

Abstract
Hepatitis C virus NS3 helicase can unwind double-stranded DNA and RNA and has been proposed to form oligomeric structures. Here we examine the DNA unwinding activity of monomeric NS3. Oligomerization was measured by preparing a fluorescently labeled form of NS3, which was titrated with unlabeled NS3, resulting in a hyperbolic increase in fluorescence anisotropy and providing an apparent equilibrium dissociation constant of 236 nm. To evaluate the DNA binding activity of individual subunits within NS3 oligomers, two oligonucleotides were labeled with fluorescent donor or acceptor molecules and then titrated with NS3. Upon the addition of increasing concentrations of NS3, fluorescence energy transfer was observed, which reached a plateau at a 1:1 ratio of NS3 to oligonucleotides, indicating that each subunit within the oligomeric form of NS3 binds to DNA. DNA unwinding was measured under multiple turnover conditions with increasing concentrations of NS3; however, no increase in specific activity was observed, even at enzyme concentrations greater than the apparent dissociation constant for oligomerization. An ATPase-deficient form of NS3, NS3(D290A), was prepared to explore the functional consequences of oligomerization. Under single turnover conditions in the presence of excess concentration of NS3 relative to DNA, NS3(D290A) exhibited a dominant negative effect. However, under multiple turnover conditions in which DNA concentration was in excess to enzyme concentration, NS3(D290A) did not exhibit a dominant negative effect. Taken together, these data support a model in which monomeric forms of NS3 are active. Oligomerization of NS3 occurs, but subunits can function independently or cooperatively, dependent upon the relative concentration of the DNA.
Helicases are ubiquitous enzymes required for virtually all cellular processes involving nucleic acids, including replication, transcription, translation, repair, and recombination (1–5). These enzymes catalyze unwinding of double-stranded DNA or RNA by converting chemical energy from ATP hydrolysis into mechanical energy for nucleic acid strand separation. However, there is considerable variability in the quaternary structure of the active forms of helicases. Some helicases function effectively in unwinding activities as monomers, whereas others are active as dimers or oligomers. For example, bacteriophage T4 gp41 helicase and Escherichia coli DnaB helicase form hexameric structures that encircle and sequester single-stranded DNA (6, 7). Indeed, a large number of helicases form and function as hexameric structures (3). PcrA, a Gram-positive bacterial helicase, translocates on single-stranded DNA as a monomer (8) and has been proposed to unwind double-stranded DNA as a monomer (9). Bacteriophage T4 Dda helicase has activity in the monomeric form (10) but demonstrates more efficient unwinding activity under conditions where multiple helicase molecules bind a given substrate molecule (11). It has been shown that E. coli Rep helicase unwinds DNA as a dimer (12). UvrD has also been proposed to perform unwinding functions most optimally as a dimer (13) despite the fact that the monomer can translocate on ssDNA2 (14). However, a functional monomeric form of UvrD has been proposed based on biochemical and crystallographic studies (15, 16). In short, the oligomerization of helicases appears to increase the efficiency of unwinding, although monomeric helicases can translocate on ssDNA. Hence, oligomerization of helicases has been intensely studied, and the role for oligomerization is under debate for some enzymes.
The hepatitis C virus-encoded NS3 (nonstructural protein 3) is a 631-amino acid residue, bifunctional enzyme containing a serine protease in the amino-terminal 180 residues and an NTPase-RNA helicase in the carboxyl-terminal 451 residues (17). Each of the two domains can be expressed separately and retain activity (18). However, the activities are interdependent, since recently it was shown that the NS3 helicase domain stimulates the serine protease activity (19). Both protease and helicase activities are required for viral replication (20). NS3 binds to a protein co-factor, NS4A, which activates the protease activity (21). Crystal structures of NS3 and NS3 helicase domain (NS3h) have been reported (22–25). NS3 helicase has a 3′ to 5′ directional bias in unwinding, it is capable of unwinding both RNA and DNA double helices, and it binds more avidly to U-rich or dT-rich nucleic acid substrates (26–28). The kinetic and physical mechanisms for full-length NS3 and NS3h activity remain an active area of investigation.
Several laboratories have reported on the oligomeric state of full-length NS3 through different methods and have arrived at differing conclusions regarding the active species, including monomer, dimer, and oligomer (29–33). Dumont et al. (29) have reported that NS3 is active as a monomer with an 11-bp kinetic step size in optical trap unwinding experiments. Serebrov and Pyle (32) have reported that the active form of NS3 helicase is a dimer with an 18-bp kinetic step size through the use of a unique set of randomly nicked substrates. Tackett et al. (33) have reported that multiple molecules of NS3 bind to a single DNA duplex and are required for optimal unwinding. Most recently, Sikora et al. (34) have shown that NS3 can form oligomeric structures and that the enzyme's optimal DNA unwinding activity correlates with formation of those structures. The question arises as to whether the monomeric form of NS3 also has DNA unwinding activity and, if so, how it relates to the oligomeric forms of the enzyme.
Several kinetic and physical methods exist for determining the oligomeric state of helicase enzymes. Poisoning the activity of an ensemble with inactive protein has been used to illustrate interactions or the lack of interactions among protein monomers for the NS3 helicase domain (35, 36). However, the study demonstrating interactions based on a dominant-negative phenotype has been reinterpreted as a functionally cooperative interaction that does not require canonical protein-protein interactions (37).
In order to address the question of whether oligomerization or cooperativity is required for DNA unwinding by hepatitis C virus (HCV) NS3, a series of experiments were performed to measure NS3 unwinding activity under a variety of conditions. Previous experiments were designed to maximize processivity by using a vast excess of enzyme over nucleic acid. In the current report, NS3 unwinding activity was measured over a range of enzyme concentrations and enzyme-to-substrate ratios in order to examine the activity under conditions that disfavor binding of multiple helicase molecules to a single substrate molecule. Such conditions should allow discernment of helicase activity that can be attributed to monomeric as compared with oligomeric forms of this enzyme.
