

Abstract
The Vitamin A metabolites, including retinoic acid (RA), form ligands for retinoic acid-related nuclear receptors and together they play pleiotropic roles in various biological processes. Recently, we described that RA also functions as a key modulator of transforming growth factor-beta (TGF-β)-driven immune deviation, capable of suppressing the differentiation of interleukin-17 secreting T helper cells (TH17) and conversely promoting the generation of Foxp3+ T regulatory (Treg) cells. This review will focus on the role of RA in the reciprocal TGF-β driven differentiation of TH17 and Treg and on the importance of such regulatory mechanism to control a functional immune system, in particular at the mucosal interface of the intestine.
Keywords: vitamin A, gut tropism, Foxp3, TH17, nuclear receptors
Natural and induced regulatory T cells
Burnet's clonal selection theory introduced the idea that through self/non-self discrimination, auto-reactive cells would be eliminated in order to achieve tolerance [1]. In the past 30 years however, it has become clear that autoreactive T cells also exist in normal individuals, and that some of these are beneficial and required to avoid the occurrence of autoimmune diseases [2-6]. Beneficial autoreactive T cells are actively selected for in the thymus, where they adapt to their phenotype and acquire regulatory functions. Because of this, they have been characterized as natural regulatory T cells (nTreg). In contrast to self-reactive thymocytes that are deleted during conventional negative selection, precursors of nTregs require strong self-agonist selection signals to gain their regulatory capacity to maintain self-tolerance (Fig.1). Regulatory T cells were initially described by Sakaguchi and coworkers as CD4+CD25+ T cells [7]. Later, Sakaguchi's and Rudensky's groups further defined the forkhead-winged helix transcription factor family member, Foxp3, as the crucial driver of the naturally-occurring regulatory T cell-lineage [8, 9]. The observation that patients with the X-linked fatal autoimmune disease, IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, lack functional Foxp3 underscores the importance of this transcription factor as the central key player in the functional regulation of nTreg [10, 11]. In mice, a similar massive and fatal lymphoproliferative disease develops spontaneously in the mouse mutant strain, scurfy, which also results from impaired Foxp3 function. Introduction of a functional Foxp3 transgene can correct the autoimmune phenotype, exemplifying the importance of Foxp3 and Treg cells in systemic immune-regulation and tolerance [10].
Figure1. Development of natural and induced regulatory T cells.
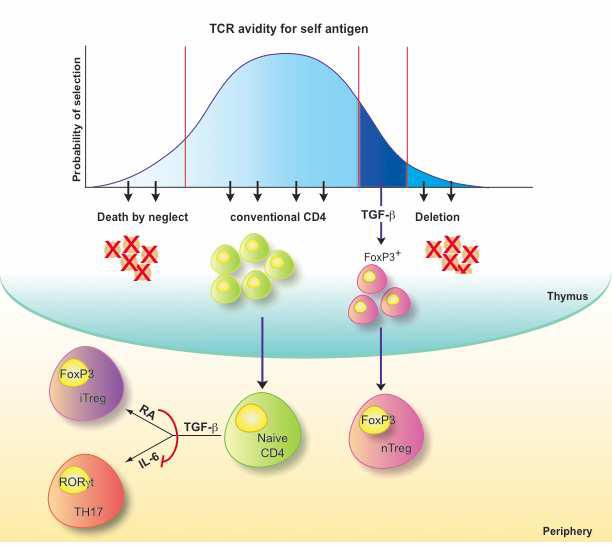
Thymus-derived “natural” regulatory T cells (nTreg) are actively selected upon high avidity TCR/self peptide-loaded MHC interactions, express the forkhead-winged helix transcription factor family member, Foxp3, and are fundamental in the process of self-tolerance, so-called “dominant tolerance”. Conventionally selected naïve Foxp3− CD4+ T cells can be “converted” into induced Foxp3+Treg (iTreg) in response to self or non-self antigens encountered post thymically in the periphery. TGF-β is required for both early nTreg development in the thymus and for peripheral induction of iTreg. TGF-β is also able to promote, in the presence of inflammatory cytokines such as IL-6, the development of pro-inflammatory TH17 cells. The vitamin A metabolite, retinoic acid (RA), is capable of inhibit the TGF-β/IL-6–driven induction of pro-inflammatory TH17 cells and simultaneously promote the TGF-β-dependent peripheral differentiation of anti-inflammatory Foxp3+ iTregs.
In addition to self-tolerance a functional immune system also needs to be able to tolerize non-self antigens that do not impose a threat to the individual. Such harmless non-self antigens are abundant in the intestine where numerous, often beneficial commensal bacteria colonize the colon and where digested food is continuously absorbed via the small intestine epithelium. An effective immune-regulation is a condition sine qua non for the gut physiology [12-15] and the importance of Treg cells to control and prevent aberrant immune responses directed towards self or non-self antigens and to establish tolerance has been demonstrated at length [16].
