

Abstract
Current evidence supports the therapeutic potential of pharmacological interventions that counter the progression of genetic disorders by promoting regeneration of the affected organs or tissues. The rationale behind this concept lies on the evidence that targeting key events downstream of the genetic defect can compensate, at least partially, the pathological consequence of the related disease. In this regard, the beneficial effect exerted on animal models of muscular dystrophy by pharmacological strategies that enhance muscle regeneration provides an interesting paradigm. In this review, we describe and discuss the potential targets of pharmacological strategies that promote regeneration of dystrophic muscles and alleviate the consequence of the primary genetic defect. Regenerative pharmacology provides an immediate and suitable therapeutic opportunity to slow down the decline of muscles in the present generation of dystrophic patients, with the perspective to hold them in conditions such that they could benefit of future, more definitive, therapies.
Keywords: Regeneration, Muscular dystrophy, Inflammation, Signaling pathways, Chromatin
1. Introduction
Muscular dystrophies (MD) include more than 30 different inherited diseases, which are caused by mutations that affect distinct genes, yet all result in muscle degeneration, impaired locomotion and, in most cases, premature death (reviewed in Dalkilic and Kunkel, 2003; Davies and Nowak, 2006). The most common MD is the duchenne muscular dystrophy (DMD)—a lethal X-linked recessive disease that affects 1 in 3500 live male births and invariably leads patients to wheel chair and death within the second decade of life. In DMD patients, mutations of the dystrophin gene result in the complete absence or, very infrequently, in the expression of a truncated, non-functional protein. The dystrophin gene is the largest one in the human genome and the coded protein localizes at the sarcoplasmic surface of the sarcolemma, where it makes interactions with a number of other structural proteins through the C-terminal domain, to form the dystrophin-associated protein complex (DAPC). Essential components of the DAPC are the dystrophin–glycoprotein complex (DGC), which includes dystrophin, dystroglycans (DG) and sarcoglycans (SCG), and other associated proteins, such as a-dystrobrevin, syntrophins, neuronal nitric oxide synthase (nNOS), growth factor receptor-bound protein-2 (Grb2), caveolin-3 and sarcospan. In addition, enzymes (LARGE) and proteins with putative enzymatic activity (e.g. fukutin related protein—FKRP) interact with DGC and catalyze DG glycosylation. The DAPC provides an essential mechanical link between the intracellular cytoskeleton and the extra-cellular matrix, thereby ensuring the structural and functional integrity of skeletal muscles after contraction. Moreover, the presence of enzymatic and signal transduction proteins suggests that post-translational modifications (i.e. glycosylation) and proper activation of downstream cascades are integral activities of the DAPC. The absence of dystrophin compromises the integrity of the DAPC, leading to an increased vulnerability of myofibers to degeneration after contraction and perturbation of downstream signaling pathways. Likewise, gene mutations that cause the absence of other components of the DAPC lead to partially overlapping clinical and pathological features in different MDs. The heterogeneity of the clinical symptoms and the variability in disease progression observed among different forms of MDs suggest that different mutations might have a peculiar impact on muscle damage. Thus, the elucidation of the events downstream to the genetic mutations helps to understand the molecular pathogenesis of individual forms of MD and to identify key targets for therapeutic interventions.
Degeneration of dystrophic muscles is followed by a number of both detrimental and compensatory events, such as the calcium influx inside the cells, necrosis, and the reactive regeneration, which tends to counterbalance the muscle loss (Fig. 1). Regeneration of dystrophic muscles is considered a physiological response (Shi and Garry, 2006), whose beneficial impact is restricted to the early stages of the disease progression. As such, it contributes to delay the onset of the most dramatic signs of the disease in children. An exhaustion of the regeneration potential of dystrophic muscles correlates with the exacerbation of the clinical manifestations. These observations provide a strong rationale behind the attempts to promote a robust and persistent regenerative response of dystrophic muscles, as a valuable therapeutic intervention. Many regeneration-associated processes, such as inflammation and fibrosis, influence the ability of dystrophic muscles to effectively regenerate. Therefore, they provide potential targets of pharmacological strategies toward countering the disease progression (reviewed in Engvall and Wewer, 2003; Tidball and Wehling-Henricks, 2005). Ultimately, the most desirable strategy consists in selective interventions toward enhancing the inflammatory signals that promote regeneration, while inhibiting the inflammation-derived pathways that cause muscle necrosis and fibrosis—the most deleterious and perhaps irreversible outcome of MDs. Although strategies that block intracellular calcium influx or target other related events are promising therapeutic avenues, in this review we will focus on pharmacological interventions that boost the reactive regeneration of dystrophic muscles toward maintaining muscle integrity, in the absence of the correction of the genetic defect.
Fig. 1.
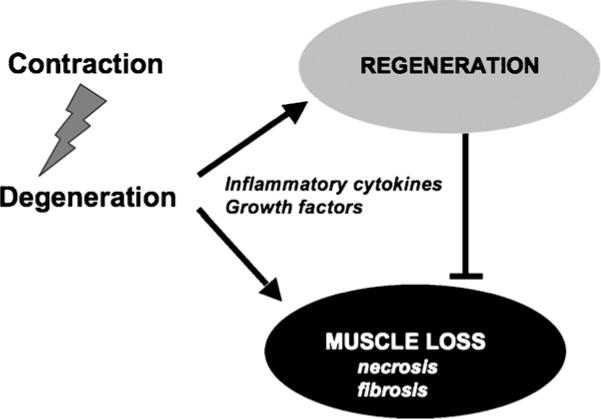
Following contraction-couple degeneration, reactive regeneration is stimulated by factors (cytokines, growth factors) released in the microenvironment and tends to counterbalance the muscle loss.
2. Muscle regeneration: regulatory networks and therapeutic targets
Regeneration of diseased or injured muscles occurs at expense of an heterogeneous population of resident muscle progenitor cells (mpc) and possibly other cells endowed with an inducible myogenic potential (Sherwood et al., 2004;reviewedin Péault et al., 2007).
2.1. Satellite cell-mediated muscle regeneration
Satellite cells are considered the prototypical mpc. Satellite cell number and regenerative capacity remain nearly constant through multiple cycles of injury and repair, suggesting that satellite cells can be renewed while maintaining the ability to regenerate myofibers. The stem cell nature of satellite cells was suggested by the ability of transplanted myofibers to generate donor-derived satellite cells that persisted in the host organism for several weeks and supported regeneration upon muscle injury, while repopulating the reserve satellite cell compartment (Collins et al., 2005). Recent evidence further revealed the heterogeneous nature of satellite cells, which include stem cell-like, self-renewing and differentiation-committed cells (Kuang et al., 2007; Cerletti et al., 2008). A functional hierarchy between these different sub-populations is established by the relative expression of genes (i.e. Pax7, MyoD and Myf5) that control satellite cell division and myogenic potential. Asymmetric division, which preferentially occurs within the satellite cell niches and is regulated by the Notch signaling (Conboy and Rando, 2002; Shinin et al., 2006), establishes the fate of satellite cells by generating self-renewing Pax7-positive/Myf5-negative populations, and differentiation-committed Pax7-negative/Myf5-positive cells (Kuang et al., 2007). Satellite cell partitioning in different cell types presumably regulates the balance between myofiber repair and maintenance of a pool of reserve cells. Since the functional exhaustion of satellite cells appears to limit the regenerative potential of diseased and aged muscles (Rando, 2006), one key challenge of regenerative medicine concerns the control of satellite cell division and transition from one population to another.
