

Abstract
The Arabidopsis thaliana disease resistance genes RPS2 and RPM1 belong to a class of plant disease resistance genes that encode proteins that contain an N-terminal tripartite nucleotide binding site (NBS) and a C- terminal tandem array of leucine-rich repeats. RPS2 and RPM1 confer resistance to strains of the bacterial phytopathogen Pseudomonas syringae carrying the avirulence genes avrRpt2 and avrB, respectively. In these gene-for-gene relationships, it has been proposed that pathogen avirulence genes generate specific ligands that are recognized by cognate receptors encoded by the corresponding plant resistance genes. To test this hypothesis, it is crucial to know the site of the potential molecular recognition. Mutational analysis of RPS2 protein and in vitro translation/translocation studies indicated that RPS2 protein is localized in the plant cytoplasm. To determine whether avirulence gene products themselves are the ligands for resistance proteins, we expressed the avrRpt2 and avrB genes directly in plant cells using a novel quantitative transient expression assay, and found that expression of avrRpt2 and avrB elicited a resistance response in plants carrying the corresponding resistance genes. This observation indicates that no bacterial factors other than the avirulence gene products are required for the specific resistance response as long as the avirulence gene products are correctly localized. We propose that molecular recognition of P. syringae in RPS2- and RPM1-specified resistance occurs inside of plant cells.
In plants, robust defense responses to invading phytopathogens often conform to a gene-for-gene relationship: resistance to a pathogen is only observed when the pathogen carries a specific avirulence (avr) gene and the plant carries a corresponding resistance (R) gene (1–3). Because avr-R gene-for-gene relationships are observed in many plant-pathogen systems and are accompanied by a characteristic set of defense responses, a common molecular mechanism underlying avr-R gene mediated resistance has been postulated (4). One simple model which explains gene-for-gene relationships is that pathogen avr genes directly or indirectly generate a specific molecular signal (ligand) that is recognized by cognate receptors encoded by plant R genes. Recent cloning of plant resistance genes and corresponding pathogen avirulence genes provided the tools for a direct test of this ligand-receptor model (5).
In the phytopathogenic interaction between the small flowering plant Arabidopsis thaliana and the bacterial phytopathogen Pseudomonas syringae, two R genes, RPS2 (6, 7) and RPM1 (8), and three corresponding avr genes, avrRpt2 (9), avrRpm1 (10), and avrB (11), have been isolated. RPS2 confers resistance to P. syringae strains expressing avrRpt2 (12, 13) and RPM1 confers resistance to P. syringae expressing avrRpm1 (14) or avrB (15). RPS2 and RPM1 belong to a major class of plant resistance genes which encode proteins containing nucleotide binding sites (NBS) and leucine-rich repeats (LRR) and which confer resistance to bacterial, fungal, or viral pathogens (5). The structural conservation among R genes is consistent with the presence of a common molecular mechanism underlying gene-for-gene mediated disease resistance.
The hypersensitive response (HR) is the most characteristic defense response associated with gene-for-gene interactions (1). The HR involves rapid plant cell death localized at the site of infection. Using the HR as a marker for disease resistance, a transient expression assay for RPS2 function was previously developed that involves biolistic introduction of an RPS2 cDNA clone into plant cells (7). In this assay, expression of a β-glucuronidase (GUS)-encoding reporter gene cointroduced with RPS2 is monitored as an indicator of the HR: when the HR causes plant cell death, low levels of GUS activity are observed.
Here we report that the RPS2 gene product is probably localized in the plant cell cytoplasm. We also report that when transiently expressed in plants, two P. syringae avr genes, avrRpt2 and avrB, can elicit an HR in a gene-for-gene specific manner. For these experiments, the transient expression assay was enhanced to make it quantitative. We propose that molecular recognition of the pathogen in RPS2- and RPM1-specified resistance occurs inside of plant cells.
MATERIALS AND METHODS
A. thaliana plants representing four different R gene genotypes were used; ecotype Columbia (Col-0) wild type (phenotype, RPS2 RPM1; genotype, RPS2/RPS2 RPM1/RPM1), rps2-101C (rps2 RPM1; rps2-101C/rps2-101C RPM1/RPM1; Col-0 background) (7, 16), ecotype Niederzenz (Nd-0) wild type (RPS2 rpm1; RPS2/RPS2 Δrpm1/Δrpm1) (8, 14); and a hybrid line derived from a cross between rps2-101C and Nd-0 with an rps2-101C/rps2-101C Δrpm1/Δrpm1 genotype (phenotype rps2 rpm1) (7). Plants were grown at 22°C with ≈80% relative humidity with a 12 hr light/12 hr dark cycle in environment-controlled growth chambers.