Although self-specific thymus-derived nTregs can suppress effector T cells with other specificity, they need to be activated first by their cognate self-antigens. In addition, since nTregs functionally differentiate in the thymus during agonist selection with self-antigens, the differentiation of nTregs in the thymus with specificity towards non-self antigens is very unlikely. Instead, a substantial number of studies have clearly demonstrated that naïve Foxp3−, primarily non-self-reactive, CD4+ T cells can be “converted” by TGF-β into induced Foxp3+Treg (iTreg) in response to self or non-self antigens encountered in the periphery [17-22] (Fig.1). Such iTreg are especially important to maintain tolerance towards antigens introduced post thymically via the intestine (Fig.2) [21, 22]. Consistent with this, our group and those of Belkaid and Powrie have clearly demonstrated that iTregs are preferentially induced at the mucosal induction sites of mesenteric lymph node (MLN) and lamina propria (LP), rather than in spleen or peripheral lymph nodes (LNs), reinforcing that the intestine is a privileged site for iTreg differentiation [23-25]. In this context, Lafaille's group has further shown, using mice that lack nTregs, that peripheral neoconverted Foxp3+ iTreg cells are effective and sufficient to mediate oral tolerance, which can be defined as diminished systemic immune responses for an antigen previously contacted via the oral route [21]. Recently these findings were extended by the demonstration that mice which cannot induce iTreg are unable to mediate oral tolerance, suggesting the iTreg are not only sufficient, by also crucial for oral tolerance induction in this model [22].
Figure 2. Role of retinoic acid in mucosal immune-regulation.
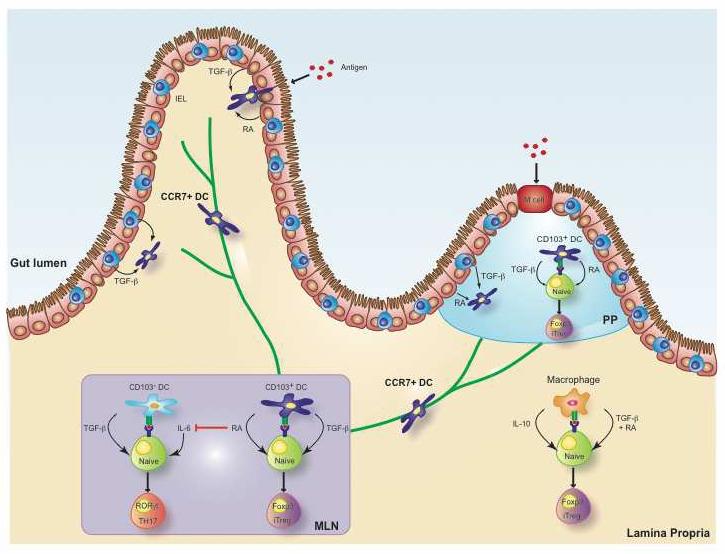
Under the influence of intestinal epithelial cells and intra-epithelial lymphocytes, lamina propria and Peyer's patch CD103+ DCs acquire their ability to produce retinoic acid (RA). LP DCs that have acquired the expression of the chemokine receptor CCR7 are able to migrate through the afferent lymphatic vessels into the mesenteric lymph nodes and influence T-cell activation and differentiation during priming of naïve T cells. Gut-derived CD103+ DCs and LP macrophages efficiently promote the generation of Foxp3+iTreg in a RA-, TGF-β-, and also IL-10 (in the case of LP macrophages) dependent manner. In contrast, release of RA by these DCs suppresses the TGF-β- and IL-6- dependent differentiation of TH17 cells.
Through the expression of tight junctions (TJs), epithelial-associated DCs are able to establish rigid contacts with the neighboring epithelial cells, while extending their dendrites to sample luminal antigens, including food particles and whole bacteria [26]. Upon activation, DC mature and migrate to regional LNs, where they can present processed antigens to naïve T cells. This migration requires the expression of the chemokine receptor, CCR7, by the mucosal DCs (Fig. 2). Consistent with this, CCR7+ but not CCR7− MLN DCs, have vacuoles containing intestinal epithelial-cell derived debris, suggesting their intestinal origin [27]. Presentation of gut luminal antigens by these mucosal DCs plays crucial roles in the development of oral tolerance and in the generation of iTregs [13, 28, 29]. Consistent with this, CCR7−/− mice are inefficient in inducing oral tolerance, likely due to the fact that antigen-carrying CCR7-deficient DCs cannot migrate to the MLN and induce T cell-dependent oral tolerance [29].
A balance between protective and suppressive immunity in the intestine
In addition to the requirement for tolerance to maintain the integrity of the barrier, the intestine, which forms the largest entry surface for pathogens, also requires the most effective immune protection, indicating that a critical but flexible immune balance has to be in place. An important molecule in this context is TGF-β, abundantly produced in the gut and crucial for both systemic and mucosal immune-regulation. Among multiple roles, TGF-β has the capacity to block TH1 and TH2 differentiation and to convert naïve CD4 cells into Foxp3-expressing iTreg cells [30]. Paradoxically, TGF-β also displays pro-inflammatory roles and in the presence of pro-inflammatory cytokines such as IL-6, IL-21, IL-1β and TNF, TGF-β promotes the conversion of naïve T cells into TH17 [31-33]. This contrasting deviation puts TGF-β as a principal controller of immune responses and underscores a central role of this cytokine in orchestrating the pro- and anti-inflammatory nature of adaptive immunity. Nowhere else is a critical regulation of this TGF-β dependent balance of more significance that at the mucosal interface of the gut, where efficient immune protection against pathogens has to coincide with maintenance of the mucosal barrier integrity.
The reciprocal developmental pathways for the generation of pro-inflammatory effector TH17 and immune suppressive iTreg, imposes a dichotomy in their generation in addition to their functional antagonism. Therefore, TGF-β-mediated iTreg cells and TH17 effectors arise in a mutually exclusive fashion, depending on whether they are activated in the presence of TGF-β alone or TGF-β together with pro-inflammatory cytokines [32]. The constant exposure to luminal antigens creates a so-called “physiological chronic inflammation” in the gut, which could easily favor the TGF-β driven effector differentiation at the expense of iTreg. Nevertheless, under normal steady state conditions, immune responses in the gut are under control and iTregs tend to preferentially accumulate at this site.
