

Abstract
A new enzyme-linked immunosorbent assay (ELISA)-based immunoglobulin G (IgG)-plus-IgM antibody detection test for severe acute respiratory syndrome (SARS) has been developed by using a cocktail of four recombinant polypeptides as the antigen. These recombinant fragments were designed as parts of two different structural proteins from SARS-associated coronavirus (SARS-CoV). One recombinant polypeptide, S251-683, was designed as part of the spike glycoprotein, and the other three polypeptides comprised almost the whole nucleocapsid protein, avoiding the last 25 C-terminal amino acids. Immunization with a cocktail of these four polypeptides yielded a specific polyclonal antibody that is able to recognize SARS-CoV-infected cells by an immunofluorescence assay. This polypeptide cocktail was also used to set up an ELISA-based IgG-plus-IgM antibody detection test, which showed 99% specificity and 90% sensitivity upon evaluation using sera from 100 healthy negative controls and 20 SARS patients. Separate immunoreactivity assays with each recombinant polypeptide demonstrated that a combination of N and S protein fragments was more suitable than the individual peptides for developing a serological assay for SARS-CoV.
A new coronavirus (CoV) (order Nidovirales, family Coronaviridae, genus Coronavirus) has been implicated as the causal agent of severe acute respiratory syndrome (SARS) (3, 20). Its single-stranded plus-sense RNA genome of ∼30 kb contains 23 putative open reading frames, including four major structural proteins that are common to all known CoVs: the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins (12). The control of an epidemic requires a rapid and accurate diagnostic technique that can promptly identify the infection and allow its treatment and the implementation of infection control measures, including the isolation of cases and the management of contacts to prevent further transmissions. The development of a sensitive and highly specific diagnostic kit that is effective at early stages of SARS is of great interest because of (i) the highly contagious and acute nature of the disease, (ii) the stress caused to individuals by unnecessary quarantines during an epidemic, and (iii) the absence of effective anti-SARS therapy. However, the level of virus excretion is comparatively low during the initial phase of SARS (15); therefore, early diagnosis requires a highly sensitive test that can detect the low levels of viral genome or viral proteins present during the first few days after SARS onset. Several molecular assays have been designed to provide an early diagnosis, based on the detection of specific RNA sequences by PCR. However, RNA extraction protocols are not straightforward and may produce RNA preparations that are not useful for reverse transcription if the protocols are not properly followed, yielding false-positive results. A specific antibody or antigen detection test would be technologically simpler and less expensive. Thus, the objective of the present study was to develop an easy-to-use diagnostic assay by evaluating the use of combinations of recombinant proteins obtained from the SARS-associated CoV (SARS-CoV) in an enzyme-linked immunosorbent assay (ELISA)-based antibody detection test.
Published studies drew our attention to the N and S proteins, which appeared to be ideal targets for an early and sensitive SARS-CoV diagnostic kit (25, 26). The S protein, a projection of the viral surface, is the major neutralizing antigen of the known CoVs (2, 6, 10, 11, 18). It plays an important role in the initial stages of infection, forming the characteristic corona of large, distinctive spikes in the viral envelope (20). The N protein has been described by Krokhin et al. as the major immunogen in SARS-CoV (12) and is the most abundant protein throughout infection (13). Previous studies have suggested that a combination of these two proteins in the same assay may be needed to confirm the specificity of presumptive SARS antibodies and to offer a sensitive serodiagnosis (8, 24). For this reason, our study focused on the natural immune responses of SARS patients to a recombinant S-protein (S251-683) and N-protein (N1, N2, and N3) cocktail used as the antigen in an ELISA-based antibody detection test.
MATERIALS AND METHODS
Bacterial strains, virus, and sera.
The bacterial strains used in this study were purchased from Invitrogen and Novagen and have been described previously (1). Amplified gene fragments were obtained by reverse transcription-PCR (RT-PCR) using a sample of genomic RNA extracted from Vero-E6 cells infected with SARS-CoV. The isolated virus was available to the research community thanks to H. W. Doerr and colleagues from the University of Frankfurt, Frankfurt, Germany (3). The RNA sample was prepared at the Robert-Koch Institute, Berlin, Germany, and was kindly provided by Matthias Niedrig, who also supplied positive sera from 6 SARS patients and performed ELISAs on 14 additional SARS-CoV-positive sera by using a mixture of the four recombinant polypeptides described in this study. Negative controls consisted of 86 serum samples from a healthy population in Saragossa (an area not exposed to SARS) and 14 serum samples from individuals with other infectious diseases.