The avrRpt2 and avrB genes were amplified by PCR using Pfu polymerase (Stratagene) from plasmids pLH12 (13) and pPSG0002 (17), respectively, using the primers, AVRT1 and AVRT2 for avrRpt2 and AVRB1 and AVRB2 for avrB: AVRT1, 5′-CGCGGATCCACCATGATGAAAATTGCTCCAGTTG-3′; AVRT2, 5′-GGAGCGCGGCCGCTTGTCATGATGCCGCCACGTG-3′; AVRB1, 5′-CGCGGATCCACCATGGGCTGCGTCTCGTC-3′; AVRB2, 5′-GGAGCGCGGCCGCTATACATTTAAAAGCAATC-3′. These primers were designed to change the sequence preceding the initiation codon into a eukaryote-type translation initiation site (18). Each PCR product was digested with BamHI and NotI and cloned into the BamHI–NotI site of the plant transient expression vector pKEx4tr (7) to obtain pExavrRpt2 and pExavrB, respectively. These plasmids were used for transient expression of the avr genes. A full-length RPS2 cDNA (clone 11) (7) in pKEx4tr was used for transient expression and in vitro translation of RPS2. The RPS2 mutant I353K was created using PCR by changing the ATC codon of isoleucine 353 into the lysine codon AAG. Similarly, the rps2-101C nonsense mutation (7) was recreated by changing nucleotide G704 to A. These RPS2 wild-type and mutant genes in pKEx4tr were cut out with PmeI and SacI, and cloned into the SmaI–SacI site of pBI1Rpro11 (a derivative of the plant transformation vector pBI121 (Clonetech) in which the cauliflower mosaic virus 35S promoter was replaced with the 1.4-kb RPS2 promoter region; F.K. and F.M.A., unpublished) to obtain pR11-I353K, pR11-101C, and pR11-X11, respectively. These plasmids were used for generating transgenic plants. RPM1 cDNA clones were isolated from an A. thaliana cDNA library (7) in pKEx4tr by hybridization screening using a DNA probe based on the sequence of RPM1 (8). Three clones were purified, and one of the two full-length clones (clone 7), whose 5′ end starts at nucleotide number −86, was used for transient expression. pKEx4tr-G (7) was used for the GUS construct. The luciferase (LUC) construct p35S-LUC (19) was a gift from M. Bustos. Generation of transgenic plants and analysis of the plants for a macroscopic HR were performed as described (7).
For transient expression, gold biolistic particles (1 μm in diameter) were coated with DNA as described (7). Note that all genes for transient expression were linked to the cauliflower mosaic virus 35S promoter. pKEx4tr-G (1.4 μg) and 2.0 μg of one of the avr gene constructs were used for coating each mg of gold particles. When an R gene was included in the assay, 0.1 μg of RPS2 clone 11 or 2.0 μg of RPM1 clone 7, respectively, was used for each mg of gold particles. In all cases, the total amount of DNA/mg of gold particles was adjusted to be the same by addition of pKEx4tr. When the LUC gene was used for a reference of transformation efficiency, 1.8 μg of p35S-LUC was used for each mg of gold particles. These LUC-coated particles were mixed with the particles coated with GUS and the relevant test gene in the ratio of 1:5.6. Bombardment was carried out with a Bio-Rad PDS-1000/He machine and in each bombardment experiment, 0.5 mg of gold particles and six leaves of A. thaliana (5–6 weeks old) were used. After a 27-hr incubation at room temperature in the dark, the leaves were either histochemically stained for GUS activity (20) or homogenized for quantitation of the GUS and LUC activities (19). The GUS and LUC activities of leaves that were not bombarded were used as blank values. The relative GUS activities (GUS activity/LUC activity) in a single series of experiments were renormalized by setting the mean value of the GUS only samples to be 100. All transient expression experiments were repeated at least twice to confirm reproducibility.
The RPS2 cDNA clone 11 digested with NotI was transcribed in vitro using T7 RNA polymerase and the resulting RPS2 RNA or β-lactamase RNA (Promega) were used for in vitro translation. The in vitro translation/translocation experiments were performed using rabbit reticulocyte lysate and canine pancreatic microsomes (Promega) with [35S]methionine according to the supplier’s instruction. Fractionation of the proteins after the reaction and a posttranslation procedure (see Fig. 3B) were performed essentially according to Sakaguchi et al. (21). The proteins were resolved by 7.5% SDS/PAGE, and the autoradiograms of the gels were obtained by a PhosphorImager. Each lane in Fig. 3 corresponds to 9 μl of reaction.
