient bone marrow (C) and an IL-10–deficient mouse receiving C57BL/6 bone marrow. Note absent staining from IL-10–deficient leukocytes (arrowheads, [C]) and IL-10–deficient bronchial epithelia (F). IL-10–positive leukocytes are also shown in (F) (arrows).
DISCUSSION
Our findings indicate that IL-10 reduces LPS-induced inflammation and airway remodeling. In a bone marrow transplant model, leukocyte expression of IL-10 was sufficient to attenuate the chronic effects of inhaled LPS, whereas structural cell IL-10 expression alone was not. Furthermore, induced expression of hIL-10 in airway epithelial cells restores the protective effect of IL-10, suggesting that for therapeutic purposes, the source of airway IL-10 may be of secondary importance. Our results support the concept that IL-10 is effective in attenuating LPS-induced airway disease, suggesting that IL-10 may be therapeutic in chronic inflammatory diseases, such as asthma and chronic obstructive pulmonary disease (COPD).
This study and previous studies from this laboratory have demonstrated that repeated episodes of acute inflammation can lead to airway remodeling. We have also previously shown that reduced inflammation can attenuate chronic airway disease (7, 26, 27). In this set of studies, chronic LPS exposure increased expression of mRNA for endogenous IL-10 by 350%, whereas induced expression of IL-10 was protective from the effects of chronic LPS inhalation. Conversely, patients with chronic airway diseases, such as asthma, COPD, and cystic fibrosis, have reduced endogenous IL-10 levels (17, 19), suggesting that IL-10 may be critical for maintaining lung architecture homeostasis in the face of environmental insult or injury. These patients are often colonized with gram-negative microorganisms, which are a steady source of LPS, and may be further susceptible to environmental sources of LPS (28). Such patients may be vulnerable to inhaled LPS in at least two ways: first, by having reduced baseline levels of IL-10; second, by failing to appropriately increase IL-10 expression after chronic exposure (16, 18). In our hands, induced expression of IL-10 in the airways significantly reduced the degree of inflammation after acute LPS exposure. More importantly, we demonstrated a significant decrease in airway remodeling, as well as pulmonary collagen deposition, after chronic (repeated) LPS exposure. Remodeling can account for the accelerated decline in pulmonary function observed in patients with asthma and those with COPD (29). Our results indicate that adequate control of acute inflammatory episodes will lead to decreased remodeling in the long term. Thus, exogenously administered IL-10 may prove to be useful as a therapeutic agent to decrease the persistent effects of chronic inflammatory airway diseases.
Hematopoietic cells are crucial to the response to inhaled LPS (7, 8). The present study demonstrates that hematopoietic cells can autoregulate their inflammatory response to LPS through IL-10 production. Teleologically this may be an adaptation to the continuous environmental exposure of the airways. Airway epithelial cells have decreased Toll-like receptor (TLR) 4 expression (30). Hematopoietic cells are plausibly the primary responder cells to inhaled LPS, and need to autoregulate their inflammatory response. In the ovalbumin model of allergic airway inflammation, adoptive transfer of regulatory T cells ameliorates inflammation in an IL-10–dependent manner (31). Our bone marrow transplant results indicate a similar hematopoietic cell–dependent effect of IL-10 on chronic LPS-induced remodeling. Additionally, we demonstrate that induced expression of IL-10 in the airway epithelial cells via an adenoviral vector decreases inflammation after LPS exposure. This implies that pharmacologic delivery of IL-10 into the airways will attenuate LPS-induced inflammation regardless of the mode of delivery.
We demonstrate that IL-10 deficiency appears to protect from LPS-induced airway hyperreactivity. The protective effect of IL-10 deficiency despite increased inflammation is particularly interesting. Previous studies in the ovalbumin model of allergic sensitization resulted in the same observation (32, 33). A possible mechanism has been recently proposed by Ameredes and colleagues (34). These investigators showed that IL-10–deficient mouse airways had enhanced NO production and decreased baseline methacholine responsiveness. In this study of chronic LPS exposure, invasive measurement (APTI) of airway resistance demonstrated a similar decrease in airway hyperreactivity, and we detected increased levels of nitrite and expression of iNOS and eNOS in IL-10–deficient mice. Although this finding does not provide definite proof about the mechanism of hyporesponsiveness in IL-10 deficiency, it does support previous observations in the allergic model of airway inflammation (35).
In summary, we have demonstrated the important role of IL-10 in the response to chronic LPS exposure of the airways. In vivo, leukocyte-derived IL-10 attenuates airway remodeling after chronic inhaled LPS exposure. Induced IL-10 expression in the lung reduces LPS-induced inflammation, both in deficient mice and in WT mice. Pulmonary IL-10 expression could provide a novel therapeutic option in nonallergic asthma.