DNA manipulations.
Covalently closed circular DNA was purified by the sodium dodecyl sulfate (SDS) lysis method (21), followed by phenol extraction and ethanol precipitation. Analytical gel electrophoreses of plasmids and restriction fragments were carried out in 0.8% (wt/vol) agarose-Tris-borate-EDTA-ethidium bromide horizontal slab gels. DNA concentrations were determined by using molar extinction coefficients of 6,500 M−1·cm−1 at 260 nm (4). The general DNA manipulation methods have been reported previously (21). Double-stranded DNA sequencing reactions were performed at the López Neyra Institute of Parasitology and Biomedicine in Granada, Spain.
Amplification of gene inserts.
The lyophilized RNA was solubilized in 100 μl of sterile deionized RNase-free water treated with diethyl pyrocarbonate to an apparent concentration of 107 genome units/ml. Appropriate oligonucleotides were synthesized at the Analytical Services department of DNA Technology; their corresponding sequences and positions are shown in Table 1. RT-PCR conditions for amplification were those recommended by the WHO Collaborative Multi-Centre Research Project on Severe Acute Respiratory Syndrome Diagnosis (http://www.who.int/csr/sars/project/en/). Each reaction mixture contained 1 μl of RNA, 10 pmol of each of the two oligonucleotide primers, deoxynucleoside triphosphates at 200 μM each, and 1× buffer for the SuperScript III RT/Platinum Taq enzyme mix, supplemented with 3 mM Mg2+. The reaction was initiated with 2 U of the SuperScript III RT/Platinum Taq enzyme mix (Invitrogen). For expression in Escherichia coli, the sequences amplified with the appropriate oligonucleotide primers were cloned into the pET-15b (Novagen) and/or pRSET (Invitrogen) expression vector. These plasmids were subsequently used to transform the E. coli expression host Rosetta BL21(DE3)pLysS.
TABLE 1.
Oligonucleotide primers used for RT-PCR amplification of recombinant fragments of nucleocapsid and spike proteins from SARS-CoVa
| Primer | Sequence | Position (bp) |
|---|---|---|
| N1-5′ | ATGTCTGATAATGGACCCAATC | 1-23 |
| N1-3′ | TCCCTGAGTTGCAACCTATAC | 394-417 |
| N2-5′ | AAAGAAGGCATCGTATGGGTTGC | 382-404 |
| N2-3′ | GCAGCAATAGCGCGAGGGCAGT | 652-673 |
| N3-5′ | GGAGGTGGTGAAACTGCCCTC | 640-660 |
| N3-3′ | ATGTCAGCCGCAGGAAGAAGAG | 1181-1202 |
| S251-683-5′ | TCAGCTGCAGCCTATTTTGTTGGCTATTTAAAG | 753-785 |
| S251-683-3′ | TCTTAGGGCCTAAGCTTGCCATGTCTAACATAC | 1334-1366 |
Specific oligonucleotide primers were designed from the sequence of the SARS-CoV genome in the GenBank nucleotide sequence database (accession number AY278741) (20).
Expression system and protein purification.
The truncated His-tagged fusion proteins, termed N1, N2, N3, and S251-683, were purified from extracts of Rosetta cells. Bacterial clones transformed with the corresponding plasmid were grown in Luria-Bertani medium containing 100 μg/ml ampicillin at 37°C. When an A600 of 0.6 was reached, 1 mM isopropyl-β-thio-d-galactopyranoside (IPTG) was added, and cultures were grown for an additional 3 h. Cells were harvested by centrifugation at 1,789 × g, and pellets that were not used immediately were frozen at −80°C. For the scale-up purification procedure, frozen pellets from 4 liters of Rosetta cells overproducing N1, N2, N3, or S251-683 were thawed and resuspended in denaturing lysis buffer (50 mM Tris-HCl [pH 7.5], 100 mM sodium phosphate, 8 M urea, 0.1% Emulphogen [polyoxyethylene 10-tridecyl ether; Sigma], 10 mM imidazole, and 0.2 mM phenylmethylsulfonyl fluoride) containing 1 M NaCl and were equilibrated at pH 8 with 10 N NaOH. Cells were lysed by sonication. The crude extract was centrifuged at 50,000 × g for 30 min at 4°C, and the supernatant was purified by using a nickel-nitrilotriacetic acid-agarose matrix column according to the manufacturer's instructions (Qiagen).