Figure 3.
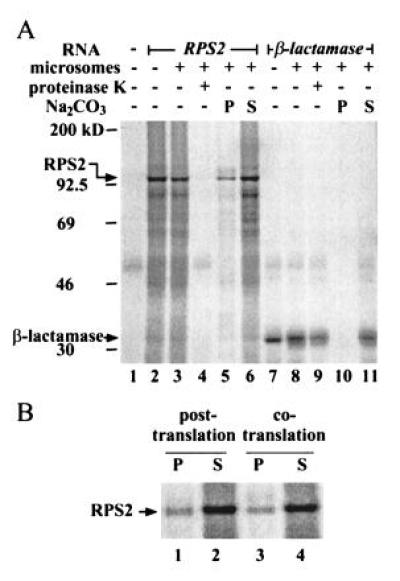
Subcellular localization of RPS2 by in vitro translation/translocation. (A) RPS2 appears to be cytoplasmic. RPS2 RNA (lanes 2–6) and β-lactamase RNA (a positive control for translocation; lanes 7–11) were translated either in the presence (lanes 3–6 and 8–11) or absence (lanes 2 and 7) of dog pancreatic microsomes. Lane 1 shows a no RNA control for translation. The reactions were treated with proteinase K (lanes 4 and 9) or fractionated by ultracentrifugation into precipitate (lanes 5 and 10) and supernatant (lanes 6 and 11) fractions after Na2CO3 treatment. The positions of molecular weight markers, RPS2, and β-lactamase are indicated on the left. (B) RPS2 detected in the precipitate fraction is an artifact. The microsomes were either included in the translation reaction as in the standard procedure (cotranslation; lanes 3 and 4) or added after the translation reaction was terminated with cycloheximide (posttranslation; lanes 1 and 2). The reactions were fractionated into precipitate (lanes 1 and 3) and supernatant fractions (lanes 2 and 4) after Na2CO3 treatment.
RESULTS
Evidence That RPS2 Is a Cytoplasmic Protein.
A direct approach for detection of R gene products in plants is technically challenging due to the low abundance of the R proteins. An anti-RPS2 antibody, which apparently has high affinity for in vitro-translated RPS2 protein, barely detected the protein in plant extracts by Western blot analysis (F.K. and F.M.A., unpublished work). Therefore, we used indirect approaches to investigate the subcellular localization of RPS2.
The computer program alom (22) predicts that one polypeptide region (amino acid residues 340–356) of RPS2 is membrane-integrated (7) whereas the corresponding region of the protein encoded by the tobacco mosaic virus resistance gene N (amino acid residues 377–393; Fig. 1) (23), which also belongs to the NBS-LRR class, is not membrane-integrated. The corresponding region of RPM1 (8) is not predicted to be membrane-integrated either (not shown). It seems unlikely that one of the corresponding polypeptide regions of functionally and structurally related proteins is membrane-integrated and the others are not. A mutation (I353K) was created in RPS2 by changing isoleucine 353 to lysine, the corresponding residue in N (Fig. 1). To analyze the activity of the I353K mutant, which alom does not predict to be membrane-integrated, transgenic rps2-101C mutant plants carrying the I353K transgene linked to the RPS2 promoter (F.K. and F.M.A., unpublished work) were constructed. As controls, transgenic rps2-101C mutant plants carrying a wild-type RPS2 transgene and an rps2-101C mutant transgene were also constructed. One-month old primary transformants were challenged with a high dose (0.5 × 107 colony-forming unit/ml) of P. syringae pv. phaseolicola (Psp) 3121 ± avrRpt2 and observed 20 hr after inoculation (Fig. 2A). When challenged with Psp 3121 carrying a vector control, none of the transgenic plants showed detectable change. When challenged with Psp 3121 carrying avrRpt2, all seven independent transgenic lines transformed with the RPS2 wild-type gene showed a typical macroscopic HR (Fig. 2A Right), indicating complementation of the mutant phenotype, whereas none of 20 independent transgenic lines transformed with the rps2-101C gene showed an HR (Fig. 2A Center). Among 24 independent lines transformed with I353K, 17 lines showed as strong an HR as the plants transformed with the wild-type gene (Fig. 2A Left), and five lines showed a weak HR (not shown). The occurrence of a small number of transgenic lines showing a weak HR may indicate that the activity of the I353K gene may be slightly weaker than the wild-type gene. Nevertheless, these results demonstrate that the I353K mutation, which would disrupt the transmembrane-integrated property of this region of RPS2, does not significantly affect RPS2 function, indicating that the polypeptide region is unlikely to be transmembrane.