2.2. Other mpc populations
A number of resident mpc populations that are distinct from satellite cells have been also described. Among them, Pax7-negative/Pax3-positive interstitial mpc have been identified (Kuang et al., 2006; Relaix et al., 2006). Intriguingly, a recent report has showed that ectopic expression of Pax3 confers to embryonic stem cells the myogenic potential in vitro and in vivo (Darabi et al., 2008), suggesting that redundancy or interchangeability between Pax3 and Pax7 in mpc should be further explored. Other less defined progenitors derived from vessels, blood and bone marrow have been described and partially characterized (Péault et al., 2007). The functional and anatomical relationship between these non-satellite mpc and satellite cells is still unclear. It is unknown if these cellular populations represent sequential, and possibly reversible, stages of progression from one common progenitor to distinct populations of myogenic cells. Or if different cell types described so far derive from distinct precursors. Likewise, the actual contribution of non-satellite mpc to myofiber turnover and repair in physiological and pathological conditions, respectively, is unclear (LaBarge and Blau, 2002; Sherwood et al., 2004). However, these alternative sources of mpc could be exploited to support therapeutic strategies to regenerate diseased or aged muscles. An excellent example is provided by myogenic cells derived from blood vessels, such as embryonic mesoangioblasts (Minasi et al., 2002) and adult pericytes (Dellavalle et al., 2007), which can repopulate diseased muscles upon transplantation and provide an optimal platform for cell-mediated gene therapy in muscular dystrophies (Sampaolesi et al., 2003, 2006).
Future studies should define the pharmacological potential of manipulating the responsiveness to extracellular signals and reveal the intracellular pathways that regulate the activity and the myogenic potential of non-satellite mpc. These studies will hopefully establish if these cells might provide an alternative source of muscle precursors when satellite cells fail to support muscle repair. The complete understanding of the functional interactions between injury-activated events, such as inflammation, fibrosis, necrosis and locally released substances with paracrine/autocrine activity will be important to select candidate targets for interventions toward implementing muscle regeneration.
2.3. Biological rationale for therapeutic effectiveness of regeneration-based strategies
How can an increased regeneration from endogenous, dystrophin deficient, mpc have a therapeutic effect in MD? Shifting the equilibrium between muscle loss and repopulation is one obvious explanation. Indeed, it is consistently observed that just increasing the size of dystrophic muscles somehow protects them from contraction-coupled degeneration (Zammit and Partridge, 2002). Moreover, cytokines and growth factors released in the regenerative environment might also produce beneficial effect on adverse processes, such as fibrosis, necrosis and proteolysis of dystrophic muscles.
Thus, strategies that promote muscle regeneration can exert independent, beneficial effects in dystrophic muscles and delay the disease progression. Because of the hurdles that still prevent the application to dystrophic patients of gene- and cell-mediated therapies, pharmacological enhancement of regeneration provides a unique, immediate and suitable resource for the treatment of the current generation of dystrophic patients.
In the next paragraphs we will describe the most effective regenerative strategies that have been reported in animal models of muscular dystrophy. We will also discuss relevant targets of pharmacological interventions that promote regeneration in dystrophic muscles and the potential application of drugs that are already available or being tested in clinical trials, in the treatment of MDs.
3. Targeting myostatin and the TGFβ signaling
Myostatin or GDF-8 (growth and differentiation factor-8) is a member of TGF-β (transforming growth factor-beta) superfamily that is highly conserved among species (reviewed in Lee, 2004). Solid evidence indicates that myostatin is a potent, negative regulator of muscle growth during development and adult life. The physiological function of myostatin likely consists in limiting an excessive growth of skeletal muscles. Spontaneous mutations of myostatin have been originally detected in cattles (McPherron and Lee, 1997) and other animals displaying an excessive skeletal muscle development and myofibers stronger and with larger size than normal (Mosher et al., 2007). A mutation at the myostatin locus that leads to the absence of myostatin expression and an abnormal muscle growth has also been reported in a child (Schuelke et al., 2004). This “hypermuscular” phenotype has been replicated in mice either by genetic ablation of the myostatin gene (McPherron et al., 1997) or by pharmacological blockade of myostatin protein (reviewed in Lee, 2004). Importantly, inactivation of myostatin in dystrophic mice exerted beneficial effects on disease progression (Wagner et al., 2002; Bogdanovich et al., 2002), suggesting that myostatin is a primary target of pharmacological interventions in MDs. Since myostatin activity results from complex interactions with other members of the TGF-β superfamily, it is reasonable to extend this concept to the entire TGF-β signaling to muscle regeneration.
In the next paragraphs, we will illustrate the different levels of regulation of myostatin activity and will describe the successful applications of interventions targeting key interactions between myostatin and other TGF-β family members, which control muscle regeneration and other processes (i.e. fibrosis) that participate to the progression of MD.
3.1. Regulation of myostatin activity by extracellular factors
Members of the TGF-β family undergo a complex regulation at the level of biosynthesis. Myostatin is synthesized as a precursor, which is processed by furin proteases to generate a dimer composed by an N-terminal propeptide that is non-covalently bound to a 110 amino acid, biologically active C-terminal fragment (McPherron et al., 1997). This complex is secreted and circulates as an inactive, latent myostatin form (Lee and McPherron, 2001). One mechanism for activating myostatin consists of a proteolytic cleavage of the propeptide at the residue Asp76 by members of the bone morphogenetic protein-1/tolloid (BMP-1/TLD) family of metalloproteinase (Wolfman et al., 2003). The importance of propeptide proteolysis in the activation of latent myostatin is highlighted by experiments showing an increased muscle mass in transgenic mice over-expressing the propeptide (Lee and McPherron, 2001). Alternatively, the carboxyl-terminal is regulated in the extracellular environment through interactions with binding proteins, such as follistatin, follistatin related gene (FLRG), and Gasp-1 (Lee, 2004). Although each of these proteins can inhibit myostatin activity in vitro, only the inhibitory activity of follistatin has been demonstrated in vivo (Lee and McPherron, 2001; Zimmers et al., 2002). Consistently, follistatin knockout mice have reduced muscle mass at birth (Matzuk et al., 1995), and transgenic expression of follistatin produced a hypermuscular phenotype that resembles that of myostatin knock out (Nakatani et al., 2007). Notably, transgenic expression of follistatin in myostatin knock out mice produces an additive effect (a quadruplication of muscle mass, in comparison to the duplication observed in myostatin knock-out mice) (Lee, 2007). This suggests that follistatin can regulate muscle mass by mechanism(s) that are independent on myostatin inactivation.
3.2. Myostatin intracellular signaling
The unbound C-terminal myostatin dimer binds to the activin type II receptors, ACVR2A and ACVR2B and elicits an intracellular signaling pathway that is typical of members of the TGFβ family (Lee, 2004). Myostatin/activin type II receptor complex engages type I receptors, such as ALK4 and/or ALK5, leading to phosphorylation of downstream signaling components—the R-Smads, Smad2 and Smad3. Phosphorylated Smad2 and 3 associate with co-Smad, Smad4, and enter the nucleus to regulate transcription of downstream genes (Lee et al., 2005). Importantly, the identity of the genes that mediate myostatin effects on muscle regeneration is still unknown.