We recently identified a metabolite of the nutrient vitamin A, retinoic acid, as a key regulator of TGF-ß–dependent immune responses, capable of inhibiting the TGF-β/IL-6–driven induction of pro-inflammatory TH17 cells and promoting the TGF-β-dependent peripheral differentiation of anti-inflammatory Foxp3+ iTregs [23] (Fig.2). Since vitamin A cannot be synthesized by the human body and it must be absorbed by the intestine from the diet, this means that this intestinal induced regulatory mechanism allows for environmental factors to modulate aberrant endogenous immune responses from the outside in. The induced immune tolerance is not necessarily irreversible and in the presence of innate danger signals RA effects might diminish or synergize with innate responses to promote or enhance protective immunity.
Retinoids and their nuclear receptors: from the diet to the nucleus
Vitamin A is initially absorbed from the diet and via the function of various enzymes, including the ubiquitous alcohol dehydrogenases (ADH) and the more restricted retinal dehydrogenases (RALDH), it is further metabolized to RA. Two isoforms of RA have been identified in vivo (all-trans-RA (at-RA) and 13-cis-RA) but the 9-cis-RA isoform has never been detected in vivo. All RA isoforms, can bind to homo- or hetero-dimers of nuclear receptors, such as retinoic acid receptors (RARs) and retinoid X receptors (RXRs). These nuclear receptors are able to function as ligand-activated transcription factors through binding to DNA response elements (RARE) [34, 35]. Depending on the type of ligand interaction as well as on the availability of co-activators (CoAs) and co-repressors (CoRs), nuclear receptor-ligand complexes are able to perform either transcriptional repression or activation of target genes [36]. Due to its role as a required heterodimer partner, RXR also influences the transcriptional control of many other nuclear receptors such as vitamin D (VDR) and thyroid hormone receptors in addition to RARs. This may have important additional repercussions in T cell differentiation, since for example, Vitamin D3, is able to regulate transcription factors, such as NF-AT, NF-κB, and, importantly, TGF-β signaling molecules, SMADs [37-39]. Specifically, Smad3 was reported to acts as a CoA for ligand-induced transactivation of VDR [39]. Finally, VD3 is also described as a potent inducer of the anti-inflammatory cytokine IL-10 [40], reinforcing the network of immune-regulation associated with retinoic acid receptors. It is noteworthy that although it was reported that RA directly enhanced LPS-induced IL-10 expression in a cell line [41], our data suggest that RA efficiently suppresses TGF-β/IL6-induced IL-10 production (Mucida, D et al., manuscript in preparation), which might be correlated with this uncommon characteristic of RXR/VDR heterodimer action [42]. RXRs can also interact with members of the orphan nuclear peroxisome proliferative activated receptor family, PPAR, which have important functions in immune-regulation that are closely linked to retinoic acid-related pathways. Recently PPAR-δ was reported to play a role in the suppression of IL-17 and IFN-γ production in colitis [43].
Immune-regulatory functions of RA and TGF-β
Similar to the vitamin A metabolites and their nuclear receptors, members of the TGF-β superfamily are pleiotropic regulators of numerous biological processes. In many of their functions, TGF-β and retinoids synergize or counteract each other reflecting an intense relationship between members of these two superfamilies of gene regulators.
In addition to their crucial roles in development, TGF-β and RA are involved at almost every level of immune differentiation and function, affecting passive immunity as well as innate and adaptive immunity. Both, TGF-β and RA are actively produced by the intestinal epithelium and both play important roles in mucosal epithelial cell differentiation and in maintaining the integrity of its barrier function (Fig 2).
TGF-β signaling and nuclear hormone receptor regulation also greatly influence multiple functions of the innate immune system. An intensive crosstalk between nuclear receptors and factors of the innate immune system, for example IFN regulatory factor 3, a key transcription factor involved in the induction of antiviral genes, may lead to repression of innate immune responses and inflammation and conversely to the repression of nuclear hormone receptor signaling [44, 45]. This crosstalk has been implicated in the pathogenesis of atherosclerosis and Reye's syndrome and other pathogen-associated metabolic and developmental disorders [45]. The cytosolic pattern recognition receptor, retinoic acid-inducible gene I (RIG-I), also plays important role in the cytosolic recognition of invading pathogens and the induction of the retinoic acid early inducible gene 1 (RAE-1), which forms a potent ligand for the NK receptor NKG2D, makes target cells highly sensitive to NK cell killing. TGF-β negatively regulates the antigen presentation, phagocytosis and expression of costimulatory molecules by DCs and macrophages and, similarly to RA, TGF-β can negatively regulate the production of inflammatory cytokines such as IL-1, TNF-α and IFN-γ, by antigen presenting cells and NK cells [41, 46-50].
In the adaptive immune system, TGF-β and RA positively regulate secretory IgA production, important in mucosal barrier function and antibacterial protection, by actively promoting IgA-class switching [51-54]. In addition to secretory/mucosal IgA-class switching, RA also induces migration and homing of B cells to the LP of the intestine [54].
On T cells, gut specific homing receptors, including α4β7 and CCR9, are induced by RA released during priming by MLN, LP and Peyer's patch (PP) DCs [55-58]. On the other hand, TGF-β, which is produced by many cells in the intestine including epithelial cells, macrophages, DCs and subsets of intraepithelial lymphocytes (IEL)[24, 59-62], induces the integrin αEβ7 (CD103). The combination of TGF-β and RA results in further upregulation of both integrins, α4β7 and αEβ7, but downmodulates the expression of CCR9 [23].