Figure 1.

The I353K mutation in the putative transmembrane region of RPS2. The putative transmembrane region of RPS2 protein (Middle) (7) is compared with the corresponding region of N (Top) (23). Identical and similar residues between the two proteins are indicated by vertical lines and colons, respectively, between the two sequences. The putative transmembrane region of RPS2, predicted by alom (22), is underlined. The corresponding region of the I353K mutant is shown at bottom. The mutated amino acid residue in I353K and the corresponding residues in N and RPS2 are shown in boldface. The numbers indicate the amino acid residue numbers in each protein.
Figure 2.

(A) The I353K RPS2 mutant gene can complement an rps2 mutant phenotype in transgenic plants. One-month old rps2-101C plants transformed with I353K (Left), with a rps2-101C mutant gene (Center), and with the RPS2 wild-type gene (Right) were inoculated at 0.5 × 107 colony-forming unit/ml with Psp 3121 carrying a vector control (pLAFR3) or carrying avrRpt2. Only one-half of each leaf (arrowhead) was inoculated. The photographs were taken 20 hr after inoculation. (B) Transient expression of avrRpt2 causes a reduction of cointroduced GUS gene expression in RPS2 plants. Leaves of RPS2 wild-type (Right) and rps2-101C mutant (Left) plants were bombarded with biolistics carrying the avrRpt2 and GUS constructs. After a 27-hr incubation, the leaves were histochemically stained for GUS activity. The cells that express GUS enzyme at a high level are visualized as blue dots on the leaves.
As a second indirect approach, we examined the localization of RPS2 in an in vitro translation/translocation system that utilizes rabbit reticulocyte lysate and dog pancreatic microsomes. In this procedure, if a protein, labeled with 35S by in vitro translation, has a signal peptide for secretion, the protein is cotranslationally transported into microsomes. Proteinase K treatment and ultracentrifugation after Na2CO3 treatment allow protein localization to be classified as cytoplasmic, membrane-integrated, or secreted (21). A cytoplasmic protein remains outside of microsomes, so that it is sensitive to proteinase K and is recovered in the supernatant after centrifugation. A secreted protein is transported into microsomes but remains soluble, so that it is protected from proteinase K and recovered in the supernatant after centrifugation. A membrane-integrated protein can be partially protected from proteinase K and it is precipitated with the membrane after centrifugation. Fig. 3A shows the results of this analysis, including β-lactamase as a positive control for translocation. β-Lactamase, which is a secreted protein, was protected from proteinase K (lane 9) and detected in the soluble fraction after Na2CO3 treatment (lane 11), demonstrating the high efficiency of the translocation system. In vitro-translated RPS2 migrated with an apparent molecular weight consistent with that calculated from its deduced amino acid sequence (105 kDa; lane 2). RPS2 was not protected from proteinase K (lane 4). The major portion of RPS2 was detected in the soluble fraction after Na2CO3 treatment (lane 6), but a significant amount was also detected in the precipitate fraction (lane 5). The amount of this precipitate remained the same in the absence of the translocation system (posttranslation, Fig. 3B), indicating that the precipitate was an artifact. These results indicate a cytoplasmic localization for RPS2. Although the results must be interpreted with caution because of the use of a heterologous in vitro system, taken together with the mutagenesis results described above, they suggest that RPS2 is a cytoplasmic protein.
When Transiently Expressed in Plants, avrRpt2 Can Elicit a Specific Resistance Response.
If RPS2 is indeed cytoplasmic and if it is the primary receptor for the avrRpt2-generated ligand, the ligand must also be present in the plant cytoplasm. A possible ligand for RPS2 is the avrRpt2 gene product itself. If AvrRpt2 protein were expressed in plant cells, it would most likely be located in the plant cytoplasm because it is a hydrophilic protein and does not have an obvious signal peptide (9). Based on these considerations, we tested whether expression of avrRpt2 in plants can elicit a specific resistance response.
Transient expression by biolistic bombardment was used for this purpose. Based on the same principle as the transient expression assay for RPS2 (7), reduced expression of a cointroduced GUS gene was used as an HR indicator. In Fig. 2B, the avrRpt2 construct was cointroduced with the GUS construct into either RPS2 wild-type (Right) or rps2-101C mutant (Left) plants, and plant cells that expressed GUS were visualized by histochemical staining (blue dots in the figure). The GUS expression in wild-type plants was reduced compared with that in the rps2 mutant plants, indicating the occurrence of the HR in an RPS2-dependent manner. Therefore, avrRpt2 can elicit a resistance response with gene-for-gene specificity when expressed in plants.