3.3. Strategies of myostatin blockade in the therapy of muscular dystrophies
The pharmacological strategies to block myostatin signaling in adult muscles are mostly based on the mechanism of regulation of myostatin biosynthesis and activity described above. Compounds capable of binding and inhibiting the C-terminal dimer have been the focus of the most recent and successful strategies. Targeting of the C-terminal dimer by neutralizing monoclonal antibody (JA16) resulted in the increase of muscle mass and function in wild type mice (Whittemore et al., 2003) and could rescue the pathological phenotype in dystrophin-deficient mdx mice (Bogdanovich et al., 2002). This latter study showed the first evidence that myostatin blockade in dystrophic mice increased the myofiber size and alleviated signs and symptoms of the disease, such as decline in strength, fiber susceptibility to degeneration and fibrosis. This original observation provided the impetus for the evaluation of the monoclonal antibody, MYO-029, in a trial with patients with muscular dystrophy that was recently published (Wagner et al., 2008). This study reported on the safety of the molecule, but did not demonstrate any sign of clinical improvement in the patients treated with MYO-029. It is possible that the selection of dystrophic patients at late stage of the disease – that is, when the regenerative response is exhausted – eliminated the myostatin substrate (e.g. regenerating muscle progenitors) and therefore preclude any appreciable effect of myostatin blockade. Inactivation of myostatin with a propeptide fused to an Fc domain, which enhances stability in vivo, also alleviated the signs of disease when injected into mdx mice (Bogdanovich et al., 2005). Similar results were observed by genetic ablation of myostatin in mdx mice, obtained by breeding myostatin knock-out and mdx mice (Wagner et al., 2002). However, the rescue of the pathological phenotype in this experimental setting was less evident, when compared to that described by Bogdanovich and colleagues. It is unclear the reason for such discrepancy, although it might depend on the different timing of myostatin blockade (during embryogenesis vs. adult life), on the magnitude of myostatin inactivation and the possibility that anti-myostatin antibodies could affect other pathways. Indeed, the injection of the soluble form of the ACVRIIB receptor fused to an Fc domain (ACVRIIB-Fc), which was designed to block myostatin activity, led to an increased muscle growth in myostatin-deficient mice (Lee et al., 2005), suggesting an action through a myostatin-independent pathway. Likewise, inactivation of myostatin by either transgenic expression of follistatin (Nakatani et al., 2007)orby single injection of adeno-associated viral (AAV) vector that delivered a follistatin-splicing variant (FS-344) (Haidet et al., 2008) increased muscle mass and strength, and reduced the histological signs of disease in dystrophic mice. Moreover, increased levels of follistatin appear to mediate the beneficial effects of two independent therapeutic strategies in mdx mice—one based on the deacetylase inhibitor delivery (Minetti et al., 2006) and the other on nitric oxide release (Brunelli et al., 2007) (see following sections).
Collectively, these data indicate that interactions between myostatin, follistatin and possibly other members of the TGF-beta pathway provide a valuable target for pharmacological treatments of DMD.
The inhibition of myostatin was also effective in alleviating the pathological phenotype of caveolin 3-deficient mice (a model of LGMD 1C) (Ohsawa et al., 2006), but not in a mouse model of lamin-deficient muscular dystrophy, which caused a more severe disease phenotype (Li et al., 2005). Likewise, antibody-mediated inactivation of myostatin failed to revert the pathological phenotype of aged δ-sarcoglycan null mice (Parsons et al., 2006). The lack of effect of myostatin inactivation in lamin-deficient and δ-sarcoglycan null mice correlates with the reduced regeneration and the more severe fibrosis observed in these animals. Once again, this suggests that myostatin blockade could be effective only at early stages of disease progression. However, differences in experimental conditions and distinct types of muscular dystrophies could also explain these results.
3.4. Effect of myostatin blockade on muscle regeneration
How does blockade of myostatin signaling interfere with the progression of muscular dystrophy? Myostatin signaling regulates both muscle regeneration and fibrosis, which are two interdependent processes. The data reported in the sections above show a correlation between the enhanced muscle regeneration and the reduced fibrosis, upon myostatin blockade in young dystrophic mice. Instead, an impaired ability to regenerate fibrotic muscles at late stages of the disease coincides with the therapeutic failure of myostatin blockade.
The negative impact of myostatin on muscle regeneration is well documented. The widespread increase in skeletal muscle mass displayed by myostatin null mice results from the combination of muscle cell hyperplasia and hypertrophy during development (McPherron et al., 1997). However, myostatin action is not limited to embryonic development, as myostatin regulates muscle mass also in adult animals (Lee and McPherron, 2001; Grobet et al., 2003). Given the strong analogies in the molecular mechanisms regulating developmental myogenesis and muscle regeneration in adult life (Snider and Tapscott, 2003), it is tempting to speculate that myostatin signaling targets conserved effectors of these processes. Indeed, recent studies revealed that excess of myostatin downregulates the expression of Pax-3, Pax7, MyoD and Myf5 (Amthor et al., 2002; McFarlane et al., 2008). Receptors for myostatin are found on numerous muscle cell lines, and the effects on muscle fiber number probably result from blockade of muscle cell proliferation and differentiation (Lee, 2004). Studies with cultured myoblasts showed that elevated concentrations of active myostatin inhibits mpc proliferation by up-regulating the cyclin-dependent kinase inhibitor p21 (Waf1/Cip1) and leading to the accumulation of unphoshorylatyed, active pRb (Thomas et al., 2000). Studies with satellite cells showed the inverse patterns of myostatin and follistatin levels in quiescent vs. activated satellite cells, with follistatin/myostatin ratio increasing during satellite cells activation (McCroskery et al., 2003). Taken together, these and other data support the notion that myostatin inhibits both proliferation and differentiation potential of satellite cells. Interestingly, the increased regeneration observed in myostatin null mice did not decline along the life span. And aged myostatin-deficient mdx mice, which have undergone multiple degeneration/regeneration cycles, continued to maintain an enhanced regenerative response and an increased muscle mass (Wagner et al., 2005). This evidence indicates that the absence of myostatin counters satellite cell exhaustion to regenerate diseased or aged muscles, and indirectly suggests an effect of myostatin on symmetric division of satellite cells to maintain a reserve pool.
The interest on therapeutic blockade of myostatin extends to the possibility to increase the size and the strength of muscles in atrophic conditions. However, recent studies reported on the excessive muscle growth but impaired force generation in two independent mouse lines that harbor mutations in the myostatin gene, constitutive null (myostatin−/−) and compact (Berlin High Line, BEHc/c) mice (Amthor et al., 2007. These data are in conflict with the evidence that mammals with spontaneous mutation of the myostatin gene show enhanced muscle performance (Mosher et al., 2007; Schuelke et al., 2007). However, it should be noted that the therapeutic benefit of myostatin inactivation in dystrophic muscles would derive from a decreased susceptibility to degeneration of bigger myofibers (Zammit and Partridge, 2002), rather than from an increased force of contraction.
3.5. TGF-β-mediated regulation of muscle regeneration
It is becoming progressively clear that the TGF-β network signaling profoundly influences proliferation and differentiation of mpc. The relative expression of the TGF-β family members (BMP4, gremlin, activin) and receptors in human mpc (side population and main population) and myofibers establishes a regulatory network that reciprocally control cell proliferation in a paracrine fashion (Frank et al., 2006). This network is implicated in the regulation of the number of mpc available for sequential waves of regeneration and is therefore a potential target for interventions aimed at ensuring long-lasting efficacy of regeneration. Quite surprisingly, the impact of TGF-β on the symmetry of mpc cell division has not been yet addressed. Furthermore, members of TGF-β family are known inhibitors of terminal differentiation of muscle cells in vitro (Liu et al., 2001), and increased levels of TGF-β in vivo limit regeneration of injured muscle by inhibiting satellite cells proliferation and differentiation (Cohn et al., 2007). In resident satellite cells of aged animals, excessive levels of TGF-β induce high pSmad3, which antagonizes Notch signaling by activating the expression of cyclin-dependent kinase inhibitors (cdks) (Carlson et al., 2008). Moreover, TGF-β signaling regulates fibrosis in different tissues, including muscles. Consistent with the importance of the TGF-β signaling in regulating both regeneration and fibrosis, Cohn et al. showed that blockade of the angiotensin II receptor 1 (AT1) with Losartan ameliorates the pathological phenotype in dystrophic mice via the inhibition of the TGF-β signaling (Cohn et al., 2007). In this study, both improvement in muscle regeneration and decreased fibrosis were observed in mdx mice after prolonged exposure to Losartan.