This work was supported by grants from the National Institute of Environmental Health Sciences (ES11375, ES07498, ES012496, ES12717, and ES011961) and the Veterans Administration Medical Center (Merit Review).
Originally Published in Press as DOI: 10.1165/rcmb.2006-0055OC on June 29, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Laitinen S, Kangas J, Husman K, Susitaival P. Evaluation of exposure to airborne bacterial endotoxins and peptidoglycans in selected work environments. Ann Agric Environ Med 2001;8:213–219. [PubMed] [Google Scholar]
- 2.Soukup JM, Becker S. Human alveolar macrophage responses to air pollution particulates are associated with insoluble components of coarse material, including particulate endotoxin. Toxicol Appl Pharmacol 2001;171:20–26. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz DA, Thorne PS, Yagla SJ, Burmeister LF, Olenchock SA, Watt JL, Quinn TJ. The role of endotoxin in grain dust–induced lung disease. Am J Respir Crit Care Med 1995;152:603–608. [DOI] [PubMed] [Google Scholar]
- 4.Thorne PS, Kulhankova K, Yin M, Cohn R, Arbes SJ, Jr., Zeldin DC. Endotoxin exposure is a risk factor for asthma: the national survey of endotoxin in United States housing. Am J Respir Crit Care Med 2005; 172:1371–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jagielo PJ, Thorne PS, Watt JL, Frees KL, Quinn TJ, Schwartz DA. Grain dust and endotoxin inhalation challenges produce similar inflammatory responses in normal subjects. Chest 1996;110:263–270. [DOI] [PubMed] [Google Scholar]
- 6.O'Grady NP, Preas HL, Pugin J, Fiuza C, Tropea M, Reda D, Banks SM, Suffredini AF. Local inflammatory responses following bronchial endotoxin instillation in humans. Am J Respir Crit Care Med 2001;163: 1591–1598. [DOI] [PubMed] [Google Scholar]
- 7.Savov JD, Gavett SH, Brass DM, Costa DL, Schwartz DA. Neutrophils play a critical role in development of LPS-induced airway disease. Am J Physiol Lung Cell Mol Physiol 2002;283:L952–L962. [DOI] [PubMed] [Google Scholar]
- 8.Hollingsworth Ii JW, Chen BJ, Brass DM, Berman K, Gunn MD, Cook DN, Schwartz DA. The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am J Respir Crit Care Med 2004;171:806–813. [DOI] [PubMed] [Google Scholar]
- 9.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol 2004;287: L143–L152. [DOI] [PubMed] [Google Scholar]
- 10.Morris GE, Whyte MK, Martin GF, Jose PJ, Dower SK, Sabroe I. Agonists of Toll-like receptors 2 and 4 activate airway smooth muscle via mononuclear leukocytes. Am J Respir Crit Care Med 2005;171:814–822. [DOI] [PubMed] [Google Scholar]
- 11.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991;174:1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasama T, Strieter RM, Lukacs NW, Burdick MD, Kunkel SL. Regulation of neutrophil-derived chemokine expression by IL-10. J Immunol 1994;152:3559–3569. [PubMed] [Google Scholar]
- 13.Olszyna DP, Pajkrt D, Lauw FN, van Deventer SJ, van Der Poll T. Interleukin 10 inhibits the release of CC chemokines during human endotoxemia. J Infect Dis 2000;181:613–620. [DOI] [PubMed] [Google Scholar]
- 14.Pajkrt D, van der Poll T, Levi M, Cutler DL, Affrime MB, van den Ende A, ten Cate JW, van Deventer SJ. Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood 1997;89:2701–2705. [PubMed] [Google Scholar]
- 15.Dokka S, Shi X, Leonard S, Wang L, Castranova V, Rojanasakul Y. Interleukin-10–mediated inhibition of free radical generation in macrophages. Am J Physiol Lung Cell Mol Physiol 2001;280:L1196–L1202. [DOI] [PubMed] [Google Scholar]
- 16.Borish L, Aarons A, Rumbyrt J, Cvietusa P, Negri J, Wenzel S. Interleukin-10 regulation in normal subjects and patients with asthma. J Allergy Clin Immunol 1996;97:1288–1296. [DOI] [PubMed] [Google Scholar]
- 17.Takanashi S, Hasegawa Y, Kanehira Y, Yamamoto K, Fujimoto K, Satoh K, Okamura K. Interleukin-10 level in sputum is reduced in bronchial asthma, COPD and in smokers. Eur Respir J 1999;14:309–314. [DOI] [PubMed] [Google Scholar]