A Quantitative Transient Expression Assay for the Resistance Response.
A problem associated with biolistic bombardment is that the efficiency of transient transformation varies to a great extent both for each bombardment and for different areas of the target in a single bombardment. To quantitate the assay, a reference for transformation efficiency must be included. This was achieved by introducing a second reporter gene, LUC (24), linked to the 35S promoter, into cells different from the cells into which the GUS and avrRpt2 genes were introduced. Gold particles were coated either with the LUC construct or with the GUS and avrRpt2 constructs, the particles were mixed and then bombarded together. Because a relatively small number of plant cells are transformed by the biolistics procedure, the cells transformed with LUC and the cells transformed with the GUS and avrRpt2 are statistically different and far apart. Because the HR is a local event (1), LUC expression is not expected to be affected very much by an HR occurring in other cells. Therefore, LUC activity represents the relative transformation efficiency in a particular bombardment and can be used as a reference to normalize the GUS activity.
Using this quantitative transient expression assay, avrRpt2 was examined in RPS2 and rps2 plants which had either an RPM1 or rpm1 phenotype (Fig. 4, second row of each panel). Irrespective of the RPM1 phenotype, expression of avrRpt2 caused ≈50% reduction of the normalized GUS activity in RPS2 plants (A and C) compared with the activity in rps2 plants (B and D). Another P. syringae avirulence gene avrB, which corresponds to the RPM1 resistance gene, was also examined (Fig. 4, third row of each panel). Similar to the results obtained with avrRpt2, irrespective of the RPS2 phenotype, expression of avrB caused more than 85% reduction of the normalized GUS activity in RPM1 plants (A and B) compared with the activity in rpm1 plants (C and D). Thus the gene-for-gene relationship for avrRpt2 and avrB is strictly conserved in this assay, confirming that the assay indeed reflects a specific resistance response. Two different P. syringae avr genes with distinct specificities can elicit a specific resistance response when expressed in plants, and no other bacterial components are required for this response.
Figure 4.
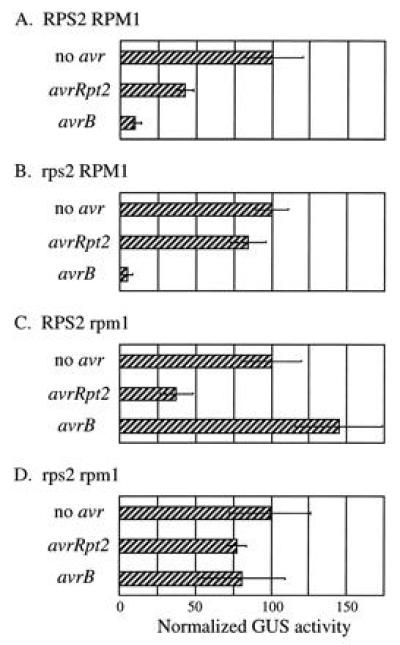
avrRpt2 and avrB can elicit a specific resistance response when expressed in plants. RPS2 RPM1 (A), rps2 RPM1 (B), RPS2 rpm1 (C), and rps2 rpm1 (D) plants were bombarded with biolistics carrying the GUS and the indicated avr gene constructs. Each bar represents the mean value of four bombardment events. (Bars = SEM.)
The reduction of GUS activity reflects both the percentage of dead cells and how quickly the cells die. The reduction of GUS activity caused by avrRpt2 was smaller than that caused by avrB (Fig. 4). This is consistent with the observation that the HR caused by avrB is faster than the one caused by avrRpt2 when the avr genes are carried by a P. syringae strain (25).
In the case of avrB expression in RPM1 wild-type plants, we often observed a statistically significant reduction of LUC reference gene expression (typically 50–70% reduction) compared with the LUC expression in controls in which no avirulence genes were expressed (not shown). Therefore, the actual reduction of relative GUS activity in the case of avrB-RPM1 interactions was even greater than that shown in Fig. 4. A similar reduction in LUC expression was not observed in any of the other assay conditions, including the case in which avrRpt2 was expressed in RPS2 wild-type plants. This difference in LUC gene expression between the avrRpt2-RPS2 and avrB-RPM1 interactions may be correlated with different gene induction patterns that are observed between these two interactions (25, 26).
The Specific Resistance Response Can Be Elicited by Transiently Expressed avr and R Genes.