3.6. Future challenges
Future studies should optimize pharmacological strategies to maximize the benefits deriving from myostatin blockade or from manipulation of the TGF-β network, and to identify the profile of dystrophic patients suitable for such an effect. This can be achieved by the complete elucidation of the mechanism by which myostatin regulates muscle regeneration and fibrosis. It is possible that the modulation of the myostatin/follistatin pathway, and more in general the TGFβ network, could have an independent impact on different parameters, such as mpc activity, inflammation and fibrosis. While the combination of these effects might result in a global positive impact on regeneration, it would be interesting to identify individual pathways that can selectively improve specific disease features—i.e. muscle fibrosis in older patients. The discovery that increased levels of follistatin produce beneficial effects in dystrophic muscles both through myostatin blockade and via myostatin-independent pathways, suggests that strategies that upregulate follistatin might have a stronger therapeutic potential than selective myostatin inhibitors. Mouse models of dystrophies that show resistance to myostatin blockade should be exploited to address this issue.
Additionally, it will be critical to develop methods that assess the magnitude of myostatin inhibition in muscles, to monitor the treatment effectiveness. Possibly, the development of small molecules inspired by the mechanism of myostatin blockade might help to increase the selectivity of the treatment.
4. Manipulation of inflammation-activated pathways
Although dystrophin mutations represent the primary cause of DMD, the inflammation that develops in dystrophic muscles exacerbates the pathology and contributes to fibrosis and necrosis. However, the abundant cellular infiltrate typically found in dystrophic muscles also provides the source of cytokines and other paracrine factors that promote regeneration. Thus, it is important to precisely discriminate between the inflammation-activated pathways that exert a positive or negative effect on regeneration.
The deleterious effect of inflammatory pathways on muscular dystrophy is supported by the beneficial effects of anti-inflammatory steroids (prednisone or deflazacort), in mouse models of disease and in dystrophic children (Muntoni et al., 2002). However, it is unclear if the inhibition of inflammation completely accounts for the beneficial effects of steroids in dystrophic patients. Furthermore, definitive evidence for a positive role of steroids on muscle regeneration is lacking. Overall, the severe side effects caused by chronic exposure to steroids limits their therapeutic usefulness and raise a special interest toward developing strategies that target specific pathways activated by inflammation in dystrophic muscles.
4.1. NF-kB inhibition: multiple targets and effects
Recent evidence points to the inflammation-activated NF-kB pathway as a selective target of therapeutic interventions. It is conceivable that the beneficial effect of steroids in the treatment of DMD might be, at least partly, due to NF-kB inhibition, leading to downregulation of NF-kB downstream targets, such as cytokines and adhesion molecules that amplify the inflammation process.
NF-kB consists of five members, RelA (p65), RelB, c-Rel, p50 and p52, which activate transcription by forming nuclear dimeric complexes in different combinations. In unstimulated cells, NF-kB members are retained in the cytoplasm through binding of the inhibitor protein (IkB). Upon stimulation by pro-inflammatory cytokines, the IkB kinase (IKK) complex, which contains catalytic subunits, IKKα and IKKβ, and the regulatory subunit, IKKγ/NEMO, phosphorylates IkB, leading to its degradation by the 26S proteasome and nuclear translocation of NF-kB members (Häcker and Karin, 2006). Post-translational modifications such as direct phosphorylation, acetylation and ubiquitination of NF-kB members and non-canonical activatory pathways can also regulate NF-kB-dependent transcription (Perkins, 2006).
Perturbation of NF-kB signaling was shown in immune cells and myofibers of dystrophic mice and patients (Acharyya et al., 2007). Consistently, ablation of one single allele of p65 could improve muscle pathology and other signs of disease in mdx mice. Furthermore, conditional deletion of Ikkβ in macrophages reduced the inflammation and necrosis in muscles of mdx mice, and deletion of Ikkβ in myofibers of mdx mice had a positive effect on regeneration. Collectively, these data support the concept that inhibition of NF-kB is a valuable strategy for the treatment of muscular dystrophy, an evidence further substantiated by the beneficial effect shown by a soluble inhibitor of IKK in mdx mice (Acharyya et al., 2007). However, it is unclear if the therapeutic action of NF-kB inhibition in dystrophic muscles relies only on the anti-inflammatory effect, or also includes derepression of NF-kB-mediated inhibition of the myogenic program in mpc. Future experiments of conditional inactivation of NF-kB in satellite cells should address this issue.
4.2. Targeting TNFα downstream pathways
Among the variety of pro-inflammatory genes induced by NF-kB, TNF-α is of special interest in MDs. TNF-α levels are up-regulated in dystrophic muscles from animal models and DMD patients (Porter et al., 2002). In turn, TNF-α is a potent NF-kB inducer, contributing to a positive feedback loop that perpetuates the negative effects of the activation of NF-kB signaling in dystrophic muscles. Pharmacological blockade of TNF-α activity in mdx mice with anti-TNF-α antibodies reduced muscle necrosis and ameliorated the histological profile of dystrophic muscles (Radley et al., 2008). Given the multitude of TNF-α activated pathways (i.e. NF-kB, JNK, p38, PW1) and the different impact they have on myoblast differentiation in vitro and on muscle regeneration in vivo, it is difficult to assign to TNF-α blockade a specific role in the recovery of dystrophic muscles. The ambiguous effect of TNF-α signaling in muscular dystrophy is well illustrated by the phenotype of TNF-α receptor p55(−/−)p75(−/−) double knockout mice, which show deficient muscle regeneration (Chen et al., 2005). The same authors demonstrated that production of TNF-α from injured muscles activates the p38 signaling—an essential event for mpc differentiation into myofibers (Forcales and Puri, 2005). In support of a positive role played by the inflammatory cells recruited to dystrophic muscles is also the finding that deficiency of urokinase plasminogen activator (uPA) exacerbates the dystrophic phenotype in mdx mice, by preventing muscle infiltration of macrophages (Suelves et al., 2007). The beneficial effect of specific inflammation-activated pathways on the regeneration of dystrophic muscles should be taken into consideration in the search for selective anti-inflammatory drugs in the treatment of DMD patients.
In this regard, Brunelli et al. recently explored in dystrophic mice the effect of combination of anti-inflammatory agents with the activation of nitric oxyde (NO) pathway (Brunelli et al., 2007), which promotes mpc regeneration via follistatin upregulation in mpc (Pisconti et al., 2006). Previous work demonstrated that the NO signaling is severely impaired in dystrophic muscles, and that restoration of NO improves the pathological phenotype of mdx mice (Wehling et al., 2001). A derivative of flurbiprofene – HCT 1026 – which combines the pro-myogenic effect of NO with nonsteroidal anti-inflammatory activity – improved morphological, biochemical and functional pathological features in mdx and α-sarcoglycan null mice. Additionally, HCT 1026 enhanced engraftment of mesoangioblasts injected in these mice, indicating that pharmacological strategies can be used to support cell-based therapies in the treatment of muscular dystrophies (Brunelli et al., 2007).
4.3. The molecular interface between inflammation and fibrosis
The close relationship between inflammation and fibrosis indicates that the investigation of the molecular events regulating these processes might open new avenues in the treatment of MDs.
A recent discovery sheds new light on the molecular link between inflammation and fibrosis by showing that fibrinogen-Mac-1 receptor binding promotes the synthesis of TGFβ in mdx macrophages, via induction of IL-1β. TGFβ in turn induces collagen production in mdx fibroblasts (Vidal et al., 2008). The search for agents that simultaneously modulate the immune response and muscle fibrosis appears of key interest for the development of regenerative strategies.
5. Boosting the IGF1 pathway
Insulin-like growth factor I (IGF-I) is a key regulator of skeletal muscle development and post-natal growth. In adult muscles, IGF1 stimulates both muscle fiber hypertrophy and proliferation of mpc by distinct intracellular pathways. IGF1 exists as systemic, circulating hormone and as locally released factor with paracrine activity. Neuro-hormonal changes control the levels of systemic IGF1, while tissue and organ-specific cues regulate the expression of local IGF1 isoforms (reviewed in Mourkioti and Rosenthal, 2005).