EXPERIMENTAL PROCEDURES
Materials—HEPES, EDTA, β-mercaptoethanol (βME), SDS, MOPS, Tris, NaCl, Na4EDTA, SDS, BSA, HEPES, acrylamide, bisacrylamide, MgCl2, KOH, ATP, formamide, xylene cyanole, bromophenol blue, urea, glycerol, and MgCl2 were purchased from Fisher. Sephadex G-25, phosphoenolpyruvate kinase/lactate dehydrogenase, NADH, ATP, and phosphoenolpyruvate were from Sigma. Poly(U) was purchased from Amersham Biosciences. DNA oligonucleotides were from Integrated DNA Technologies and purified by preparative gel electrophoresis (38). [γ-32P]ATP was purchased from PerkinElmer Life Sciences. T4 polynucleotide kinase was obtained from New England Biolabs. Recombinant full-length NS3, NS3(D290A), and NS3-tetra-Cys were derived from 1b replicon consensus sequence and expressed and purified as SUMO fusion proteins.3 The sumoylation was removed during the purification process by incubating the crudely purified protein with Ulp 1 protease. NS3 helicase domain (NS3h) derived from the same Con 1b sequence was expressed and purified as previously described (39). A form of NS3, NS3-tetra-Cys, was engineered to have a tetracysteine cassette (Cys-Cys-Pro-Gly-Cys-Cys) located at the C terminus of the protein by using the QuikChange mutagenesis kit (Stratagene). NS3-tetra-Cys and FlAsH-EDT2 (Invitrogen) were incubated at 4 °C for 5 h with gentle agitation. After dialysis to remove free dye, ∼85% of the protein was found to have bound the dye. The resulting labeled protein was referred to NS3-FlAsH.
Multiple Turnover DNA Unwinding—NS3 was prepared in 25 mm MOPS (pH 7.0), 10 mm NaCl, 0.1 mm EDTA (pH 8.0), 2 mm βME, and 0.1 mg/ml BSA. DNA substrate (15 nt/30 bp, the longer strand radiolabeled with 32P) was added to 100 nm, and the mixture was incubated at 37 °C for 5 min. The sequence of the 45-nt oligonucleotide was 5′-GACTGACGCTAGGCTGACAGGACGTACTACT15-3′. The sequence of the 30-nt complementary strand was 5′-GTAGTACGTCCTGTCAGCCTAGCGTCAGTC-3′. The unwinding reaction was initiated by the addition of 5 mm ATP, 10 mm MgCl2, and a 30-fold excess of DNA trap to bind the displaced strand and prevent reannealing to the radiolabeled product. At appropriate time points, a 10-μl aliquot of the reaction mixture was transferred to a centrifuge tube containing 200 mm EDTA, 0.7% SDS, 0.1% bromphenol blue, 0.1% xylene cyanol, and 6% glycerol. The double- and single-stranded DNA were resolved via native 20% polyacrylamide gel. The radiolabeled substrate and product were detected using a PhosphorImager (Amersham Biosciences). Quantitation was performed with ImageQuant software (GE Healthcare), and the ratio of single- to double-stranded DNA was plotted as a function of time. Data were fit to a straight line using Kaleidagraph (Synergy Software, Reading, PA). Identical experiments were performed at 100 and 500 nm NS3wt incubated with varying amounts of ATPase-deficient NS3(D290A) with 100 nm and 1.25 μm substrate, respectively.
Single Turnover DNA Unwinding—NS3 (250 nm NS3wt with variable NS3(D290A)) and DNA substrate (2 nm 15 nt/30 bp, the longer strand radiolabeled with 32P) were prepared in 25 mm MOPS (pH 7.0), 10 mm NaCl, 0.1 mm EDTA (pH 8.0), 2 mm βME, and 0.1 mg/ml BSA. The protein(s) was co-incubated for 90 min at 37 °C. Substrate was then incubated with the protein(s) for 5 min. Reaction components were incubated at 37 °C using a circulating water bath. The unwinding reaction was initiated by the rapid addition of 5 mm ATP, 10 mm MgCl2 (unless otherwise specified), 30-fold excess DNA trap, and 10-fold excess poly(U) protein trap using an RQF-3 Rapid Quench Flow instrument (KinTek Corp., Austin, TX). The reaction was quenched after 0.1–30 s by adding 200 mm EDTA and 0.7% SDS. Bromphenol blue (0.1%), 0.1% xylene cyanol, and 6% glycerol were added to each, and the double- and single-stranded DNA were resolved via native 20% polyacrylamide gel. The radiolabeled substrate and product were detected using a PhosphorImager, and quantitation was performed with ImageQuant software (GE Healthcare). Data were converted to concentration of ssDNA product as described (33).
Fluorescence Anisotropy Assay—Experiments were performed at 37 °C using a PerkinElmer Life Sciences Victor3V 1420 multilabel counter. Measurements were integrated over a 0.2-s detection period. The instrument lamp energy was at the maximum setting, and filters were set to 485 nm for excitation and 535 nm for emission. The polarizing aperture was set to normal, and the excitation aperture was set to 4 mm. NS3-tetra-Cys was labeled with FlAsH and prepared as described (40). The labeled protein was referred to as NS3-FlAsH. A solution of NS3-FlAsH (50 nm) was titrated against an increasing concentration of NS3wt or NS3h in buffer containing 25 mm MOPS, pH 7.0, 10 mm NaCl, 0.1 mm EDTA, 1 mm βME, and 0.1 mg/ml BSA. Triplicate samples were incubated at 37 °C for 90 min prior to measuring fluorescence polarization. Data were plotted as the average of three independent experiments with S.D. values.
Fluorescence Resonance Energy Transfer Assay—Measurements were made using a SLM Aminco-Bowman Series 2 luminescence spectrometer. The temperature was regulated at 37 °C with a circulating water bath. The excitation wavelength was set to 550 nm with a band pass of 0.5 nm. The emission wavelength was set to 668 nm with a band pass of 8 nm. Buffer consisted of 25 mm MOPS, pH 7.0, 10 mm NaCl, 0.1 mm EDTA, 1 mm βME, and 0.1 mg/ml BSA. 5′-Cy3-labeled and 3′-Cy5-labeled, noncomplementary, 8-base oligonucleotides were added to the buffer to a final concentration of 500 nm each. The sequences for the probes were 5′-GTCACACT-Cy5–3′ and 5′-Cy3-AGCATCAG-3′. NS3wt or NS3h was then titrated into solution, and the intensity of emission at 668 nm was detected. Data for NS3wt were plotted as the average of three experiments with S.D. values. The experiment was repeated with each oligonucleotide probe at 2.5 μm, and NS3wt was titrated into the solution.