TGF-β and RA play also major roles in functional differentiation of T cells. In addition to the RA mediated reciprocal differentiation of TGF-β dependent iTreg/TH17 cells, TGF-β was shown to be crucial for early nTreg development as well, indicating that TGF-β is important for both nTreg and iTreg differentiation [63]. Similar to TGF-β, RA also strongly inhibits the TH1 cytokine IFN-γ synthesis, but in contrast, RA has the capacity to stimulate TH2 differentiation under certain conditions [64]. The effect of RA on TH2 development is dependent on the stage of T helper differentiation and RA enhances IL-4 production only when added after the initial activation of naïve CD4 cells in vitro [65]. When added at the time of the initial stimulation, RA rather suppresses both TH1 and TH2 differentiation, indicating that RA could have divergent effects depending on the context of exposure [66, 67].
Reciprocal regulation of TGF-β mediated TH17/Treg differentiation by RA
iTreg cells and TH17 cells arise in a mutually exclusive way, depending on whether they are generated in the presence of TGF-β or TGF-β and the pro-inflammatory cytokine IL-6. This remarkable pro- and anti-inflammatory immune deviation is of particular relevance at the mucosal surface of the intestine and the recent finding that RA plays a key role in controlling this critical TGF-β dependent immune balance, implies an important role for RA to mediate mucosal immune regulation (Fig. 2). Consistent with this, several groups, including ours, demonstrated that iTregs were preferentially induced by mucosal DCs rather than spleen or peripheral LN DCs, reinforcing that the intestine is a privileged site for Treg induction (Fig. 2) [24, 25]. Among the mucosal DCs, CD103+ DCs seem to be especially capable of releasing RA during priming. Coombes et al. showed that RA-production by CD103+ but not CD103− MLN DCs, efficiently converted naïve CD4+ T cells into FoxP3+ T cells in a TGF-β-dependent fashion [24]. In contrast however, Sun et al. described that whereas LP CD103+ DCs could induce Foxp3+ T cells in the absence of exogenous TGF-β, both CD103+ and CD103− LP DC-populations were highly efficient at inducing Treg cells in the presence of exogenous TGF-β [25]. More recently, Denning et al. extended and added complexity to these findings. They reported that LP macrophages are also potent regulatory antigen presenting cells, able to convert naïve T cells into Foxp3+Treg cells in an IL-10-, RA- and TGF-β-dependent manner. On the other hand, LP CD11b+CD11c+ DCs are pro-inflammatory, inducing IL-17-producing T cells but few Tregs. Denning et al. also reported that LP CD11b−CD11c+ cells express high levels of CD103 however they did not detect any increased ability for these CD103+CD11b− DCs to induce Tregs [60].
In contrast to the enhanced Foxp3 induction by MLN DCs, we showed that they had less capacity to promote TH17 differentiation as compared to their spleen counterparts. In the presence of exogenous RA, however, spleen DCs readily induce iTregs but show much reduced capacity to promote TH17 differentiation. Conversely, the addition of RAR inhibitors impaired Treg differentiation by MLN DCs but enhanced the TH17 induction. Using an infectious mouse model, we further showed that exogenous RA inhibits the induction of TH17 cells in vivo whereas injection of RAR antagonists resulted in a decrease of Foxp3+Treg cells in the LP [23]. These observations clearly demonstrated the capacity of RA to directly control the reciprocal differentiation of TGF-β driven Treg and TH17 cells.
The abundant production of TGF-β and RA in the mucosa and the ability of RA to promote TGF-β-dependent iTregs could thus directly be related to the high frequency of Foxp3-expressing Treg cells in the intestine of normal mice. Furthermore, the synergistic effect of TGF-β and RA to promote anti-inflammatory but suppress pro-inflammatory immune responses might be central for oral tolerance induction and therefore, mucosal and systemic immune regulation [23, 25]. It is also of importance to note that RA mediated the differentiation of a stable TGF-β induced iTreg lineage in vitro, which upon transfer efficiently prevented colitis induction in vivo indicating that in addition to functional iTregs, RA mediates epigenetic imprinting that is critical for establishment of a stable Treg lineage in vivo.
The RA mediated enhanced TGF-β induced Foxp3 expression was drastically reduced in the presence of anti-IL-2 antibodies or when IL-2-deficient naïve CD4 T cells were used in vitro. In the later case, the addition of exogenous IL-2 restored RA-effects [23]. Conversely, the RA-mediated suppression of IL-17 production by either CD4 or CD8 T cells was inhibited in the presence of anti-IL-2 antibodies or when IL-2 deficient naïve T cells were used [23]. Although it was previously shown that the addition of exogenous IL-2 also promotes TGF-β induced iTreg differentiation in vitro, the direct effects of RA and exogenous IL-2 appear distinct. Whereas the combination of IL-2 and TGF-β induce mostly CD103−Foxp3+ Treg cells, RA and TGF-β induced preferentially CD103+Foxp3+ cells. In addition, since RA also induces the expression of the gut homing receptors, CCR9 and a4b7, in combination, RA and TGF-β readily induce iTregs with a gut-specific tissue tropism.
The observation that in the absence of DCs, RA also mediates reciprocal TGF-β dependent differentiation of naïve T cells in vitro, indicates that RA directly affects the transcriptional control within the T cells. We showed that indeed RA directly inhibits retinoic acid orphan receptor gamma T (RORγt) which is induced by TGF-β and further enhanced by IL-6 signaling and which is required for efficient TH17 differentiation [68]. It is not known, however, whether RA antagonistic effects on IL-6 signaling extend to the recently described IL-21 pathway of TH17 differentiation [69-71]. In addition to the negative regulation of IL-17 transcription, RA also has direct positive regulatory effects for the transcription of Foxp3.