A first level of IGF1 regulation is provided by extra-cellular IGF ligands – the IGF-binding proteins (IGFBPs) – which form binary complex with IGF1. Seven IGFBPs have been identified so far, some of them exerting positive and others negative effects (Juul, 2003). In circulation, 95% of free IGF1 is bound to ternary complex consisting of IGF1, IGF1-BP3, and acide labile subunit. A small percentage of free IGF-1 is found in circulation to activate muscle through its receptor.
5.1. IGF1 signaling in muscles
Most of the information on IGF1 function as an anabolic factor derives from the knowledge gained on the receptor-activated intracellular signaling.
IGF1-receptor binding stimulates downstream tyrosine kinases, which in turn activate a signaling network toward multiple cellular responses, including myoblast proliferation and survival, differentiation, muscle fiber hypertrophy and metabolic responses (reviewed in Rotwein, 2003 and in Mourkioti and Rosenthal, 2005). One critical upstream element of the IGF1 pathway is the phosphatidylinositol 3-kinase (PI3K), which activates serine/threonine kinases AKT 1 and 2, from which the signal diverges to regulate distinct cascades involved in specific responses, via AKT-mediated phosphorylation of downstream substrates. AKT-mediated activation of mammalian target of Rapamycin (mTOR) and inhibition of glycogen synthase kinase (GSK3) promote hypertrophy by stimulating protein synthesis; AKT-mediated phosphorylation of FOXO (Forkhead box-containing protein, O-subfamily) contributes to the hypertrophic effect, by inhibiting the activation of E3-ubiquitin ligases, which trigger protein degradation; AKT-dependent activation of Bcl2 and p21 expression supports the survival effect of IGF1. Recently, the AKT-mediated phosphorylation of p300/CBP acetyltransferases was reported to recruit these chromatin-modifying enzymes to MyoD-responsive loci, thereby promoting local hyper-acetylation and muscle gene transcription (Serra et al., 2007). Additional pathways elicited by the IGF1-receptor include ERK activation, which promotes proliferation, the calcium-dependent activation of calcineurin, which contribute to muscle hypertrophy, and the de-repression of MEF2-dependent transcription (Musarò et al., 1999).
Increased serum concentration of IGF1, overexpression of IGF1-downstream effectors in muscle cultures or muscle-restricted expression of IGF1 in transgenic mice invariably result in a hypertrophic muscle phenotype (Shavlakadze et al., 2005). It is likely that this phenotype results from the combined activation of multiple IGF1 downstream pathways. However, it is clear that enhanced regeneration via recruitment of mpc and increased protein synthesis in myofibers independently contribute to the hypertrophic phenotype induced by the muscle-specific IGF1 transgene. Recently, the ability of IGF1 to modulate the population of inflammatory cells and the cytokines released in the regenerative environment has been reported (Pelosi et al., 2007). This study links the immuno-modulatory activity of IGF1 with its ability to enhance regeneration and decrease fibrosis in injured muscles.
5.2. Pharmacological manipulation of IGF1 pathway in the treatment of muscular dystrophies
The beneficial effect of IGF1 over-expression in dystrophic muscles was shown by the seminal work of Barton et al. reporting on the amelioration of the dystrophic phenotype in transgenic MDX mice expressing muscle-specific IGF1 (Barton et al., 2002). These mice showed increased muscle mass and force, and decreased signs of diseases, such as fibrosis and necrosis. Importantly the fibrosis and myonecrosis in the diaphragm, which are typically associated to aged mdx mice, were reduced in mdx IGF1 transgenic mice. This evidence has inspired several IGF1-based interventions for the treatment of muscular dystrophy, via distinct routes of IGF1 administration. Of note, the ability of IGF1 to antagonize the atrophic process elicited by steroids in myofibers, by blockade of Foxo-dependent induction of E3-ubiquiting ligases (Sandri et al., 2004), could be used to reduce one of the more severe side effects of these drugs in dystrophic patients.
5.3. Current challenges and future perspective for IGF1 therapy of DMD
A number of issues need to be addressed before the translation of IGF1 therapy in the clinical practice for DMD treatment. First of all, it is unknown the effect of long-term treatment with IGF1. Second, it is unclear if the IGF1 isoforms act on different receptors and what type of receptor is preferentially expressed in quiescent and activated satellite cells. Likewise, the response of non-satellite mpc to IGF1 has not been elucidated. Furthermore, the influence of IGFBPs on therapeutic administration of IGF1 and the optimal dose of IGF1 administration at different stages of MDs are still unknown. Once these and other aspects of IGF1 signaling will be elucidated, it is possible that undesired side effects on metabolic parameters and the risk of oncogenic events associated with IGF1 administration could be eliminated.
Interestingly, although increased levels of IGF1 and myostatin blockade exert both the same effect on muscle fibers (increased regeneration and hypertrophy) the underlying mechanisms seem to be different. Future studies should address possible interactions between these two pathways and potential synergistic effects in the therapy of neuromuscular disorders.
6. Chromatin targeting by epigenetic drugs
Despite the identification of candidate target pathways for pharmacological enhancement of muscle regeneration in the treatment of MD, a major limitation in the manipulation of these pathways consists in the lack of selectivity. For example, blockade or activation of intracellular signaling pathways at the receptor or cytosolic level often influences multiple downstream pathways and results in undesired side effects. Conceivably, the identification of chromatin targets of individual signaling pathways would restrict the effect of pathway modulation to specific subsets of genes. Thus, a major challenge in regenerative medicine is to understand how the cues released in the regeneration environment are converted into the epigenetic changes that regulate expression of particular genes. These studies hold the promise to reveal selective targets for direct manipulation of mpc at the chromatin level. Since most of the epigenetic changes that control gene expression are reversibly regulated by chromatin-modifying enzymes, a key task is to elucidate the mechanism by which intracellular pathways control the function of these enzymes.
6.1. Chromatin signaling regulating muscle regeneration
Muscle regeneration results from the proliferation of mpc that differentiate into mature myofibers. A dramatic reprogramming of mpc underlies the sequential activation and repression of genes involved in lineage determination and maintenance, proliferation, migration, morphological and biochemical differentiation (Palacios and Puri, 2006). As such, the most desirable strategy to implement muscle regeneration should consist in the possibility to independently manipulate each of these stages, with the final goal of achieving the highest number of differentiation-competent mpc, while maintaining the integrity of the reserve pool of mpc. This goal can be, in theory, achieved by reversibly targeting specific subsets of genes that regulates individual stages of regeneration. In this perspective, it is important to elucidate the signaling that imparts to the chromatin of mpc the epigenetic modifications responsible for repression or activation of different subsets of genes.
The activation of the myogenic program by muscle bHLH proteins (MyoD, Myf5, myogenin and MRF4) and MEF2 family factors (MEF2A, B, C and D) entails the sequential displacement and recruitment of chromatin-modifying enzymes, which determine the chromatin conformation repressive or permissive for transcription (reviewed in Puri and Sartorelli, 2000; Tapscott, 2005; and illustrated in Fig. 2). Histone acetylation – a modification that promotes gene expression – is induced by the recruitment of histone acetyltransferases p300/CBP and PCAF by MyoD and MEF2 proteins, and correlates with the transcription of differentiation genes. Moreover, acetylation of MyoD (and possibly other muscle bHLH proteins) and MEF2 factors contributes to stimulate transcription of target genes (Palacios and Puri, 2006). Conversely, the unscheduled expression of differentiation genes during myoblast proliferation is prevented by the interactions between histone deacetylases (HDACs) with MyoD and MEF2 members (McKinsey et al., 2002). Thus, the balance between acetylation and deacetylation is a critical determinant of muscle gene transcription and might be considered a valuable target of interventions toward manipulating mpc ability to sustain muscle regeneration. Recent studies have shown that additional chromatin-modifying enzymes are recruited to the chromatin of muscle genes to activate gene transcription. In particular, the recruitment of SWI/SNF chromatin remodeling complexes is necessary for the activation of the differentiation program in muscle cells (de la Serna et al., 2001).