The results of the transient expression of the avirulence genes prompted us to test whether the assay works when both avr and R genes are transiently expressed. In the following experiments, rps2 rpm1 double mutant plants were used for biolistic bombardment. The avr gene, R gene, and GUS gene constructs were used to coat one set of gold particles and the LUC gene construct was used to coat another set. As shown in Fig. 5A, when avrRpt2 and RPS2 were transiently expressed together, reduction of GUS activity was observed, but was not observed when RPS2 was expressed together with avrB. Similarly, reduction of GUS activity was observed when RPM1 was transiently expressed together with avrB, but not when RPM1 was expressed together with avrRpt2 (Fig. 5B). This strict conservation of the gene-for-gene specificity indicates that the transient expression assay can be used for functional analysis of both avr and R genes.
Figure 5.
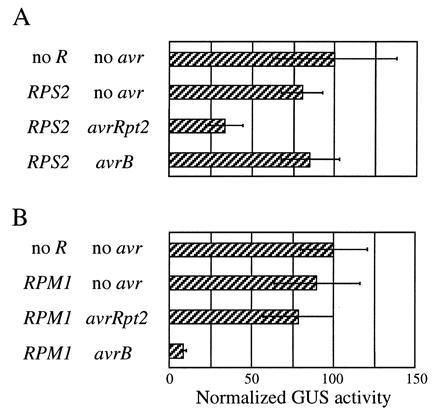
Both avirulence and resistance genes can be analyzed by transient expression. rps2 rpm1 plants were bombarded with biolistics carrying the GUS gene, the indicated R gene, and the indicated avr genes. Each bar represents the mean value of four bombardment events. (Bars = SEM.)
As in the experiment described in Fig. 4, the avrB-RPM1 interaction resulted in greater reduction of the GUS activity than the avrRpt2-RPS2 interaction, when both the avr and R genes were transiently expressed (Fig. 5). A significant reduction of LUC reference gene expression specific to the avrB-RPM1 interaction was also observed in this experiment (not shown).
DISCUSSION
To determine where the molecular recognition occurs between avr-generated signals and R-gene mediated responses in gene-for-gene relationships, we investigated the subcellular localization of RPS2 and the effects of directly expressing avr genes in plants. Mutational analysis of the putative transmembrane region of RPS2 and in vitro translation/translocation analysis suggested that RPS2 is a cytoplasmic protein. RPM1 is also likely to be cytoplasmic because RPS2 and RPM1 belong to the same class of R genes (8). Expression in plants of avrRpt2 and avrB, avr genes corresponding to RPS2 and RPM1, respectively, elicited resistance responses in a gene-for-gene specific manner. This indicates that these avr gene products are the only bacterial components required for the gene-for-gene interaction as long as they are correctly localized. Judging from their amino acid sequences (9, 11), the avr gene products are also likely to be cytoplasmic when expressed in plants. Therefore, we propose that the molecular recognition involved in the RPS2- and RPM1-specified resistance takes place inside of plant cells.
A corollary of the notion that R gene mediated recognition of avr gene signals occurs in the cytoplasm is that avr gene products themselves directly interact with the corresponding R gene products although this notion does not exclude other possibilities. For example, the avr gene products could be enzymes that modify a plant product into specific ligands. However, we prefer the direct interaction model for the following reasons. First, it has been speculated that the LRR of an NBS-LRR gene product is a determinant of avr gene specificity (27). Because LRR structures are generally involved in protein-protein interactions (28), it is likely that the ligands would be proteins. Second, in most cases, the genetically identifiable factors involved in specificity determination in resistance are a single R gene and a single corresponding avr gene (1). If avr genes generate signals through indirect mechanisms, more genes required for the specificity determination might have been identified. This view of an NBS-LRR resistance gene product as an intracellular receptor is compatible with the fact that tobacco mosaic virus resistance gene N is also an NBS-LRR gene (23) because the products of the viral genes, one of which should be the corresponding avirulence gene, accumulate inside of plant cells.
For the molecular recognition to occur inside plant cells, the avr gene products must be transported into plant cells from bacteria. For most P. syringae avr genes, including the two avr genes used in this study, the bacterial hrp (hypersensitive response and pathogenicity) gene cluster is required to elicit the specific resistance response in plants (29, 30). Some of the genes in the hrp cluster encode components of a type III protein secretion system (31). Bacterial pathogens of mammals, such as Salmonella, Shigella, and Yersinia, apparently use the type III secretion pathway to transfer proteins important for pathogenicity directly into mammalian cells (32). By analogy, P. syringae could also use a type III secretion system to directly transfer avr gene products into plant cells. The hrp cluster is also required for pathogenicity (33), suggesting that the type III secretion system may also transfer virulence gene products into plant cells.