Protein manipulations.
The apparent molecular mass of each polypeptide was determined by SDS-polyacrylamide gel electrophoresis (PAGE). The concentration of proteins was determined by UV absorbance at 280 nm using molar extinction coefficients, calculated as described by Gill and von Hippel (5).
Polyclonal antibody against SARS-CoV.
The polyclonal antibody was prepared by immunizing a rabbit with an intramuscular injection of 200 μg of protein (50 μg of each recombinant polypeptide: N1, N2, N3, and S251-683) in 1 ml of an emulsion containing 50% Freund's complete adjuvant. The same dose was repeated after 15 days, and the same dose prepared in Freund′s incomplete adjuvant was injected at 15 days after the second dose. The serum used corresponded to a blood sample drawn 3 months after the third injection.
IFA.
The immunofluorescence assay (IFA) used SARS-CoV-infected Vero-E6 cells from a commercial SARS-CoV IFA kit (Euroimmun). Briefly, the rabbit polyclonal serum obtained was first purified by protein A affinity chromatography (HiTrap protein A HP; Amersham BioLabs) and then labeled with fluorescein isothiocyanate (Sigma). A 1:1,000 dilution of the fluorescein-labeled rabbit polyclonal antibody was incubated for 30 min with SARS-CoV-infected cells. Noninfected cells were used as a control. After a wash, the reaction was visualized by fluorescence microscopy.
ELISA measurement.
Microtiter plates were coated with a mixture of the four recombinant polypeptides, diluted in phosphate-buffered saline (PBS) at a concentration of 1 to 5 μg/ml each (3 μg/ml N1, 2 μg/ml N2, 1 μg/ml N3, and 5 μg/ml S251-683), and were incubated overnight at room temperature. Plates were blocked with newborn calf serum, washed, and then incubated with sera from SARS patients or healthy controls (both at 1:100) in PBS containing newborn calf serum for 45 min at 37°C. After a wash, a 1:100,000 dilution of peroxidase-conjugated goat anti-human immunoglobulin G (IgG) plus IgM (Jackson) was added and incubated at 37°C for 30 min. Finally, the peroxidase reaction was visualized by using a tetramethylbenzidine-hydrogen peroxide solution as a substrate (Neogen Corporation). ELISAs with the single recombinant proteins, each applied to a plate, were also performed on six SARS serum samples. The same antigen dilutions were used, and the protocol described above was followed.
Western blotting.
Briefly, 0.25 μg of each purified polypeptide was loaded onto an SDS-PAGE (12%) gel and transferred to a 0.2-μm-pore-size nitrocellulose membrane using a Bio-Rad Mini Trans-Blot transfer unit. The membrane was first blocked with PBS containing 0.1% Tween 20 and 5% nonfat dry milk at room temperature for 2 h and then incubated with the specific primary antibody diluted 500-fold with washing buffer (PBS with 0.1% Tween 20). The membrane was washed five times and further incubated with horseradish peroxidase-labeled anti-rabbit IgG (Sigma). The proteins were visualized by a peroxidase reaction using a tetramethylbenzidine-hydrogen peroxide solution as the substrate.
Calculations.
The sensitivity of the assays was calculated as (number of samples with true-positive results)/(number of samples with true-positive results + number of samples with false-negative results). The specificity of the assays was calculated as (number of samples with true-negative results)/(number of samples with true-negative results + number of samples with false-positive results).
Statistical analysis.
MedCalc software (version 9.5.2.0; MedCalc Software, Mariakerke, Belgium) was used to analyze the ELISA results, and data were plotted in an interactive dot diagram.
RESULTS
RT-PCR amplification and expression and purification of the recombinant proteins.
The cDNA synthesized by RT-PCR was cloned into the expression vectors. Four different polypeptides were prepared; three corresponded to the nucleocapsid protein of the virus, and the fourth corresponded to the spike protein. The amplified fragments were first cloned into pRSET, but several problems related to overexpression of the heterologous genes appeared. Therefore, three of the fragments (N1, N2, and S251-683) were cloned into pET-15b, a T7 system expression vector, which increased production by as much as 10 to 15% (22). Lysates of the induced cells displayed the heterologous proteins, which were not present in noninduced cells (data not shown). Densitometric scanning of stained gels showed that the levels of induced proteins were approximately 10, 5, 15, and 12%, respectively, of the levels of soluble protein in E. coli containing the S251-683, N1, N2, and N3 expression constructs.