Fig. 2.
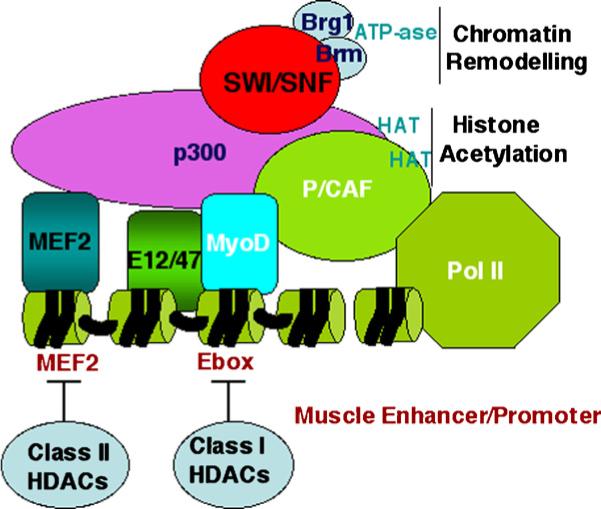
Schematic representation of the components of the myogenic transcriptosome. The activity of muscle-regulatory factors (myogenic bHLH and MEF2 proteins) is controlled by different chromatin-modifying enzymes.
How is the recruitment of chromatin-modifying enzymes directed in response to external cues? Recent evidence indicates the importance of extracellular signal-activated pathways in regulating the assembly of these enzymes into MyoD- and MEF2-associated complexes (reviewed in Forcales and Puri, 2005). A signal-dependent dissociation of class II HDAC-MEF2 interactions occurs via HDAC phosphorylation (McKinsey et al., 2002). The kinase responsible for this event has not been clearly identified, although Berdeaux et al. reported on the HDAC5 phosphorylation by the salt inducible kinase (SIK1) as a potential mechanism to directly activate MEF2 activity (Berdeaux et al., 2007). The recruitment of acetyltransferases and SWI/SNF chromatin remodeling complex into the myogenic transcriptosome is coordinated by two parallel, pro-myogenic cascades, the p38 pathway and the IGF1 signaling. p38 kinases alpha and beta phosphorylate multiple components of the myogenic transcriptosome, including MEF2A and C, the MyoD heterodimerization partner E47 and the structural component of the SWI/SNF complex, BAF60 (Wu et al., 2000; Simone et al., 2004; Rampalli et al., 2007; reviewed in Lluis et al., 2006). These phosphorylations link the activation of the p38 signaling with the hetero-dimerization of MyoD and E2A proteins, the chromatin recruitment of SWI/SNF and MLL-1 methyltransferase-containing complex (Tritorax), which in turn establish the epigenetic conditions for gene transcription. IGF1-activated AKT1 and 2 kinases promote interactions between MyoD and the acetyltransferases p300 and PCAF, by phosphorylating two serines located at the C/H3 region of p300 (Serra et al., 2007). These data indicate that the simultaneous activation of the p38 and IGF1/AKT pathways stimulates the assembly of the muscle transcriptosome. Notably, while these two pathways converge on the chromatin of muscle genes, they also have independent effects on cell proliferation—p38 pathway induces cell cycle arrest and IGF1 conveys mitogenic signals (Fig. 3). Thus, selective interruption of these pathways at the chromatin level might be used to separate distinct cellular activities and generate population of mpc particularly suitable for regeneration—i.e. proliferating mpc with pre-assembled transcriptosome at muscle loci (Serra et al., 2007).
Fig. 3.
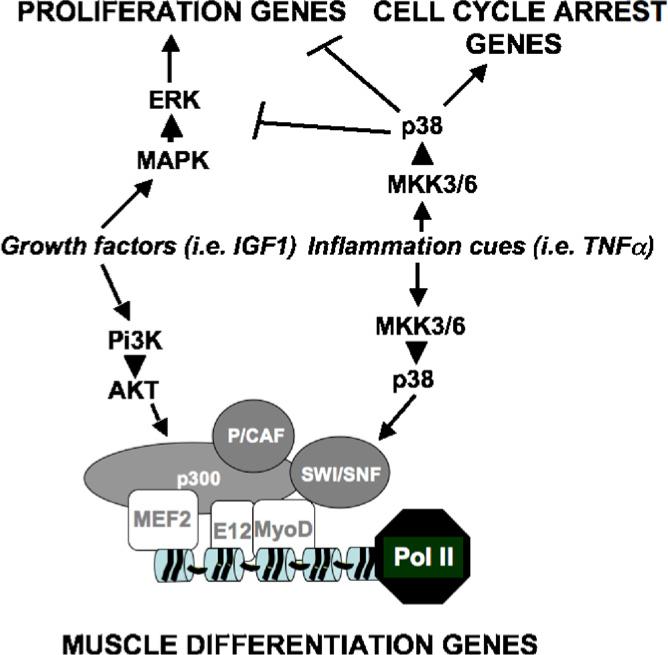
Pi3K-AKT and p38 signaling pathways elicited by factors released in the regenerative environment converge on the chromatin of muscle genes, but have independent effects on the cell cycle in mpc. Conceivably, targeting the chromatin interface by which these pathways regulate subsets of genes involved in different biological activity might produce optimal pharmacological effects—i.e. expansion of mpc with the chromatin poised for activation of muscle gene expression.
6.2. Regulation of muscle regeneration by epigenetic drugs
The availability of compounds that inhibit HDACs (deacetylase inhibitors) prompted an interest toward manipulating skeletal myogenesis by targeting the acetylation of histone and non-histone (e.g. MyoD, MEF2) proteins. The general assumption that HDAC inhibition would indiscriminately cause a global hyperacetylation in treated cells is challenged by the cell specific distribution of HDACs along the genome at various stages of skeletal myogenesis. In undifferentiated myoblasts HDAC distribution is, at least partly, determined by the interactions with MyoD and MEF2 proteins, whereas in myotubes the increased affinity of HDAC for components of co-repressor complexes (i.e. pRb) leads to redistribution to the chromatin of cell cycle genes (Puri et al., 2001; McKinsey et al., 2002). This concept inspired experiments that demonstrated a stage specific effect of deacetylase inhibitors in muscle cells; only undifferentiated myoblasts that were exposed to deacetylase inhibitors and subsequently cultured in conditions permissive for differentiation showed a dramatic enhancement of differentiation leading to formation of hypernucleated myotubes with an increased size (Iezzi et al., 2002). Accordingly, only specific subsets of genes were induced by these compounds in treated myoblasts in vitro (Iezzi et al., 2004). And in vivo experiments showed an effect of deacetylase inhibitor only in regenerating muscles of treated mice (Iezzi et al., 2004; Minetti et al., 2006). Among the target genes identificated in myoblasts, the myostatin antagonist, follistatin, is an essential mediator of deacetylase inhibitor ability to promote muscle regeneration (Iezzi et al., 2004). This evidence established an indirect link between deacetylase inhibitors and myostatin blockade, and inspired experiments testing the effect of deacetylase inhibitors on dystrophic mice. Systemic delivery of deacetylase inhibitors in young (1.5-month-old) MDX and alpha-sarcoglycan null mice increased the myofiber size and conferred resistance to contraction-coupled degeneration. This effect correlated with follistatin upregulation and increased ability of mpc to form myotubes ex vivo, and was eliminated by RNAi-mediated downregulation of follistatin. Importantly, muscles from treated mice showed increased force, drastic reduction of fibrosis, cellular infiltrate and normal architecture, as compared to muscle of untreated dystrophic mice (Minetti et al., 2006). The interpretation of this effect was that deacetylase inhibitor-mediated upregulation of follistatin enhanced the ongoing regeneration in dystrophic muscles, via myostatin antagonism, leading to increased size and resistance to degeneration. This is the first example of a beneficial effect in dystrophic muscles treated with drugs targeting epigenetic events.