Recently, it was shown that CD11chiCD11bhi LP DCs, when stimulated by TLR5-ligand flagellin, promoted a modest differentiation of antigen-specific TH17 and TH1 cells and addition of RAR-antagonist LE540 inhibited this IL-17 production [72]. Unexpectedly, LPS did not induce measurable IL-17 production, but addition of a low-dose (1nM) RA induced some IL-17 production in the presence of LPS whereas a higher dose of RA (10μM), inhibited IL-17 production. Based on this, it was concluded that low-dose RA induces TH17-development, whereas a high dose of RA is suppressive for IL-17 expression [72]. It is important, however, to point out here that previous work, including ours, describing the suppressive effects of RA on TH17 development in the absence of TLR stimulation, reported RA-mediated suppression of IL-17 in all doses examined, ranging from 1nM to 100nM [23] and 10μM [66]. A typical dose-response curve of TH17-suppression by RA was also observed by Kattah and coworkers using a range from 1nM to 1μM in human cells [73]. In addition, the release of physiological amounts of RA by mucosal DCs in T cell/DC co-cultures also inhibited IL17-production and enhanced Foxp3 induction, whereas addition of RAR-antagonist in these cultures reversed this effect [23-25]. It does seem that “physiological amounts” of RA released by gut-derived DCs suppress rather than induce IL-17 production but enhance TGF-β mediated Foxp3 induction. It is possible, however, that innate stimuli, such as TLR-signals, may induce contrasting effects of RA on DCs and/or T cells [74].
Transcriptional regulation of Foxp3 and IL-17 by RA
The transcription factors STAT5 and STAT3 are important for the transcription of Foxp3 and IL-17, respectively [68, 75, 76]. The enhanced expression of Foxp3 in the presence of RA suggests a potential relationship between STAT5 and RARs in a similar fashion as the cooperation between STAT3 and RORγt (Fig.3). It is therefore perhaps not a coincidence that RORγt displays strong homology with the RARs and also appears to function in the context of transcriptional activators and repressors [77]. Consistent with STAT5 and RAR cooperation they can physically interact with each other in vivo to promote RAR-mediated transcription (Fig. 3) [78]. Furthermore, the STAT5 consensus-binding site directly overlaps with RAR-response elements, which may lead to coordinated transcription activity rather than competition for the same site [78]. The cooperation between STAT5 and RARs results in STAT5-enhanced responsiveness of the RARs to RA induced transcription of target genes [78]. RAR and STAT5 can also bind the same repressor of transcription, silencing mediator of thyroid and retinoid receptors, SMRT, which can be released upon RA engagement. The RA mediated effects on Treg and TH17 differentiation may thus reflect an intense communication between the STAT and RAR families of transcription factors for the differentiation of T lymphocytes [79].
Figure3. Transcriptional regulation of Foxp3 and RORγt/IL-17 mediated by RA.
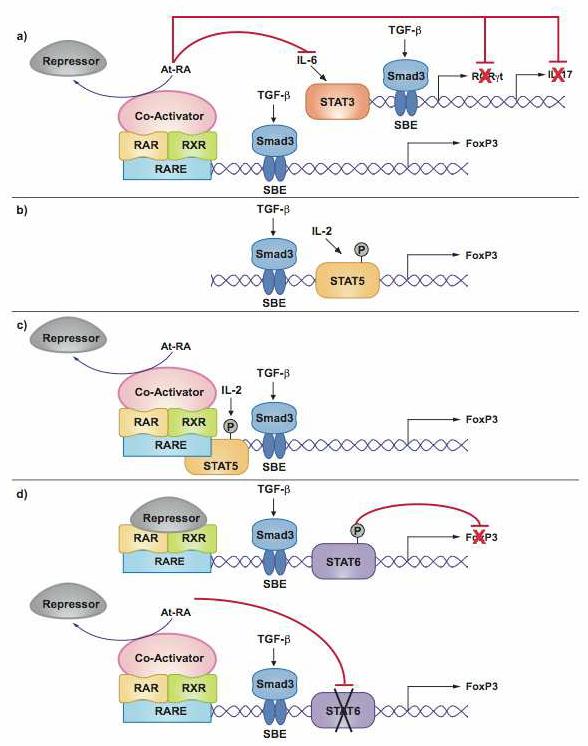
Retinoic acid receptors (RARs) and retinoid X receptors (RXRs) are nuclear receptors able to function as ligand-activated transcription factors through binding to retinoic acid DNA response elements (RARE). Upon ligand (at-RA) binding, RAR/RXRs release co-repressors (CoRs) and recruit co-activators (CoAs). This figure shows possible mechanisms by which RA binding to its nuclear receptors can affect the induction of Foxp3 and IL-17. A. RA could enhance TGF-β signaling via the TGF-β signaling molecules, Smad2 and 3 and SBE (Smad-binding elements) and thus indirectly increase TGF-β-dependent Foxp3 induction. In contrast, RA may also repress different target genes in the TH17-development pathway, including STAT3, RORγt and IL-17. Conversely, the suppression of these molecules could also result in enhanced TGF-β induced Foxp3 expression. B. The induction of Foxp3 expression in naïve T cells requires both TGF-β signaling and IL-2-dependent STAT5 phosphorylation and binding in the Foxp3 promoter. C. STAT5 and RAR interaction may lead to coordinated transcription activity of target genes, therefore enhancing TGF-β-induced Foxp3 expression. D. IL-4 is able to inhibit TGF-β-mediated Foxp3 induction via IL-4 receptor activated STAT6, which directly binds to the Foxp3 promoter. RA binding to RARα/RXRα heterodimers can interfere with the silencing capacity of STAT6 resulting in increased Foxp3 induction.