To demonstrate effects of avirulence gene expression in plants, we used transient expression using biolistic bombardment. The principle of the assay, in which reduction of a cointroduced reporter gene expression is used as an indicator of the HR, was initially developed by us to demonstrate complementation of an rps2 mutant phenotype by an RPS2 cDNA clone (7). In this study, we showed that the transient expression assay can also be used to monitor the activity of a cloned avr gene by delivering the avr gene constructs biolistically into plant cells. (Fig. 2B). While this work was in progress, Gopalan et al. also reported that avrB can elicit a specific resistance response when expressed in plants using a transient expression assay that is essentially the same as the assay used in Fig. 2B (29).
One shortcoming of the biolistic mediated transient expression assay is that the transformation efficiency varies to a large extent, both with respect to independent bombardments and to different areas of the target. To compensate for this variability, our original assay for RPS2 function involved infection of only one half of each leaf with P. syringae carrying avrRpt2; the uninfected half of the leaf served as a reference for the transformation efficiency (7). However, the assay for avr genes used in Fig. 2B did not include an internal reference for transformation efficiency. We therefore modified the transient expression assay by including a second reporter gene (LUC) delivered on a separately coated set of biolistic particles in the same bombardment event (Fig. 4). Using this quantitative assay, we have unequivocally demonstrated that transient expression of avrRpt2 or avrB in plants elicits an HR in a gene-for-gene specific manner (Fig. 4).
We further extended the application of the functional transient expression assay by simultaneously assaying for both avr and R gene functions (Fig. 5). This rapid assay will be applicable to studies of many other gene-for-gene plant-pathogen systems for three reasons. First, biolistic transient transformation is applicable to many plant species. Second, the HR is a characteristic resistance response in gene-for-gene resistance. Third, simultaneous bombardment of avr and R genes is not limited by pathogen type. This assay may also be used for a rapid determination of whether a particular R gene can function in a heterologous host.
Acknowledgments
We thank Mauricio Bustos for his help during F.K.’s lab set-up period and for p35S-LUC, Julie Stone for information on in vitro translation/translocation systems, Maw-Shenq Chern for technical advice on the LUC quantitation, Jeffery Dangl for providing the RPMI sequence prior to publication, and Jane Glazebrook for critical reading of the manuscript. This study was supported in part by National Institutes of Health Grant 48707 and by a grant from Monsanto Company awarded to F.M.A. and by a start-up fund to F.K. from University of Maryland Baltimore County. F.K. was a recipient of a Summer Faculty Fellowship and R.T.L. was supported by Graduate Research Assistantship from University of Maryland Graduate School, Baltimore.
Footnotes
Abbreviations: avr gene, avirulence gene; R gene, resistance gene; HR, hypersensitive response; NBS, nucleotide binding site; LRR, leucine-rich repeats; GUS, β-glucuronidase; LUC, luciferase.
References
- 1.Keen N T. Plant Mol Biol. 1992;19:109–122. doi: 10.1007/BF00015609. [DOI] [PubMed] [Google Scholar]
- 2.Lamb C J, Lawton M A, Dron M, Dixon R A. Cell. 1989;56:215–224. doi: 10.1016/0092-8674(89)90894-5. [DOI] [PubMed] [Google Scholar]
- 3.Flor H H. Annu Rev Phytopathol. 1971;9:275–296. [Google Scholar]
- 4.Gabriel D W, Rolfe B G. Annu Rev Phytopathol. 1990;28:365–391. [Google Scholar]
- 5.Staskawicz B J, Ausubel F M, Baker B J, Ellis J G, Jones J D G. Science. 1995;268:661–667. doi: 10.1126/science.7732374. [DOI] [PubMed] [Google Scholar]
- 6.Bent A F, Kunkel B N, Dahlbeck D, Brown K L, Schmidt R, Giraudat J, Leung J, Staskawicz B J. Science. 1994;265:1856–1860. doi: 10.1126/science.8091210. [DOI] [PubMed] [Google Scholar]