RESULTS
Fluorescence Anisotropy of NS3-FlAsH with NS3wt or NS3h—Previous work has indicated that NS3 interacts with itself. Results have indicated a dimeric form of the enzyme (30, 32), whereas other studies have suggested that larger oligomers can also form (34). As a biophysical method of investigating the interaction of NS3 with itself, a fluorescence anisotropy assay was employed. Fluorescence anisotropy is a technique that reports on the size and shape of a species in solution (41). If the size of a species increases upon interactions with a binding partner, then the degree of fluorescence polarization increases. Conversely, if a species is broken into smaller units upon mixing, then polarization decreases. To conduct these experiments, we prepared a variant of NS3 containing a peptide (Cys-Cys-Pro-Gly-Cys-Cys) on the C terminus (NS3-tetra-Cys) that binds tightly to a fluorescein derivative termed FlAsH (42, 43). The labeled protein, NS3-FlAsH, was found to behave similarly to wild-type NS3 with regard to DNA unwinding (supplemental Fig. 1). NS3-FlAsH was titrated with NS3wt and NS3h in unwinding assay buffer (Fig. 1). The results show an increase in anisotropy when the labeled protein is mixed with NS3wt, whereas little or no change is noted when it is mixed with NS3h. Fitting the data to the quadratic equation results in an apparent Kd value of 236 ± 52 nm. This result provides direct support for NS3 oligomerization in solution and highlights the importance of the protease domain in mediating NS3-NS3 interactions, as was previously reported (30). However, one consideration is that the observed equilibrium dissociation constant in Fig. 1 may be reflective of multiple equilibria, including monomers associating to form dimers, dimers associating to form tetramers, and so on, in light of previous studies indicating that quaternary structures larger than dimers are likely to exist at concentrations of NS3 of ≥1 μm (34).
FIGURE 1.

Fluorescence anisotropy of NS3-FlAsH titrated with wild-type NS3 and NS3h. Shown is polarization of 50 nm NS3-FlAsH with wild-type NS3 (•) and NS3h (▪) in unwinding buffer. The data were fit to the quadratic equation using Kaleidagraph software. The resulting observed Kd value from this fit was 236 ± 52 nm.
NS3 Behaves as an Oligomer in Solution That Is Capable of Binding Multiple Nucleic Acid Strands—To explore whether NS3 oligomers can bind to DNA, a fluorescence resonance energy transfer (FRET) experiment was designed to simultaneously report on the oligomerization of the protein as well as the number of active DNA binding sites within the oligomer. Binding of DNA to NS3 as a function of NS3 concentration was examined by using oligonucleotides that were designed to bind in a 1:1 stoichiometry with NS3. The nucleic acid binding site of NS3 has previously been shown to accommodate 8 nucleotides (25). Therefore, Cy3- and Cy5-labeled 8-mers were used as probes to minimize the possibility of multiple NS3 monomers binding to an individual nucleic acid sequence. The Cy3 and Cy5 FRET pair was chosen based on their successful application in protein-nucleic acid measurements. The Förster radius for this pair has been determined to be 61–65 Å (44). Hence, upon titration of the mixture of oligonucleotides with NS3, FRET should be observed if the oligonucleotides bind to adjacent sites within an oligomeric form of NS3 as depicted in Fig. 2A.
FIGURE 2.

Fluorescence resonance energy transfer between Cy3- and Cy5-labeled oligonucleotides bound by NS3. A, 8-mer oligonucleotides labeled by Cy3 or Cy5 fluorophores were titrated with NS3wt or NS3h. If the protein forms oligomeric structures that are capable of binding each oligonucleotide in adjacent sites, then FRET will result. B, scanning emission spectra for samples containing the two oligonucleotide probes (each at 500 nm) in the absence (○) or presence (•) of 1.5 μm NS3wt. The excitation wavelength was 550 nm. C, the donor fluor (Cy3) was excited at 550 nm, and emission of acceptor (Cy5) at 668 nm was measured as a function of NS3 (▪) or NS3h (•) concentration. Data for NS3 are reported as an average of three experiments with S.D. values. D, the experiment in C was repeated at a 2.5 μm concentration of each oligonucleotide probe and titrated with NS3wt. E, fluorescence data from C and D were normalized and are shown relative to the protein/oligonucleotide ratio.
The concentration of each nucleic acid was initially set at 500 nm in order to be above the equilibrium dissociation constant for ssDNA binding under these conditions (7 nm; supplemental Fig. 2). The raw emission spectra for the oligonucleotide probes alone and in the presence of 1.5 μm NS3 are shown in Fig. 2B. The emission from the Cy3 fluorophore is reduced in the spectrum generated from the sample containing NS3 with a concomitant increase in the signal from the Cy5 label, as expected for a FRET signal. The emission was measured as a function of increasing NS3 concentration and is plotted in Fig. 2C. Interestingly, the FRET signal reached a maximum at a ∼1:1 ratio of protein to nucleic acid strands, which indicates that all of the protein is capable of binding nucleic acid substrate. Additionally, NS3h, which does not display the oligomeric characteristics of full-length NS3, did not exhibit an increase in FRET acceptor signal as protein was titrated into a solution containing labeled oligonucleotides. The sigmoidal shape of the curve in Fig. 2C suggests that positive cooperativity occurs due to protein-protein interactions, protein-nucleic acid interactions, or both. If protein-protein interactions are solely responsible for the sigmoidal shape, then repeating the titration at higher nucleic acid concentration (and hence higher protein concentration) may reduce the sigmoidicity. The concentration of each oligonucleotide probe was increased to 2.5 μm, and the titration was repeated. As with the experiment performed at lower DNA concentration, the shape of the binding curve was sigmoidal, and the FRET signal reached a maximum point when the nucleic acid strand/protein ratio was 1:1 (Fig. 2D). Overlaying the scaled data from Fig. 2, C and D (Fig. 2E) shows that both curves demonstrate a very similar sigmoidal increase in signal, suggesting that the initial phase of the curves may due to a threshold that must be exceeded for the development of FRET signal. Thus, the equilbria that are responsible for the shape of the FRET signal as a function of NS3 concentration are likely to involve protein-protein and protein-nucleic acid interactions. There are several clear results from this experiment: 1) NS3 forms oligomeric species consisting of, at a minimum, dimers in the presence of nucleic acids; 2) adjacent monomers within the oligomeric structure are capable of binding nucleic acid substrates; and 3) all of the protein is capable of binding to DNA even after oligomerization.