A high protein yield was obtained by treating the bacterial pellets with a buffer containing 8 M urea, followed by strong probe sonication. Briefly, a clarified extract of E. coli Rosetta BL21(DE3) overproducing N1, N2, N3, or S251-683 was adsorbed in batch mode to an affinity resin, purified by using a 7-ml column (diameter, 0.7 cm; height, 5 cm) equilibrated with buffer A (50 mM Tris-HCl [pH 7.5], 100 mM sodium phosphate, 8 M urea, 0.1% Emulphogen, 10 mM imidazole, and 1 M NaCl) at pH 8, and washed with 10 column volumes of buffer B (50 mM Tris-HCl [pH 7.5], 100 mM sodium phosphate [pH 7.0], 8 M urea, 0.1% Emulphogen, 20 mM imidazole, and 1 M NaCl) at pH 8. Recombinant proteins were eluted with 5 column volumes of buffer C (50 mM Tris-HCl [pH 7.5], 100 mM sodium phosphate [pH 7.0], 8 M urea, 0.1% Emulphogen, 250 mM imidazole, and 1 M NaCl) at a flow rate of 6 ml/h. This procedure yielded highly purified proteins. A single band was obtained for each protein, as shown in Fig. 1.
FIG. 1.
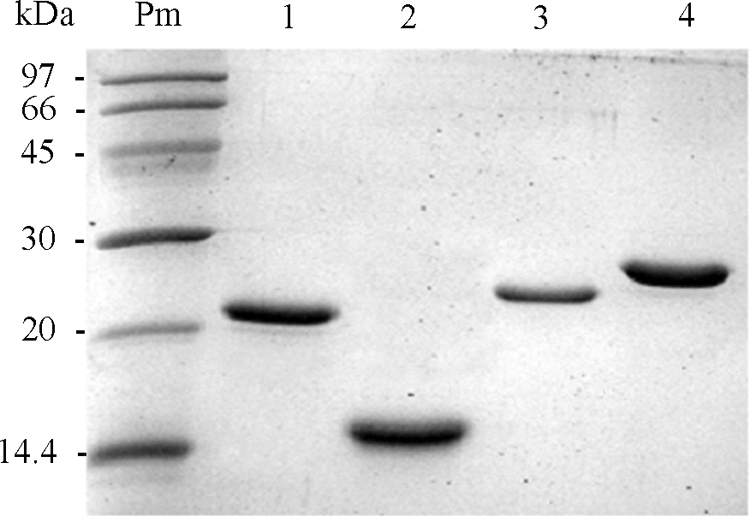
Pure recombinant polypeptides from SARS-CoV. Two to 5 μg of each purified recombinant protein was loaded onto an SDS-PAGE gel. Proteins were analyzed by SDS-PAGE and dyed with Coomassie blue. The molecular masses of the markers are given on the left. Lanes: Pm, markers; 1, N1 (18 kDa); 2, N2 (15 kDa); 3, N3 (20 kDa); 4, S251-683 (27 kDa).
N1, N2, N3, and S251-683 peptides showed similar antigenicities in an immunized rabbit.
Western blot analysis of the polyclonal serum from the rabbit immunized against the mixture of the four recombinant polypeptides showed that all recombinant fragments reacted with the antibody in a very similar manner (Fig. 2). The polyclonal antibody was also subjected to a separate ELISA for each recombinant polypeptide, and the results obtained were consistent with those of Western blotting, with S251-683 showing a slightly greater antigenic capacity than the N protein fragments (in order of highest to lowest antigenicity, N2, N3, and N1) (data not shown). Several other pathogen antigens were also tested with the rabbit polyclonal serum (Chlamydophila pneumoniae, Mycoplasma pneumoniae, Coxiella burnetii, cytomegalovirus, adenovirus, influenza A virus, influenza B virus, respiratory syncytial virus, herpesvirus), and no cross-reaction was detected (data not shown).
FIG. 2.