- 7.Mindrinos M, Katagiri F, Yu G-L, Ausubel F M. Cell. 1994;78:1089–1099. doi: 10.1016/0092-8674(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 8.Grant M R, Godiard L, Straube E, Ashfield T, Lewald J, Sattler A, Innes R W, Dangl J L. Science. 1995;269:843–846. doi: 10.1126/science.7638602. [DOI] [PubMed] [Google Scholar]
- 9.Innes R W, Bent A F, Kunkel B N, Bisgrove S R, Staskawicz B J. J Bacteriol. 1993;175:4859–4869. doi: 10.1128/jb.175.15.4859-4869.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dangl J L, Ritter C, Gibbon M J, Mur L A J, Wood J R, Goss S, Mansfield J, Taylor J D, Vivian A. Plant Cell. 1992;4:1359–1369. doi: 10.1105/tpc.4.11.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamaki S, Dahlbeck D, Staskawicz B, Keen N T. J Bacteriol. 1988;170:4846–4854. doi: 10.1128/jb.170.10.4846-4854.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong X, Mindrinos M, Davis K R, Ausubel F M. Plant Cell. 1991;3:61–72. doi: 10.1105/tpc.3.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whalen M C, Innes R W, Bent A F, Staskawicz B J. Plant Cell. 1991;3:49–59. doi: 10.1105/tpc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debener T, Lehnackers H, Arnold M, Dangl J L. Plant J. 1991;1:289–302. doi: 10.1046/j.1365-313X.1991.t01-7-00999.x. [DOI] [PubMed] [Google Scholar]
- 15.Bisgrove S R, Simonich M T, Smith N M, Sattler A, Innes R W. Plant Cell. 1994;6:927–933. doi: 10.1105/tpc.6.7.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu G-L, Katagiri F, Ausubel F M. Mol Plant–Microbe Interact. 1993;6:434–443. doi: 10.1094/mpmi-6-434. [DOI] [PubMed] [Google Scholar]
- 17.Staskawicz B, Dahlbeck D, Keen N, Napoli C. J Bacteriol. 1987;169:5789–5794. doi: 10.1128/jb.169.12.5789-5794.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozak M. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chern M-S, Bobb A J, Bustos M M. Plant Cell. 1996;8:305–321. doi: 10.1105/tpc.8.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jefferson R A. Plant Mol Biol Rep. 1987;5:387–405. [Google Scholar]
- 21.Sakaguchi M, Tomiyoshi R, Kuroiwa T, Mihara K, Omura T. Proc Natl Acad Sci USA. 1992;89:16–19. doi: 10.1073/pnas.89.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klein P, Kanehisa M, DeLisi C. Biochim Biophys Acta. 1985;815:468–476. doi: 10.1016/0005-2736(85)90375-x. [DOI] [PubMed] [Google Scholar]
- 23.Whitham S, Dinesh-Kumar S P, Choi D, Hehl R, Corr C, Baker B. Cell. 1994;78:1101–1105. doi: 10.1016/0092-8674(94)90283-6. [DOI] [PubMed] [Google Scholar]
- 24.de Wet J R, Wood K V, Helinski D R, DeLuca M. Proc Natl Acad Sci USA. 1985;82:7870–7873. doi: 10.1073/pnas.82.23.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reuber T L, Ausubel F M. Plant Cell. 1996;8:241–249. doi: 10.1105/tpc.8.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritter C, Dangl J L. Plant Cell. 1996;8:251–257. doi: 10.1105/tpc.8.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence G J, Finnegan E J, Ayliffe M A, Ellis J G. Plant Cell. 1995;7:1195–1206. doi: 10.1105/tpc.7.8.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobe B, Deisenhofer J. Nature (London) 1993;366:751–756. doi: 10.1038/366751a0. [DOI] [PubMed] [Google Scholar]
- 29.Gopalan S, Bauer D W, Alfano J R, Loniello A O, He S Y, Collmer A. Plant Cell. 1996;8:1095–1105. doi: 10.1105/tpc.8.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pirhonen M U, Lidell M C, Rowley D L, Lee S W, Jin S, Liang Y, Silverstone S, Keen N T, Hutcheson S W. Mol Plant–Microbe Interact. 1996;9:252–260. doi: 10.1094/mpmi-9-0252. [DOI] [PubMed] [Google Scholar]
- 31.Huang H C, Lin R H, Chang C J, Collmer A, Deng W L. Mol Plant–Microbe Interact. 1995;8:733–746. doi: 10.1094/mpmi-8-0733. [DOI] [PubMed] [Google Scholar]
- 32.Rosqvist R, Hakansson S, Forsberg A, Wolf-Watz H. EMBO J. 1995;14:4187–4195. doi: 10.1002/j.1460-2075.1995.tb00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindgren P B, Peet R C, Panopoulos N J. J Bacteriol. 1986;168:512–522. doi: 10.1128/jb.168.2.512-522.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]