DNA Unwinding at Varying Concentrations of NS3 Indicates no Increase in Specific Activity under Multiple Turnover Conditions— Formation of NS3 oligomeric structures has been correlated with increasing DNA and RNA unwinding activity by showing that the burst amplitude for product formation increases with increasing enzyme concentration (34). The reported experiments were conducted under single turnover conditions in which the enzyme concentration was similar to or greater than the DNA substrate concentration. The results indicated that if a monomeric form of NS3 is active, then its processivity is relatively low compared with oligomeric forms.
DNA unwinding can be measured under multiple turnover conditions in which the substrate concentration is similar to or greater than the enzyme concentration. Multiple turnover conditions effectively increase the “signal” for measuring DNA unwinding by allowing multiple opportunities for unwinding to occur. If NS3 must oligomerize to function, then the specific activity of the enzyme should increase as the enzyme concentration increases. To determine if the active species of NS3 changes with a dependence on enzyme concentration, we measured unwinding of a DNA substrate under multiple turnover conditions over a range of enzyme concentrations, both below and above the apparent Kd value observed by anisotropy. If NS3-NS3 interactions are necessary for the observed DNA unwinding activity, then an increase in specific activity with increasing enzyme concentration should be observed, as long as DNA unwinding is rate-limiting in the reaction. An increase in the unwinding rate with increasing enzyme concentration was observed (Fig. 3A), but no increase in specific activity was measured (Fig. 3B). The rate constant for dissociation of NS3 from the 15-nt/30-bp substrate is ∼1 s–1.4 NS3 can unwind the 30-bp substrate with a rate constant of 0.4 s–1 under optimal conditions, in which enzyme concentration is much greater than the DNA substrate concentration (see below). Hence, dissociation of NS3 from DNA is unlikely to limit the rate of the multiple turnover catalysis. Therefore, the lack of any change in the specific activity of the enzyme can best be explained if the enzyme species that is responsible for unwinding does not change over the concentration range of the experiment. NS3 at concentrations that are very low (2.5 nm) clearly unwinds DNA. These concentrations are well below the apparent KD value for protein-protein interactions (Fig. 1); hence, the simplest conclusion is that the monomeric form of NS3 can unwind DNA. However, the lack of an increase in specific activity at increasing concentrations of NS3 was surprising, because the data in Figs. 1 and 2 show clearly that the enzyme's quaternary structure changes over this concentration range. For comparison, DNA unwinding was investigated for NS3h under similar multiple-turnover conditions. No increase in specific activity was observed for NS3h (Fig. 3C) consistent with behavior of a monomeric enzyme.
FIGURE 3.

DNA unwinding by NS3 or NS3h under multiple turnover conditions with excess concentrations of DNA. A, NS3-catalyzed unwinding of 250 nm 15-nt/30-bp partial duplex DNA by 2.5 (•), 5 (○), 10 (▪), 15 (□), 20 (▴), 25 (▵), 35 (♦), 50 (⋄), 75 (×), 100 (+), 175 (▴), 250 (▿), 350 (▾), and 500 nm (▹) NS3 under multiple turnover conditions. B, specific activity of unwinding of 15-nt/30-bp partial duplex DNA as a function of NS3 enzyme concentration. C, specific activity for DNA unwinding of 15-nt/30-bp partial duplex DNA as a function of NS3h enzyme concentration.
NS3 Activity Is Susceptible to Dominant Negative Effects under Excess Enzyme Conditions in Vitro—DNA unwinding by NS3 was measured under excess enzyme, single turnover conditions in the absence or presence of increasing amounts of NS3(D290A). A D290A mutation in the conserved DEX(H/D) motif of NS3 abolishes the ATPase activity and consequently the ATP-dependent unwinding activity of the enzyme. We previously reported that the DNA binding affinity of the NS3(D290A) was very similar to that of the wild type protein (45) (supplemental Fig. 3). Under these conditions, the amplitudes of the unwinding reactions were decreased, and the observed rates for unwinding were also decreased (Fig. 4A and Table 1). The observed rates were plotted as a function of the ATPase-deficient protein concentration (Fig. 4B). These data were fit to the following equation (35, 46),
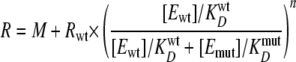 |
(Eq. 1) |
where R is the observed rate constant for unwinding, Rwt is the rate constant of unwinding for wild-type protein alone, M is the rate constant of unwinding for heterogeneous complexes of wild-type and mutant enzymes, [Ewt] and [Emut] are the concentrations of wild-type and mutant proteins, respectively, and KwtD and KmutD are the equilibrium binding constants of wild-type and mutant NS3 to fluorescein-labeled T15 ssDNA (supplemental Fig. 3). The exponential value, n, represents the number of units present in the active complex. The best fit of these data produced a value for n of 11 ± 6 (Fig. 4B). The data deviate from a simple model of competition for substrate between active and inactive protein, which is given by Equation 2.
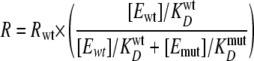 |
(Eq. 2) |
Simulated data based on Equation 2 are shown as the dashed line in Fig. 4B. The result indicates that, under single turnover conditions, the reduction in observed rate constants was not due solely to competitive interactions for substrate between the wild-type and mutant proteins, indicating that the inactive protein was interfering with an active oligomer in a dominant-negative fashion. This result demonstrates that multiple molecules of NS3 are responsible for product formation under excess enzyme, single turnover conditions, in agreement with previous reports (33, 34). Furthermore, this result is similar to investigations of NS3 helicase domain activity (35).
FIGURE 4.