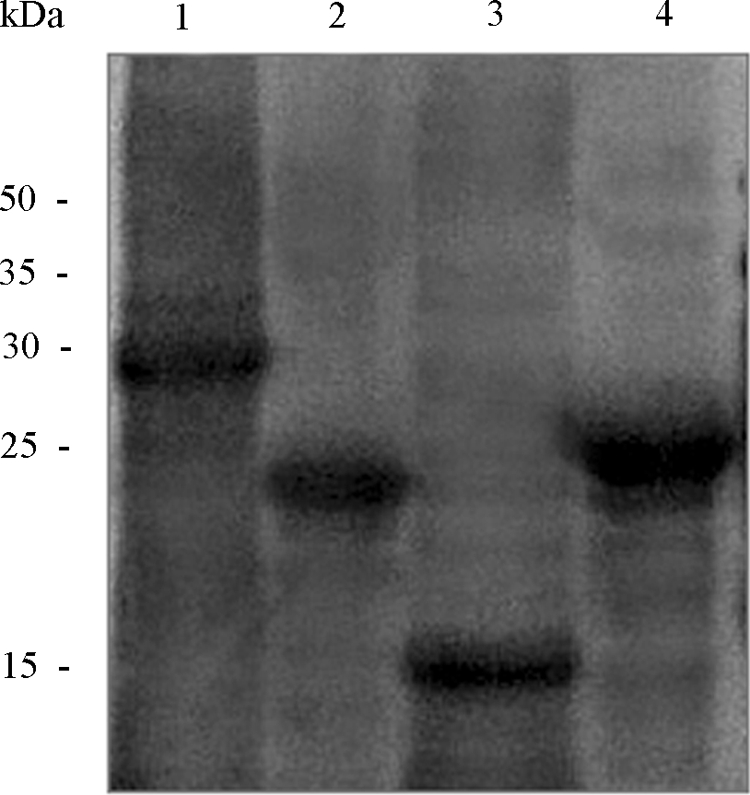
Western blot analysis of the heterologous SARS-CoV proteins expressed. Western blot analysis was performed with the rabbit polyclonal serum in order to determine the presence of epitopes in the four SARS-CoV recombinant polypeptides obtained. The gel was loaded with 0.25 μg of each protein, and a 1:500 dilution of the polyclonal antibody was used. The recombinant polypeptides were loaded as follows: lane 1, S251-683; lane 2, N1; lane 3, N2; lane 4, N3.
The polyclonal antibody induced by the polypeptide mixture showed high reactivity to SARS-CoV-infected cells.
The rabbit polyclonal serum was incubated with SARS-CoV-infected and noninfected cells and visualized by immunofluorescence. Figure 3 shows that cells infected with the virus had intact nuclear membranes and fluorescence signals located in the cytoplasm. No immunofluorescence was observed in noninfected cells.
FIG. 3.

Immunofluorescence of Vero-E6 cells infected with SARS-CoV as determined using the polyclonal antibody. Photographs show SARS-CoV-infected cells (A) and noninfected cells (B) incubated with the polyclonal serum obtained by immunization with a mixture of the four recombinant proteins. Apple-green fluorescence marks the binding sites of the rabbit antibodies; the red color corresponds to Evans blue, used as a counterstain to prevent nonspecific reactions.
Recombinant-protein-based ELISA for antibody detection.
A mixture of the four recombinant polypeptides was used as a coating antigen to develop an ELISA-based IgG-plus-IgM test for the detection of S- and N-specific antibodies against SARS-CoV. The assay specificity was studied by using serum samples from 86 healthy IFA-validated controls (dilution, 1:100), and only 1 was above the selected cutoff value, giving a specificity of 99%. Diluted (1:100) sera from 20 SARS patients (clinically diagnosed and IFA validated) were studied by using the S- and N-based ELISA, and 2 samples were below the cutoff value, giving a sensitivity of 90% (Fig. 4). The specificity of the assay was also tested using sera from 14 individuals infected with non-SARS-related respiratory viruses (adenovirus, respiratory syncytial virus, influenza A virus, influenza B virus, and parainfluenza virus), antinuclear antibodies, or rheumatoid factor. These sera were also validated by IFA as negative-control samples for SARS-CoV. No cross-reactivity was detected, since none of the serum samples were positive (Table 2).
FIG. 4.

Scatter chart of absorbance values in sera from SARS patients and healthy controls for IgG plus IgM antibodies obtained by the newly developed ELISA. The test results were plotted as optical densities at 450 nm. The cutoff line for positive diagnosis is drawn at a value that equals the sum of the mean and two times the standard deviation for healthy samples.
TABLE 2.