In addition to direct inhibition of RORγt by RA, it is also possible that RA indirectly suppresses TH17 differentiation via the enhanced transcription of Foxp3. Indeed, it was recently shown that RORγt and Foxp3 can be co-expressed in naive CD4+ T cells upon TGF-β exposure and that TGF-β-induced Foxp3 inhibits IL-17 induction by direct suppression of RORγt [80-82]. Consistent with additional STAT-independent effects of RA, it was shown that with in vitro culture of STAT3 conditional knockout CD4 T cells (in this case, IL-17 production is abolished [76]), RA still enhanced Foxp3 expression in the presence of TGF-β, suggesting that RA can induce Foxp3 independently of IL-6 signaling. Conversely, using STAT5 conditional knockout CD4 T cells (Foxp3 expression is decreased), 1μM RA still suppressed IL-17 production in the presence of TGF-β and IL-6. It is important to note, however, that in this study, total, but not naïve, CD4 STAT5-deficient T cells were used and exogenous IL-2 was added, which could mask the effects of RA on naïve T cell differentiation [66]. Finally, and in contrast to our observations, it was shown in that study that also in the presence of anti-IL2 antibodies, RA could still inhibit IL-17 production (although less efficiently) and enhance TGF-β induced Foxp3 expression [66]. It is possible that the high dose of RA used (1μM) [66] could bypass the requirement for IL-2 observed in our previous study [23].
In addition to IL-6, TH2 cytokine IL-4, also inhibits TGF-β-mediated Foxp3 induction via IL-4 receptor activated STAT6, which directly binds to the Foxp3 promoter (Fig.3) [67]. RA on the other hand can reverse this IL-4-mediated inhibition of Foxp3 expression by engaging RARα/RXRα heterodimers that interfere with the silencing capacity of STAT6 on Foxp3 induction [67].
Finally, the synergy between RA and TGF-β is likely controlled by the Smad signaling molecules (Fig.3) that act downstream of the TGF-β receptor, and/or with the transcription factor Runx3, which is mediating the TGF-β inducted CD103 expression and which physically interacts with Smads to cooperate in TGF-β mediated signaling [83].
Concluding remarks
A functional immune system is central for efficient protection of the organism against invasive pathogens as well as invasive transformed autologous cells. Aberrant immune responses directed towards harmless non-self agents or normal endogenous cells can lead to severe immune pathology and autoimmunity. Many regulatory mechanisms are in place and in particular Foxp3+ regulatory T cells are indispensable to prevent excessive and self-destructive immune responses. The intestine, which forms the largest entry surface for invading pathogens but also contains a vast load of harmless antigens derived from colonizing commensal bacteria and absorbed nutrients, imposes a unique challenge for the immune system. In particular, the ability to mount a highly efficient protective immune response needs to coincide with the steady state prerequisite of tolerance towards innocuous antigens. An improper balance between inflammatory and suppressive immunity can jeopardize mucosal homeostasis and destroy the integrity of the mucosal barrier. The intake of vitamin A as part of the food absorption by the intestine and the ability of the vitamin A metabolite, retinoic acid, to drive the differentiation of induced Foxp3+ regulatory T cells and to suppress the differentiation of pro-inflammatory effector cells, provides an expedient mechanism to secure tolerance when protective immune responses are not advantageous. The plasticity of the system and the ability of innate and pro-inflammatory signals to override the RA controlled immune suppression provides the mucosal immune system with a key control to allow for protective immunity in the face of steady state immune tolerance.
Acknowledgments
This work was supported by fellowships from the Diabetes and Immune Disease National Research Institute (DM) and the Crohn's and Colitis Foundation of America (YP) and the NIH grant RO1 AI050265-06 (HC). This is manuscript 1039 from the La Jolla Institute for Allergy and Immunology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burnet MF. The Clonal Selection Theory of Immunity. 1st. edition The Vanderbilt and Cambridge University Presses; Nashville/London: 1959. [Google Scholar]
- 2.Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in post-thymectomy autoimmune oophoritis in mice. II. Requirement of Lyt-1 cells in normal female mice for the prevention of oophoritis. J Exp Med. 1982;156(6):1577–86. doi: 10.1084/jem.156.6.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161(1):72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in postthymectomy autoimmune oophoritis in mice. I. Requirement of Lyt-1 effector cells for oocytes damage after adoptive transfer. J Exp Med. 1982;156(6):1565–76. doi: 10.1084/jem.156.6.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taguchi O, Kontani K, Ikeda H, Kezuka T, Takeuchi M, Takahashi T, et al. Tissue-specific suppressor T cells involved in self-tolerance are activated extrathymically by self-antigens. Immunology. 1994;82(3):365–9. [PMC free article] [PubMed] [Google Scholar]
- 6.Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78(3):399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64. [PubMed] [Google Scholar]
- 8.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4(+)CD25(+) regulatory T cells. Nat Immunol. 2003;4(4):330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 9.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 10.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 11.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 12.Cheroutre H. Starting at the beginning: new perspectives on the biology of mucosal T cells. Annu Rev Immunol. 2004;22:217–46. doi: 10.1146/annurev.immunol.22.012703.104522. [DOI] [PubMed] [Google Scholar]