Effect of an ATPase-deficient variant on wild type NS3 activity under excess enzyme conditions. A, unwinding of 2 nm 15-nt/30-bp partial duplex DNA by 250 nm wild type NS3 alone (•), or in the presence of 25 (⋄), 125 (▴), 250 (▿), or 500 (▴) nm NS3(D290A). Data were fit to a single exponential by using Kaleidagraph software, and the kinetic parameters are reported in Table 1. B, observed unwinding rate constants from A were plotted as a function of NS3(D290A) concentration. The solid line represents the fit of the data to Equation 1. The dashed line simulates expected rates based on a competition between wild type NS3 and NS3(D290A) for binding to the DNA substrate based on Equation 2.
TABLE 1.
Kinetic parameters from single turnover unwinding of 2 nm DNA substrate by NS3 in the presence of an ATPase-deficient variant of NS3 (data from Fig. 4)
Reported error represents the S.E. of the best fit of the data to a single exponential using the program Kaleidagraph.
| NS3 | NS3(D290A) | Observed unwinding rate | Amplitude |
|---|---|---|---|
| nm | nm | s-1 | % |
| 250 | 0 | 0.42 ± 0.04 | 70 ± 2 |
| 250 | 25 | 0.17 ± 0.01 | 60 ± 1 |
| 250 | 125 | 0.13 ± 0.01 | 42 ± 2 |
| 250 | 250 | 0.08 ± 0.02 | 32 ± 3 |
| 250 | 500 | 0.11 ± 0.05 | 16 ± 3 |
NS3 Activity Is Not Susceptible to Dominant Negative Effects under Conditions of Excess DNA in Vitro—The data in the single turnover unwinding experiments suggested that an oligomeric species of NS3 was responsible for optimal DNA unwinding (Fig. 4B). However, results from the multiple turnover DNA unwinding experiment conducted over a broad range of enzyme concentrations indicated that there was no change in the active species (Fig. 3). One possible explanation for all of these data is that NS3 forms oligomeric structures but that individual subunits are capable of functioning alone when DNA is in excess of enzyme. It is possible that above the KD value for protein-protein interaction, NS3 exists as an oligomer but that individual subunits function independently in the presence of excess DNA substrate. To test this hypothesis, DNA unwinding experiments were performed with full-length NS3 in the presence of inactive NS3(D290A) under multiple turnover conditions and with excess nucleic acid concentration over enzyme concentration. Experiments were conducted at NS3wt concentrations of 100 and 500 nm (Fig. 5, B and C, respectively). The ATPase-deficient form of NS3 had no significant effect on the rate of unwinding by wild type NS3 under these conditions, although the concentration of protein was above the apparent KD value for protein-protein interactions (Fig. 5, B and D). This indicates that protein complexes that form between the variant enzyme and the wild type enzyme do not lead to reduced activity under these conditions. The simplest explanation for the absence of any dominant negative effect is that NS3-NS3 interactions are not required for the unwinding activity observed in the presence of excess DNA substrate.
FIGURE 5.

Effect of an ATPase-deficient form of NS3 on wild type NS3 activity under excess DNA conditions. A, unwinding of 250 nm 15-nt/30-bp partial duplex DNA by 100 nm wild type NS3 in the presence of 0 (○), 10 (□), 25 (⋄), 50 (▵), 100 (•), and 150 nm (▪) NS3(D290A) under multiple turnover conditions. B, unwinding rates from A plotted as a function of NS3(D290A) concentration. C, unwinding of 1.25 μm 15-nt/30-bp partial duplex DNA by 500 nm NS3 in the presence of increasing concentration of NS3(D290A) under multiple turnover conditions. D, unwinding 250 nm 15-nt/30-bp partial duplex DNA by 100 nm NS3h under multiple turnover conditions as a function of NS3h(D290A) concentration.
For comparison, we measured NS3h-catalyzed DNA unwinding of the 15-nt/30-bp substrate under multiple turnover conditions in the presence of an ATPase-deficient form of NS3h containing the same mutation as the full-length protein, NS3h(D290A). As was observed with full-length NS3, no dominant negative result was observed for NS3h in the presence of excess substrate and increasing concentrations of NS3h(D290A) (Fig. 5D).
DISCUSSION
Helicases have been shown to exist in different oligomeric states in order to carry out their activities on nucleic acid substrates. When oligomeric structures are required for optimal activity, a question that remains is whether the monomeric form of the helicase retains activity. Work from several laboratories has led to differing proposals regarding the oligomeric state of NS3. These proposals include NS3 functioning as a monomer (29, 47), as a dimer (30–32), and as an oligomer (33, 34). The data in this report also appear to provide conflicting results. Despite the biophysical appearance of oligomeric behavior in solution, some of the kinetic investigations support the postulate that the monomer is the least complex, active species of NS3.
A model was developed to reconcile the kinetic and biophysical data and is shown in Fig. 6. In this model, NS3 exists in equilibrium between monomeric and oligomeric forms, each capable of binding to and unwinding DNA. Under conditions where the protein is in excess of the substrate, multiple NS3 subunits bind to the substrate, and their cooperative action gives rise to rapid and processive formation of ssDNA product. Under conditions where the DNA substrate is in excess of the protein, the oligomeric form is capable of binding multiple nucleic acid strands, which is strongly supported by data in Fig. 2. When NS3 active sites are saturated with DNA substrate or with annealing trap, the observed activity is similar to that of monomeric NS3, although NS3 exists as an oligomer. Thus, interpretation of the kinetic data for DNA unwinding in terms of monomers or oligomers is dependent on the relative concentrations of enzyme and DNA. The results in this report support the conclusion that NS3 exists in equilibrium between monomeric and oligomeric species in which the monomeric units are capable of independent or cooperative function, depending on the relative concentration of nucleic acid.
FIGURE 6.

Model for NS3 oligomeric behavior in the presence of limiting or excess DNA substrate. NS3 exists in equilibrium between monomeric and oligomeric forms in solution. The monomeric form can bind and unwind DNA. Increasing NS3 concentration favors oligomerization. Upon the addition of a nucleic acid substrate, NS3 binds along the substrate and exhibits processive unwinding activity when the enzyme concentration is in excess of the substrate concentration. When the concentration of nucleic acid is in excess of the enzyme, NS3 is saturated by substrate (or the single-stranded DNA that is included as an annealing trap). Under conditions in which nucleic acid is saturating, the observed activity can be attributed to active NS3 monomeric subunits. B, NS3h exists as a monomer in solution. Under conditions of excess enzyme concentration over DNA concentration, multiple NS3h monomers can bind to the same substrate molecule, resulting in functional cooperativity. When the concentration of DNA exceeds the concentration of NS3h, only one molecule of NS3h binds to the DNA substrate.