The increasing knowledge on the mechanisms that regulate reversible epigenetic modifications that control subsets of genes involved in specific stages of muscle regeneration holds the promise to develop more specific epigenetic drugs for the treatment of MDs. Likewise, the elucidation of the regulatory networks that control the chromatin modifications underlying determination of the myogenic lineage and maintenance of the differentiated phenotype in myofibers might lead to the development of pharmacological strategies to regenerate diseased muscles from stem cells or from myofibers.
6.3. Future challenges
Deacetylase inhibitors are already available in the clinical practice, and are therefore attractive drugs for an immediate translation into clinical trial for the treatment of dystrophic children. At the same time a number of questions related to their mechanism of action remain unanswered. It is unlikely that their effect relies only on follistatin upregulation; however, the identification of additional genes implicated in their therapeutic effect is a key task. Likewise, knowing the identity of the mpc sub-population(s) that mediate such an effect and their availability in long-term treatment might increase selectivity and long-term effectiveness of deacetylase inhibitors in the treatment of MDs. Additional studies should also establish the molecular basis by which deacetylase inhibitors, which increase histone acetylation non-specifically, selectively regulate gene expression in specific cellular sub-types.
7. Future prospects
The increasing knowledge on the cellular effectors of muscle repair and the epigenetic signals that regulate the transcription of genes involved in key regeneration stages will possibly inspire new strategies for selective gene manipulation in mpc.
The interventions that implement muscle regeneration appear particularly applicable to patients at early stages of muscular dystrophies, when muscles are in the regenerative stage. In patients at later stages of disease progression, secondary events, such as collagen deposition, fibrosis, sclerosis and fat infiltration, preclude an efficient regeneration and limit the success of cell transplantation. A future challenge will be the identification of complementary strategies that restore either the regeneration ability or the efficiency of cell transplantation in muscles of patients at advanced stages of disease progression. A recent work shows that co-expression of an angiogenic factor (placenta growth factor—PIGF) and a metalloproteinase (matrix metalloproteinase-9—MMP-9) restore the vascular network, reduce collagen deposition and allow efficient cell therapy in muscles of aged dystrophic mice (Gargioli et al., 2008). Likewise, follistatin delivery ameliorates the phenotype of old dystrophic mdx mice, in which fibrosis has developed (Haidet et al., 2008) and improves the efficiency of stem cell-mediated regeneration of dystrophic mice (Benabdallah et al., 2008). These findings further emphasize the notion that follistatin is a versatile therapeutic target. The identification of the endogenous source (i.e. specific cell type) of follistatin within dystrophic muscles and deciphering the “epigenetic signature” that confer to follistatin locus the selective responsiveness to therapeutic interventions are key challenges of future studies. This concept should be extended to other candidate target genes, to establish a global epigenetic network that underlies successful therapeutic strategies
Acknowledgments
PLP is an Associate Telethon Scientist of Dulbecco Telethon Institute. CM and GM are supported by fellowships from Parent Project Italy and AFM.
Abbreviations
- MD
muscular dystrophies
- DMD
duchenne muscular dystrophy
- DAPC
dystrophin-associated protein complex
- DGC
dystrophin-glycoprotein complex
- DG
dystroglycans
- SCG
sarcoglycans
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- HDAC
histone deacetylase
- HAT
histone acetyltransferase
- Grb2
growth factor receptor-bound protein-2
- FKRP
fukutin related protein
- mpc
muscle progenitor cells
- GDF-8
growth and differentiation factor-8
- TGF-β
transforming growth factor-β
- FLRG
follistatin related genes
- ACVR2, ACVR2B
activin type II receptor
- ALK4, ALK5
activin like kinase 4, 5
- LGMD1C
limb gridle muscular dystrophy type 1C
- cdks
cyclin-dependent kinase inhibitors
- BMP4
bone morpogenetic protein 4
- AT1
angiotensin receptor 1
- NF-kB
nuclear factor-kappa B
- IkB
inhibitor of NF-kB
- IKK
IkB kinase
- TNF-α
tumor necrosis factor-α
- IGF-I
insulin-like growth factor I
- FOXO
forkhead box-containing protein, O-subfamily
- PI3K
phosphatidylinositol 3-kinase
- AKT
serine-threonine kinase
- GSK3
glycogen synthesis kinase
- rAAV
recombinant adeno-associated virus
- bHLH
beta helix-loop-helix
- PCAF
p300/CBP-associated factor
- JNK
Jun N-terminal kinase
- PIGF
angiogenic factor-placenta growth factor
- MMP-9
matrix metalloproteinase-9
References
- Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amthor H, Huang R, McKinnell I, Christ B, Kambadur R, Sharma M, et al. The regulation and action of myostatin as a negative regulator of muscle development during avian embryogenesis. Dev Biol. 2002;251:241–57. doi: 10.1006/dbio.2002.0812. [DOI] [PubMed] [Google Scholar]
- Amthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA. 2007;104:1835–40. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157(1):137–48. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabdallah BF, Bouchentouf M, Rousseau J, Bigey P, Michaud A, Chapdelaine P, et al. Inhibiting myostatin with follistatin improves the success of myoblast transplantation in dystrophic mice. Cell Transplant. 2008;17(3):337–50. doi: 10.3727/096368908784153913. [DOI] [PubMed] [Google Scholar]
- Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, Shelton GD, et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med. 2007;13(5):597–603. doi: 10.1038/nm1573. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–21. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19:543–9. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- Brunelli S, Sciorati C, D'Antona G, Innocenzi A, Covarello D, Galvez BG, et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA. 2007;104:264–9. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008 June 15; doi: 10.1038/nature07034. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerletti M, Jurga S, Witczak CA, Hirshman MF, Shadrach JL, Goodyear LJ, et al. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell. 2008;134(1):37–47. doi: 10.1016/j.cell.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SE, Gerken E, Zhang Y, Zhan M, Mohan RK, Li AS, et al. Role of TNF-{alpha} signaling in regeneration of cardiotoxin-injured muscle. Am J Physiol Cell Physiol. 2005;5:C1179–87. doi: 10.1152/ajpcell.00062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–10. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13(3):231–8. doi: 10.1016/s0959-437x(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Darabi R, Gehlbach K, Bachoo RM, Kamath S, Osawa M, Kamm KE, et al. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med. 2008 January 20; doi: 10.1038/nm1705. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;10:762–73. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- de la Serna IL, Carlson KA, Imbalzano AN. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat Genet. 2001;27:187–90. doi: 10.1038/84826. [DOI] [PubMed] [Google Scholar]
- Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–67. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- Engvall E, Wewer UM. The new frontier in muscular dystrophy research: booster genes. FASEB J. 2003;17:1579–84. doi: 10.1096/fj.02-1215rev. [DOI] [PubMed] [Google Scholar]
- Forcales SV, Puri PL. Signaling to the chromatin during skeletal myogenesis: Novel targets for pharmacological modulation of gene expression. Sem Cell Dev Biol. 2005;16(45):596–611. doi: 10.1016/j.semcdb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Frank NY, Kho AT, Schatton T, Murphy GF, Molloy MJ, Zhan Q, et al. Regulation of myogenic progenitor proliferation in human fetal skeletal muscle by BMP4 and its antagonist Gremlin. J Cell Biol. 2006;175:99–110. doi: 10.1083/jcb.200511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargioli C, Coletta M, De Grandis F, Cannata SM, Cossu G. PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat Med. 2008 doi: 10.1038/nm.1852. doi:10.1038/nm.1852. [DOI] [PubMed] [Google Scholar]
- Grobet L, Pirottin D, Farnir F, Poncelet D, Royo LJ, Brouwers B, et al. Modulating skeletal muscle mass by postnatal, muscle-specific inactivation of the myostatin gene. Genesis. 2003;35:227–38. doi: 10.1002/gene.10188. [DOI] [PubMed] [Google Scholar]
- Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;357:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C, et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA. 2008;105:4318–22. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi S, Cossu G, Nervi C, Sartorelli V, Puri PL. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc Natl Acad Sci USA. 2002;99:7757–62. doi: 10.1073/pnas.112218599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6:673–84. doi: 10.1016/s1534-5807(04)00107-8. [DOI] [PubMed] [Google Scholar]