- 13.Faria AM, Weiner HL. Oral tolerance. Immunol Rev. 2005;206(1):232–59. doi: 10.1111/j.0105-2896.2005.00280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaz NM. Immunological tolerance and dogma. Med Hypotheses. 1979;5(9):1037–43. doi: 10.1016/0306-9877(79)90052-5. [DOI] [PubMed] [Google Scholar]
- 15.Vaz NM, Faria AM, Verdolin BA, Silva Neto AF, Menezes JS, Carvalho CR. The conservative physiology of the immune system. Braz J Med Biol Res. 2003;36(1):13–22. doi: 10.1590/s0100-879x2003000100003. [DOI] [PubMed] [Google Scholar]
- 16.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–71. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 17.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172(9):5149–53. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- 19.Apostolou I, Von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199(10):1401–8. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curotto De Lafaille MA, Lino A, Kutchukhidze N, Lafaille J. CD25− T Cells Generate CD25+Foxp3+ Regulatory T Cells by Peripheral Expansion. J Immunol. 2004;173(12) doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- 21.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115(7):1923–33. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29(1):114–26. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317(5835):256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 24.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204(8):1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2(4):361–7. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 27.Jang MH, Sougawa N, Tanaka T, Hirata T, Hiroi T, Tohya K, et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol. 2006;176(2):803–10. doi: 10.4049/jimmunol.176.2.803. [DOI] [PubMed] [Google Scholar]
- 28.Hanson DG, Vaz NM, Maia LC, Hornbrook MM, Lynch JM, Roy CA. Inhibition of specific immune responses by feeding protein antigens. Int Arch Allergy Appl Immunol. 1977;55(16):526–32. doi: 10.1159/000231966. [DOI] [PubMed] [Google Scholar]
- 29.Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J Exp Med. 2006;203(3):519–27. doi: 10.1084/jem.20052016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 31.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 33.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 34.Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature. 1992;355(6359):446–9. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang XK, Lehmann J, Hoffmann B, Dawson MI, Cameron J, Graupner G, et al. Homodimer formation of retinoid X receptor induced by 9-cis retinoic acid. Nature. 1992;358(6387):587–91. doi: 10.1038/358587a0. [DOI] [PubMed] [Google Scholar]
- 36.de Lera AR, Bourguet W, Altucci L, Gronemeyer H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat Rev Drug Discov. 2007;6(10):811–20. doi: 10.1038/nrd2398. [DOI] [PubMed] [Google Scholar]
- 37.Alroy I, Towers TL, Freedman LP. Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Mol Cell Biol. 1995;15(10):5789–99. doi: 10.1128/mcb.15.10.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu XP, Bellido T, Manolagas SC. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1995;92(24):10990–4. doi: 10.1073/pnas.92.24.10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yanagisawa J, Yanagi Y, Masuhiro Y, Suzawa M, Watanabe M, Kashiwagi K, et al. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 1999;283(5406):1317–21. doi: 10.1126/science.283.5406.1317. [DOI] [PubMed] [Google Scholar]
- 40.Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195(5):603–16. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Allen C, Ballow M. Retinoic acid enhances the production of IL-10 while reducing the synthesis of IL-12 and TNF-alpha from LPS-stimulated monocytes/macrophages. J Clin Immunol. 2007;27(2):193–200. doi: 10.1007/s10875-006-9068-5. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Martinez R, Zambrano A, Castillo AI, Aranda A. Vitamin D-dependent recruitment of corepressors to vitamin D/retinoid X receptor heterodimers. Mol Cell Biol. 2008;28(11):3817–29. doi: 10.1128/MCB.01909-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JW, Bajwa PJ, Carson MJ, Jeske DR, Cong Y, Elson CO, et al. Fenofibrate represses interleukin-17 and interferon-gamma expression and improves colitis in interleukin-10-deficient mice. Gastroenterology. 2007;133(1):108–23. doi: 10.1053/j.gastro.2007.03.113. [DOI] [PubMed] [Google Scholar]
- 44.Fensterl V, Grotheer D, Berk I, Schlemminger S, Vallbracht A, Dotzauer A. Hepatitis A virus suppresses RIG-I-mediated IRF-3 activation to block induction of beta interferon. J Virol. 2005;79(17):10968–77. doi: 10.1128/JVI.79.17.10968-10977.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chow EK, Razani B, Cheng G. Innate immune system regulation of nuclear hormone receptors in metabolic diseases. J Leukoc Biol. 2007;82(2):187–95. doi: 10.1189/jlb.1206741. [DOI] [PubMed] [Google Scholar]
- 46.Geissmann F, Revy P, Regnault A, Lepelletier Y, Dy M, Brousse N, et al. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol. 1999;162(8):4567–75. [PubMed] [Google Scholar]
- 47.Tridandapani S, Wardrop R, Baran CP, Wang Y, Opalek JM, Caligiuri MA, et al. TGF-beta 1 suppresses [correction of supresses] myeloid Fc gamma receptor function by regulating the expression and function of the common gamma-subunit. J Immunol. 2003;170(9):4572–7. doi: 10.4049/jimmunol.170.9.4572. [DOI] [PubMed] [Google Scholar]
- 48.Nandan D, Reiner NE. TGF-beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J Immunol. 1997;158(3):1095–101. [PubMed] [Google Scholar]
- 49.Bogdan C, Paik J, Vodovotz Y, Nathan C. Contrasting mechanisms for suppression of macrophage cytokine release by transforming growth factor-beta and interleukin-10. J Biol Chem. 1992;267(32):23301–8. [PubMed] [Google Scholar]