Multiple turnover DNA unwinding experiments conducted by varying the concentration of wild-type NS3 exhibited no increase in specific activity (Fig. 3). Further, when a fixed amount of wild-type NS3 was mixed with increasing concentrations of an ATPase-deficient variant, no reduction in unwinding activity was observed under multiple turnover conditions (Fig. 5). These observations, taken together, can be interpreted as if NS3 behaves functionally as a monomeric enzyme when the DNA concentration is in excess of the protein concentration. It should be noted that under multiple turnover conditions, a large excess of annealing trap was present in the reaction mixture, thereby making the total nucleic acid concentration in great excess to the enzyme concentration.
Under conditions where substrate is limiting, NS3 appears to behave as an oligomeric enzyme. Previous reports, as well as the single turnover experiments reported here, revealed that NS3 can be a highly proficient helicase when the protein concentration is in excess of the substrate (33, 48). As shown in Fig. 2A, 70% of a 30-bp substrate was unwound within 20 s. However, the addition of NS3(D290A) to the wild-type enzyme caused a dramatic drop in the observed rate constant for unwinding (Fig. 4B). These results are very similar to the results obtained by Levin et al. (35) for NS3h. The dashed line in Fig. 4B represents the expected rates if the interaction between NS3wt and NS3(D290A) was a simple competition for nucleic acid substrates. The data were not reflective of a competition for binding substrate but showed that the species responsible for unwinding was a large, oligomeric species consisting of 11 ± 6 units of NS3. A recent report proposed that some DEAD-box proteins function optimally as “local strand separators” as opposed to translocating helicases (49). NS3 contains a DExH-box, but when enzyme concentration is saturating relative to substrate, it may behave in a similar manner as DEAD-box proteins.
The model in Fig. 6 can also explain the effects that inactive NS3(D290A) exerts on NS3wt under the different assay conditions. In both multiple turnover (excess DNA) conditions and single turnover (excess enzyme) conditions, the concentration of the active species is fixed, and the concentration of the inactive species is varied. In the case of multiple turnover conditions, the substrate concentration is such that the available active sites are saturated. If inactive protein binds substrate, the binding event does not influence the ensemble unwinding rate because the active NS3 monomers are responsible for the observed unwinding. Under excess enzyme conditions, multiple molecules of NS3 are responsible for unwinding any one duplex. In this scenario, the maximal rate for unwinding is only achieved when all of the protein bound to the substrate is active. As NS3(D290A) is added to the reaction, the variant enzyme cannot contribute to active DNA unwinding, leading to the reduction in the observed rate constant for unwinding.
The NS3 helicase domain does not form oligomeric structures under the conditions examined here. However, an ATPase-deficient form of NS3h can poison DNA unwinding activity, as reported by the Patel laboratory (35). The explanation for this behavior is that NS3h exhibits functional cooperativity, in which multiple molecules of enzyme can bind and act on the same DNA substrate molecule, as shown in Fig. 6B. When enzyme concentration is in excess of DNA substrate concentration, NS3h can exhibit kinetic behavior that is consistent with an oligomeric protein, but it is actually multiple monomers acting on the same substrate. When the DNA substrate concentration is in excess of the enzyme concentration, only one molecule of NS3h binds to the DNA substrate. Under such conditions, the addition of an ATPase-deficient form of NS3h does not poison the reaction, as shown in Fig. 5D. Hence, the apparent discrepancy in the results is due to the behavior of NS3h, which is dependent on the relative concentration of DNA substrate. NS3h has also been shown recently to be capable of functioning as a monomer, at least in the presence of a single-stranded binding protein (50).
Mechanism(s) for NS3 translocation and unwinding have recently been proposed by two groups using single molecule approaches. The mechanisms are proposed for monomeric forms of NS3. Dumont et al. (29) used high resolution optical tweezers to examine RNA unwinding by NS3. A model for ATP hydrolysis-dependent stepping was proposed that includes a large step of 11 ± 3 base pairs that is made up of substeps of 3.6 ± 1.3 base pairs. Myong et al. (51) used single-molecule fluorescence analysis to deduce additional substeps of 1 bp that were coupled directly to ATP hydrolysis and resulted in a “spring-loaded” movement of the enzyme of 3 bp. The work in the current report clearly supports a functional, monomeric form of NS3. It should be noted that the concentrations of NS3 utilized in the single molecule experiments were much lower than the apparent KD value for protein-protein interactions reported here (Fig. 1). Hence, the proposed models for DNA or RNA unwinding put forth by others are not disputed by the data in this report. It remains to be determined whether oligomerization of NS3 provides an additional or alternative component to the proposed mechanochemical mechanisms for monomeric NS3.
It is likely that HCV genome replication is accomplished by a multiprotein complex, including, but not necessarily limited to, both helicase and polymerase enzymes. HCV NS3 helicase and NS5B polymerase have been shown to interact and modulate each other's activity in in vitro assay systems (40, 45, 52–54). This influence may be indicative of a stronger functional interdependence in vivo. Additionally, NS3 associates with the NS4A cofactor, which serves as a membrane anchor for NS3 (55). The HCV replication complex is localized within an ER-derived membrane compartment within infected cells (56). This probably provides a lipophilic replication environment that is difficult to recapitulate in vitro and probably influences the local concentrations of the relevant enzymes and their substrates in a manner that is difficult to predict.