- Juul A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm IGF Res. 2003;13:113–70. doi: 10.1016/s1096-6374(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Kuang S, Chargé SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172(1):103–13. doi: 10.1083/jcb.200508001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129(5):999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBarge MA, Blau HM. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell. 2002;111:589–601. doi: 10.1016/s0092-8674(02)01078-4. [DOI] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–11. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–22. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ. Quadrupling muscle mass in mice by targeting TGF-beta signaling pathways. PLoS ONE. 2007;8:e789. doi: 10.1371/journal.pone.0000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZF, Shelton GD, Engvall E. Elimination of myostatin does not combat muscular dystrophy in dy mice but increases postnatal lethality. Am J Pathol. 2005;2:491–7. doi: 10.1016/S0002-9440(10)62271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;22:2950–66. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluis F, Perdiguero E, Nebreda AR, Munoz-Canoves P. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 2006;16:36–44. doi: 10.1016/j.tcb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995;374:360–3. doi: 10.1038/374360a0. [DOI] [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–47. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane C, Hennebry A, Thomas M, Plummer E, Ling N, Sharma M, et al. Myostatin signals through Pax7 to regulate satellite cell self-renewal. Exp Cell Res. 2008;314(2):317–29. doi: 10.1016/j.yexcr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Signaling chromatin to make muscle. Curr Opin Cell Biol. 2002;14:763–72. doi: 10.1016/s0955-0674(02)00389-7. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12457–61. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, et al. The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Dev Dis. 2002;129:2773–83. doi: 10.1242/dev.129.11.2773. [DOI] [PubMed] [Google Scholar]
- Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–50. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;5:e79. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourkioti F, Rosenthal N. IGF-1, inflammation and stem cells: interactions during muscle regeneration. Trends Immunol. 2005;10:535–42. doi: 10.1016/j.it.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Fisher I, Morgan JE, Abraham D. Steroids in Duchenne muscular dystrophy: from clinical trials to genomic research. Neuromuscul Disord. 2002;12(Suppl 1):S162–5. doi: 10.1016/s0960-8966(02)00101-3. [DOI] [PubMed] [Google Scholar]
- Musarò A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999;400(6744):581–5. doi: 10.1038/23060. [DOI] [PubMed] [Google Scholar]
- Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, et al. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2007 September 24; doi: 10.1096/fj.07-8673com. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Ohsawa Y, Hagiwara H, Nakatani M, Yasue A, Moriyama K, Murakami T, et al. Muscular atrophy of caveolin-3-deficient mice is rescued by myostatin inhibition. J Clin Invest. 2006;11:2924–34. doi: 10.1172/JCI28520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios D, Puri PL. The epigenetic network regulating muscle development and regeneration. J Cell Physiol. 2006;207(1):1–11. doi: 10.1002/jcp.20489. [DOI] [PubMed] [Google Scholar]
- Parsons SA, Millay DP, Sargent MA, McNally EM, Molkentin JD. Age-dependent effect of myostatin blockade on disease severity in a murine model of limb-girdle muscular dystrophy. Am J Pathol. 2006;168(6):1975–8. doi: 10.2353/ajpath.2006.051316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;5:867–77. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]
- Pelosi L, Giacinti C, Nardis C, Borsellino G, Rizzuto E, Nicoletti C, et al. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 2007;7:1393–402. doi: 10.1096/fj.06-7690com. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–30. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- Pisconti A, Brunelli S, Di Padova M, De Palma C, Deponti D, Baesso S, et al. Follistatin induction by nitric oxide through cyclic GMP: a tightly regulated signaling pathway that controls myoblast fusion. J Cell Biol. 2006;172(2):233–44. doi: 10.1083/jcb.200507083. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum Mol Genet. 2002;11:263–72. doi: 10.1093/hmg/11.3.263. [DOI] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA binding, interacting proteins and post-transcriptional modifications. J Cell Physiol. 2000;185:155–73. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Puri PL, Iezzi S, Stiegler P, Chen TT, Shiltz L, Muscat G, et al. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell. 2001;8:885–97. doi: 10.1016/s1097-2765(01)00373-2. [DOI] [PubMed] [Google Scholar]
- Radley HG, Davies MJ, Grounds MD. Reduced muscle necrosis and long-term benefits in dystrophic mdx mice after cV1q (blockade of TNF) treatment. Neuromuscul Disord. 2008;18(3):227–38. doi: 10.1016/j.nmd.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Rampalli S, Li L, Mak E, Ge K, Brand M, Tapscott SJ, et al. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat Struct Mol Biol. 2007 December;14(12):1150–6. doi: 10.1038/nsmb1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080–6. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, Tajbakhsh S, et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol. 2006 January;172(1):91–110. doi: 10.1083/jcb.200508044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotwein P. Insulin-like growth factor action and skeletal muscle growth, an in vivo perspective. Growth Horm IGF Res. 2003;6:303–5. doi: 10.1016/j.ghir.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Sampaolesi M, Blot S, D'Antona G, Granger N, Tonlorenzi R, Innocenzi A, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–9. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D'Antona G, Pellegrino MA, et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–92. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350(26):2682–8. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Serra C, Palacios D, Mozzetta C, Forcales S, Ripani M, Morantte I, Jones D, Du K, Jahla U, Simone C, Puri PL. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/Pi3K/AKT pathways during muscle differentiation. Mol Cell. 2007;28(2):200–13. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shavlakadze T, Winn N, Rosenthal N, Grounds MD. Reconciling data from transgenic mice that overexpress IGF-I specifically in skeletal muscle. Growth Horm IGF Res. 2005;1:4–18. doi: 10.1016/j.ghir.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Sherwood RI, Christensen JL, Conboy IM, Conboy MJ, Rando TA, Weissman IL, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–54. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20(13):1692–708. doi: 10.1101/gad.1419406. [DOI] [PubMed] [Google Scholar]
- Shinin V, Gayraud-Morel B, Gomès D, Tajbakhsh S. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol. 2006;7:677–8. doi: 10.1038/ncb1425. [DOI] [PubMed] [Google Scholar]
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet. 2004;36:738–43. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- Snider L, Tapscott SJ. Emerging parallels in the generation and regeneration of skeletal muscle. Cell. 2003 June;113(7):811–2. doi: 10.1016/s0092-8674(03)00474-4. [DOI] [PubMed] [Google Scholar]
- Suelves M, Vidal B, Serrano AL, Tjwa M, Roma J, López-Alemany R, et al. uPA deficiency exacerbates muscular dystrophy in MDX mice. J Cell Biol. 2007;178(6):1039–51. doi: 10.1083/jcb.200705127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–95. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem. 2000;275(51):40235–43. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M. Damage and inflammation in muscular dystrophy: potential implications and relationships with autoimmune myositis. Curr Opin Rheumatol. 2005;6:707–13. doi: 10.1097/01.bor.0000179948.65895.1a. [DOI] [PubMed] [Google Scholar]
- Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22(13):1747–52. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–6. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA. 2005;102(7):2519–24. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–71. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155(1):123–33. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittemore LA, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun. 2003;300:965–71. doi: 10.1016/s0006-291x(02)02953-4. [DOI] [PubMed] [Google Scholar]
- Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, et al. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci USA. 2003;100:15842–6. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–64. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammit PS, Partridge TA. Sizing up muscular dystrophy. Nat Med. 2002;12:1355–6. doi: 10.1038/nm1202-1355. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–8. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]