- 18.Moss RB, Bocian RC, Hsu YP, Dong YJ, Kemna M, Wei T, Gardner P. Reduced IL-10 secretion by CD4+ T lymphocytes expressing mutant cystic fibrosis transmembrane conductance regulator (CFTR). Clin Exp Immunol 1996;106:374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soltys J, Bonfield T, Chmiel J, Berger M. Functional IL-10 deficiency in the lung of cystic fibrosis (cftr(−/−)) and IL-10 knockout mice causes increased expression and function of B7 costimulatory molecules on alveolar macrophages. J Immunol 2002;168:1903–1910. [DOI] [PubMed] [Google Scholar]
- 20.Stampfli MR, Cwiartka M, Gajewska BU, Alvarez D, Ritz SA, Inman MD, Xing Z, Jordana M. Interleukin-10 gene transfer to the airway regulates allergic mucosal sensitization in mice. Am J Respir Cell Mol Biol 1999;21:586–596. [DOI] [PubMed] [Google Scholar]
- 21.Dokka S, Malanga CJ, Shi X, Chen F, Castranova V, Rojanasakul Y. Inhibition of endotoxin-induced lung inflammation by interleukin-10 gene transfer in mice. Am J Physiol Lung Cell Mol Physiol 2000;279: L872–L877. [DOI] [PubMed] [Google Scholar]
- 22.Quinn TJ, Taylor S, Wohlford-Lenane CL, Schwartz DA. IL-10 reduces grain dust-induced airway inflammation and airway hyperreactivity. J Appl Physiol 2000;88:173–179. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Su Q, Grewal IS, Schilz R, Flavell RA, Wilson JM. Transient subversion of CD40 ligand function diminishes immune responses to adenovirus vectors in mouse liver and lung tissues. J Virol 1996;70: 6370–6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollingsworth JW, 2nd, Cook DN, Brass DM, Walker JK, Morgan DL, Foster WM, Schwartz DA.The role of Toll-like receptor 4 in environmental airway injury in mice. Am J Respir Crit Care Med 2004;170:126–132. [DOI] [PubMed] [Google Scholar]
- 25.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from experimental asthma by an endogenous bronchodilator. Science 2005;308:1618–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savov JD, Brass DM, Lawson BL, McElvania-Tekippe E, Walker JK, Schwartz DA, Brass DM, Savov JD, Whitehead GS, Maxwell AB, et al. Toll-like receptor 4 antagonist (E5564) prevents the chronic airway response to inhaled lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol 2005;289:L329–L337. [DOI] [PubMed] [Google Scholar]
- 27.Brass DM, Savov JD, Whitehead GS, Maxwell AB, Schwartz DA. LPS binding protein is important in the airway response to inhaled endotoxin. J Allergy Clin Immunol 2004;114:586–592. [DOI] [PubMed] [Google Scholar]
- 28.Michel O, Ginanni R, Duchateau J, Vertongen F, Le Bon B, Sergysels R. Domestic endotoxin exposure and clinical severity of asthma. Clin Exp Allergy 1991;21:441–448. [DOI] [PubMed] [Google Scholar]
- 29.Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med 2001;164:S28–S38. [DOI] [PubMed] [Google Scholar]
- 30.Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. Response of human pulmonary epithelial cells to lipopolysaccharide involves TLR4-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 2004;279: 2712–2718. [DOI] [PubMed] [Google Scholar]
- 31.Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J Exp Med 2005;202: 1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Justice JP, Shibata Y, Sur S, Mustafa J, Fan M, Van Scott MR. IL-10 gene knockout attenuates allergen-induced airway hyperresponsiveness in C57BL/6 mice. Am J Physiol Lung Cell Mol Physiol 2001;280:L363–L368. [DOI] [PubMed] [Google Scholar]
- 33.Makela MJ, Kanehiro A, Borish L, Dakhama A, Loader J, Joetham A, Xing Z, Jordana M, Larsen GL, Gelfand EW. IL-10 is necessary for the expression of airway hyperresponsiveness but not pulmonary inflammation after allergic sensitization. Proc Natl Acad Sci USA 2000; 97:6007–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ameredes BT, Sethi JM, Liu HL, Choi AM, Calhoun WJ. Enhanced nitric oxide production associated with airway hyporesponsiveness in the absence of IL-10. Am J Physiol Lung Cell Mol Physiol 2005;288: L868–L873. [DOI] [PubMed] [Google Scholar]
- 35.Ameredes BT, Zamora R, Sethi JM, Liu HL, Kohut LK, Gligonic AL, Choi AM, Calhoun WJ. Alterations in nitric oxide and cytokine production with airway inflammation in the absence of IL-10. J Immunol 2005;175:1206–1213. [DOI] [PubMed] [Google Scholar]