The tendency for NS3-NS3 interactions observed in solution may reflect in vivo interaction properties of NS3 in any of several ways. First, NS3 may be only one component of a multiprotein HCV molecular motor system. The helicase interaction with nucleic acid may be stabilized by one or more unknown cofactors in vivo, or the HCV polymerase may provide some of the motive force necessary for processive unwinding of double-stranded intermediates (57). Second, NS3 may interact extensively with the lipid membrane environment at the site of replication. Removing the protein from this environment would probably predispose it to nonspecific hydrophobic interactions in the aqueous environment common to most in vitro activity assays. Third, NS3 may act as a protein complex scaffold during replication, interacting with several different viral and host proteins (58). The absence of these natural interaction partners in in vitro assay systems may result in formation of biologically nonrelevant protein interactions. It is not clear whether the conditions that favor greatest activity in vitro are also the conditions that are operable in vivo. There is currently no evidence that NS3 is required to be highly processive in vivo. Work reported by Quinkert et al. (59) showed that the concentration of NS3 greatly exceeds the concentration of HCV RNA in isolated replication complexes. Therefore, it is possible that multiple NS3 molecules operate on each strand of HCV RNA.
The overall conclusions of this report are that NS3 forms oligomeric structures in vitro but that the monomeric form is active and that monomeric subunits are capable of retaining independent function within the context of the oligomer. Conditions in which the enzyme is saturated with nucleic acid substrate favor NS3 function as a monomeric subunit. Conditions that favor oligomerization in the presence of limiting substrate allow multiple NS3 subunits to function on a single substrate molecule, thereby producing a more processive form of the helicase in vitro.
Supplementary Material
Acknowledgments
We thank Qixin Wang for helpful discussions.
This work was supported by grants from the National Institutes of Health (AI060563 to K.D.R. and C.E.C.), Arkansas IDeA Network of Biomedical Research Excellence Research Grant (P20RR016460), and the Arkansas Biosciences Institute. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
Footnotes
The abbreviations used are: ssDNA, single-stranded DNA; MOPS, 4-morpholinepropanesulfonic acid; BSA, bovine serum albumin; FRET, fluorescence resonance energy transfer; HCV, hepatitis C virus.
D. Sikora, B. Sikora, and K. D. Raney, manuscript in preparation.
K. Raney and D. Sikora, manuscript in preparation.
References
- 1.Delagoutte, E., and von Hippel, P. H. (2002) Q. Rev. Biophys. 35 431–478 [DOI] [PubMed] [Google Scholar]
- 2.Lohman, T. M., and Bjornson, K. P. (1996) Annu. Rev. Biochem. 65 169–214 [DOI] [PubMed] [Google Scholar]
- 3.Patel, S. S., and Picha, K. M. (2000) Annu. Rev. Biochem. 69 651–697 [DOI] [PubMed] [Google Scholar]
- 4.Soultanas, P., and Wigley, D. B. (2001) Trends Biochem. Sci. 26 47–54 [DOI] [PubMed] [Google Scholar]
- 5.von Hippel, P. H., and Delagoutte, E. (2001) Cell 104 177–190 [DOI] [PubMed] [Google Scholar]
- 6.Dong, F., Gogol, E. P., and von Hippel, P. H. (1995) J. Biol. Chem. 270 7462–7473 [DOI] [PubMed] [Google Scholar]
- 7.Jezewska, M. J., Rajendran, S., Bujalowska, D., and Bujalowski, W. (1998) J. Biol. Chem. 273 10515–10529 [DOI] [PubMed] [Google Scholar]
- 8.Dillingham, M. S., Wigley, D. B., and Webb, M. R. (2002) Biochemistry 41 643–651 [DOI] [PubMed] [Google Scholar]
- 9.Velankar, S. S., Soultanas, P., Dillingham, M. S., Subramanya, H. S., and Wigley, D. B. (1999) Cell 97 75–84 [DOI] [PubMed] [Google Scholar]
- 10.Nanduri, B., Byrd, A. K., Eoff, R. L., Tackett, A. J., and Raney, K. D. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 14722–14727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrd, A. K., and Raney, K. D. (2005) Biochemistry 44 12990–12997 [DOI] [PubMed] [Google Scholar]
- 12.Cheng, W., Hsieh, J., Brendza, K. M., and Lohman, T. M. (2001) J. Mol. Biol. 310 327–350 [DOI] [PubMed] [Google Scholar]
- 13.Maluf, N. K., Fischer, C. J., and Lohman, T. M. (2003) J. Mol. Biol. 325 913–935 [DOI] [PubMed] [Google Scholar]
- 14.Fischer, C. J., Maluf, N. K., and Lohman, T. M. (2004) J. Mol. Biol. 344 1287–1309 [DOI] [PubMed] [Google Scholar]
- 15.Lee, J. Y., and Yang, W. (2006) Cell 127 1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mechanic, L. E., Hall, M. C., and Matson, S. W. (1999) J. Biol. Chem. 274 12488–12498 [DOI] [PubMed] [Google Scholar]
- 17.Kim, D. W., Gwack, Y., Han, J. H., and Choe, J. (1995) Biochem. Biophys. Res. Commun. 215 160–166 [DOI] [PubMed] [Google Scholar]
- 18.Failla, C., Tomei, L., and De, F. R. (1995) J. Virol. 69 1769–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beran, R. K., and Pyle, A. M. (2008) J. Biol. Chem. 283 29929–29937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolykhalov, A. A., Mihalik, K., Feinstone, S. M., and Rice, C. M. (2000) J. Virol. 74 2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallinari, P., Paolini, C., Brennan, D., Nardi, C., Steinkuhler, C., and De, F. R. (1999) Biochemistry 38 5620–5632 [DOI] [PubMed] [Google Scholar]
- 22.Kang, L. W., Cho, H. S., Cha, S. S., Chung, K. M., Back, S. H., Jang, S. K., and Oh, B. H. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 121–123 [DOI] [PubMed] [Google Scholar]