- 50.Aukrust P, Muller F, Ueland T, Svardal AM, Berge RK, Froland SS. Decreased vitamin A levels in common variable immunodeficiency: vitamin A supplementation in vivo enhances immunoglobulin production and downregulates inflammatory responses. Eur J Clin Invest. 2000;30(3):252–9. doi: 10.1046/j.1365-2362.2000.00619.x. [DOI] [PubMed] [Google Scholar]
- 51.McIntyre TM, Kehry MR, Snapper CM. Novel in vitro model for high-rate IgA class switching. J Immunol. 1995;154(7):3156–61. [PubMed] [Google Scholar]
- 52.Borsutzky S, Cazac BB, Roes J, Guzman CA. TGF-beta receptor signaling is critical for mucosal IgA responses. J Immunol. 2004;173(5):3305–9. doi: 10.4049/jimmunol.173.5.3305. [DOI] [PubMed] [Google Scholar]
- 53.Tokuyama H, Tokuyama Y. Retinoids enhance IgA production by lipopolysaccharide-stimulated murine spleen cells. Cell Immunol. 1993;150(2):353–63. doi: 10.1006/cimm.1993.1203. [DOI] [PubMed] [Google Scholar]
- 54.Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314(5802):1157–60. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 55.Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424(6944):88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 56.Papadakis KA, Landers C, Prehn J, Kouroumalis EA, Moreno ST, Gutierrez-Ramos JC, et al. CC chemokine receptor 9 expression defines a subset of peripheral blood lymphocytes with mucosal T cell phenotype and Th1 or T-regulatory 1 cytokine profile. J Immunol. 2003;171(1):159–65. doi: 10.4049/jimmunol.171.1.159. [DOI] [PubMed] [Google Scholar]
- 57.Svensson M, Marsal J, Ericsson A, Carramolino L, Broden T, Marquez G, et al. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J Clin Invest. 2002;110(8):1113–21. doi: 10.1172/JCI15988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–38. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 59.Denning TL, Granger SW, Mucida D, Graddy R, Leclercq G, Zhang W, et al. Mouse TCRalphabeta+CD8alphaalpha intraepithelial lymphocytes express genes that down-regulate their antigen reactivity and suppress immune responses. J Immunol. 2007;178(7):4230–9. doi: 10.4049/jimmunol.178.7.4230. [DOI] [PubMed] [Google Scholar]
- 60.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8(10):1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 61.Barnard JA, Beauchamp RD, Coffey RJ, Moses HL. Expression of TGF-β1 by intestinal epitelial cells. ProcNatlAcadSciUSA. 1989;86:1578–83. doi: 10.1073/pnas.86.5.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dignass AU, Stow JL, Babyatsky MW. Acute epithelial injury in the rat small intestine in vivo is associated with expanded expression of transforming growth factor alpha and beta. Gut. 1996;38(5):687–93. doi: 10.1136/gut.38.5.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9(6):632–40. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 64.Cantorna MT, Nashold FE, Hayes CE. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol. 1994;152(4):1515–22. [PubMed] [Google Scholar]
- 65.Iwata M, Eshima Y, Kagechika H. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol. 2003;15(8):1017–25. doi: 10.1093/intimm/dxg101. [DOI] [PubMed] [Google Scholar]
- 66.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111(3):1013–20. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takaki H, Ichiyama K, Koga K, Chinen T, Takaesu G, Sugiyama Y, et al. STAT6 inhibits TGF-beta 1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008 doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17(+) T Helper Cells. Cell. 2006;126(6):1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 69.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007 doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007 doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 71.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007 doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 72.Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol. 2008 doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 73.Kattah MG, Wong MT, Yocum MD, Utz PJ. Cytokines secreted in response to Toll-like receptor ligand stimulation modulate differentiation of human Th17 cells. Arthritis Rheum. 2008;58(6):1619–29. doi: 10.1002/art.23497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saurer L, McCullough KC, Summerfield A. In vitro induction of mucosa-type dendritic cells by all-trans retinoic acid. J Immunol. 2007;179(6):3504–14. doi: 10.4049/jimmunol.179.6.3504. [DOI] [PubMed] [Google Scholar]
- 75.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 76.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–63. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 77.Winoto A, Littman DR. Nuclear hormone receptors in T lymphocytes. Cell. 2002;109(Suppl):S57–66. doi: 10.1016/s0092-8674(02)00710-9. [DOI] [PubMed] [Google Scholar]
- 78.Si J, Collins SJ. IL-3-induced enhancement of retinoic acid receptor activity is mediated through Stat5, which physically associates with retinoic acid receptors in an IL-3-dependent manner. Blood. 2002;100(13):4401–9. doi: 10.1182/blood-2001-12-0374. [DOI] [PubMed] [Google Scholar]
- 79.Nakajima H, Brindle PK, Handa M, Ihle JN. Functional interaction of STAT5 and nuclear receptor co-repressor SMRT: implications in negative regulation of STAT5-dependent transcription. EMBO J. 2001;20(23):6836–44. doi: 10.1093/emboj/20.23.6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453(7192):236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem. 2008;283(25):17003–8. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- 82.Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29(1):44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yarmus M, Woolf E, Bernstein Y, Fainaru O, Negreanu V, Levanon D, et al. Groucho/transducin-like Enhancer-of-split (TLE)-dependent and -independent transcriptional regulation by Runx3. Proc Natl Acad Sci U S A. 2006;103(19):7384–9. doi: 10.1073/pnas.0602470103. [DOI] [PMC free article] [PubMed] [Google Scholar]