- 23.Yao, N., Hesson, T., Cable, M., Hong, Z., Kwong, A. D., Le, H. V., and Weber, P. C. (1997) Nat. Struct. Biol. 4 463–467 [DOI] [PubMed] [Google Scholar]
- 24.Yao, N., Reichert, P., Taremi, S. S., Prosise, W. W., and Weber, P. C. (1999) Structure 7 1353–1363 [DOI] [PubMed] [Google Scholar]
- 25.Kim, J. L., Morgenstern, K. A., Griffith, J. P., Dwyer, M. D., Thomson, J. A., Murcko, M. A., Lin, C., and Caron, P. R. (1998) Structure 6 89–100 [DOI] [PubMed] [Google Scholar]
- 26.Gwack, Y., Kim, D. W., Han, J. H., and Choe, J. (1996) Biochem. Biophys. Res. Commun. 225 654–659 [DOI] [PubMed] [Google Scholar]
- 27.Gwack, Y., Kim, D. W., Han, J. H., and Choe, J. (1997) Eur. J. Biochem. 250 47–54 [DOI] [PubMed] [Google Scholar]
- 28.Tai, C. L., Chi, W. K., Chen, D. S., and Hwang, L. H. (1996) J. Virol. 70 8477–8484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumont, S., Cheng, W., Serebrov, V., Beran, R. K., Tinoco, I., Jr., Pyle, A. M., and Bustamante, C. (2006) Nature 439 105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khu, Y. L., Koh, E., Lim, S. P., Tan, Y. H., Brenner, S., Lim, S. G., Hong, W. J., and Goh, P. Y. (2001) J. Virol. 75 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Locatelli, G. A., Spadari, S., and Maga, G. (2002) Biochemistry 41 10332–10342 [DOI] [PubMed] [Google Scholar]
- 32.Serebrov, V., and Pyle, A. M. (2004) Nature 430 476–480 [DOI] [PubMed] [Google Scholar]
- 33.Tackett, A. J., Chen, Y., Cameron, C. E., and Raney, K. D. (2005) J. Biol. Chem. 280 10797–10806 [DOI] [PubMed] [Google Scholar]
- 34.Sikora, B., Chen, Y., Lichti, C. F., Harrison, M. K., Jennings, T. A., Tang, Y., Tackett, A. J., Jordan, J. B., Sakon, J., Cameron, C. E., and Raney, K. D. (2008) J. Biol. Chem. 283 11516–11525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levin, M. K., and Patel, S. S. (1999) J. Biol. Chem. 274 31839–31846 [DOI] [PubMed] [Google Scholar]
- 36.Preugschat, F., Danger, D. P., Carter, L. H., III, Davis, R. G., and Porter, D. J. (2000) Biochemistry 39 5174–5183 [DOI] [PubMed] [Google Scholar]
- 37.Levin, M. K., Wang, Y. H., and Patel, S. S. (2004) J. Biol. Chem. 279 26005–26012 [DOI] [PubMed] [Google Scholar]
- 38.Morris, P. D., Tackett, A. J., and Raney, K. D. (2001) Methods 23 149–159 [DOI] [PubMed] [Google Scholar]
- 39.Tackett, A. J., Wei, L., Cameron, C. E., and Raney, K. D. (2001) Nucleic Acids Res. 29 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jennings, T. A., Chen, Y., Sikora, D., Harrison, M. K., Sikora, B., Huang, L., Jankowsky, E., Fairman, M. E., Cameron, C. E., and Raney, K. D. (2008) Biochemistry 47 1126–1135 [DOI] [PubMed] [Google Scholar]
- 41.Lundblad, J. R., Laurance, M., and Goodman, R. H. (1996) Mol. Endocrinol. 10 607–612 [DOI] [PubMed] [Google Scholar]
- 42.Griffin, B. A., Adams, S. R., and Tsien, R. Y. (1998) Science 281 269–272 [DOI] [PubMed] [Google Scholar]
- 43.Griffin, B. A., Adams, S. R., Jones, J., and Tsien, R. Y. (2000) Methods Enzymol. 327 565–578 [DOI] [PubMed] [Google Scholar]
- 44.Rasnik, I., Myong, S., Cheng, W., Lohman, T. M., and Ha, T. (2004) J. Mol. Biol. 336 395–408 [DOI] [PubMed] [Google Scholar]
- 45.Piccininni, S., Varaklioti, A., Nardelli, M., Dave, B., Raney, K. D., and McCarthy, J. E. (2002) J. Biol. Chem. 277 45670–45679 [DOI] [PubMed] [Google Scholar]
- 46.Morris, P. D., Tackett, A. J., Babb, K., Nanduri, B., Chick, C., Scott, J., and Raney, K. D. (2001) J. Biol. Chem. 276 19691–19698 [DOI] [PubMed] [Google Scholar]
- 47.Porter, D. J., Short, S. A., Hanlon, M. H., Preugschat, F., Wilson, J. E., Willard, D. H., Jr., and Consler, T. G. (1998) J. Biol. Chem. 273 18906–18914 [DOI] [PubMed] [Google Scholar]
- 48.Pang, P. S., Jankowsky, E., Planet, P. J., and Pyle, A. M. (2002) EMBO J. 21 1168–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang, Q., Del, C. M., Lambowitz, A. M., and Jankowsky, E. (2007) Mol. Cell 28 253–263 [DOI] [PubMed] [Google Scholar]
- 50.Rajagopal, V., and Patel, S. S. (2008) J. Mol. Biol. 376 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Myong, S., Bruno, M. M., Pyle, A. M., and Ha, T. (2007) Science 317 513–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dimitrova, M., Imbert, I., Kieny, M. P., and Schuster, C. (2003) J. Virol. 77 5401–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishido, S., Fujita, T., and Hotta, H. (1998) Biochem. Biophys. Res. Commun. 244 35–40 [DOI] [PubMed] [Google Scholar]
- 54.Zhang, C., Cai, Z., Kim, Y. C., Kumar, R., Yuan, F., Shi, P. Y., Kao, C., and Luo, G. (2005) J. Virol. 79 8687–8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolk, B., Sansonno, D., Krausslich, H. G., Dammacco, F., Rice, C. M., Blum, H. E., and Moradpour, D. (2000) J. Virol. 74 2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gosert, R., Egger, D., Lohmann, V., Bartenschlager, R., Blum, H. E., Bienz, K., and Moradpour, D. (2003) J. Virol. 77 5487–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stano, N. M., Jeong, Y.-J., Donmez, I., Tummalapali, P., Levin, K. M., and Patel, S. S. (2005) Nature 435 370–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Restrepo-Hartwig, M. A., and Ahlquist, P. (1996) J. Virol. 70 8908–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quinkert, D., Bartenschlager, R., and Lohmann, V. (2005) J. Virol. 79 13594–13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.