Analysis of the sensitivity and specificity of a recombinant S- and/or N-based ELISA for detection of IgG and IgM antibodies in samples of SARS-CoV-positive and -negative sera
| Fragment | Result with the following SARS patient serum samplea:
|
No. of SARS-CoV-negative serum samples testing positiveb
|
Sensitivity (%)c | Specificity (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | Blood donor sera (n = 86) | Cross-reaction sera (n = 14) | |||
| N1 | − | − | − | + | − | + | 1 | 0 | 33.3 | 99 |
| N2 | + | + | − | − | + | − | 0 | 0 | 50 | 100 |
| N3 | + | + | − | − | − | − | 0 | 0 | 33.3 | 100 |
| Sd | + | + | + | − | − | − | 0 | 0 | 50 | 100 |
| Mixture | + | + | + | + | + | + | 1 | 0 | 100 | 99 |
+ and −, results above and below the cutoff, respectively.
Negative controls comprised 86 serum samples from healthy blood donors, of which 1 gave a false-positive result, and 14 SARS-CoV-negative sera from individuals infected with non-SARS-related respiratory viruses, antinuclear antibodies, or rheumatoid factor, which were used for cross-reaction experiments.
Data correspond to analyses of the individual recombinant polypeptides and the combination of all of them for only six SARS-CoV-positive serum samples.
Fragment S251-683 of spike protein.
All control serum samples (n = 100) (data not shown) but only 6 SARS patient serum samples could be tested using the recombinant polypeptides separately (Table 2). Regarding their individual sensitivities, S251-683 was more immunogenic than the N polypeptides, and N2 was more immunogenic than N3 and N1, which were equal to each other. With respect to their specificities, recombinant fragment N1 was responsible for the false-positive result obtained.
DISCUSSION
Four recombinant polypeptides corresponding to two different structural proteins were evaluated for use in a SARS-CoV diagnostic kit.
In this study, a fragment of spike glycoprotein between amino acids 251 and 683 was selected by means of bioinformatic analysis (19). The S fragment overlaps with several regions reported to contain linear epitopes. Thus, Lu et al. (14) identified the C domain (bp 1323 to 2100) (S438-680) as the major immunodominant domain in S protein, and Zhou et al. (27) found that S-protein residues 485 to 625 elicited neutralizing antibodies against the virus.
A cocktail of these recombinant antigens was used to produce a polyclonal antibody in order to test their capacities to trigger an immune reaction that recognized the native antigen. It was demonstrated that this antibody was able to detect SARS-CoV by immunofluorescence of infected cells, showing that the functional antigenicities of the recombinant polypeptides were similar to those of their native counterparts.
These recombinant polypeptides were then used to develop an ELISA-based IgG-plus-IgM antibody detection test. Only 20 sera from clinically diagnosed SARS patients were available for these studies. Assays performed with the combination of all four recombinant polypeptides, using sera from healthy donors with no exposure to SARS-CoV as controls, showed a sensitivity of 90% and a specificity of 99%. One possible explanation for the two false-negative results may be that some essential linear antigenic sites are located outside the selected spike protein fragment, since polypeptide S251-683 covers less than one-third of the S protein sequence. Furthermore, the immunogenicity of the recombinant S protein in SARS-CoV may be lower than that of its native counterpart due to effects of the expression system used to overproduce the recombinant polypeptide, with the possible loss of essential conformation- and/or glycosylation-dependent epitopes. The N protein is reported to be free of glycosylation sites and does not appear to change its immunological characteristics, even when expressed in a prokaryotic system (17). Nevertheless, the heavy glycosylation of the spike protein with mannose and/or hybrid oligosaccharides (7) may make it a suitable candidate for use in a eukaryotic rather than a bacterial system.
However, the separate analysis of each polypeptide with six SARS-CoV-positive serum samples showed that the S251-683 fragment (reactive with three of six sera) was less immunogenic than the nucleocapsid protein (reactive with five of six sera due to the additive results of the three fragments). However, the spike protein fragment reacted with one serum sample (sample 3) that did not react with the N antigen. Similar findings have been published by Woo et al. (24) and Yu et al. (25), who concluded that a recombinant N-based IgG ELISA was more sensitive than a recombinant S-based IgG ELISA for the serodiagnosis of SARS-CoV. ELISAs with polypeptides obtained from the nucleocapsid protein showed that N2 was the most immunogenic region. It should be taken into account that these studies used only six positive serum samples; therefore, these results should be confirmed with a more representative number of SARS-CoV-positive sera.
No direct serology was performed against other human CoV strains in the cross-reaction assays, but a large percentage of the serum samples from our negative controls probably contain antibodies against these highly prevalent viruses (16). Thus, human CoVs are reported to be responsible for around 30% of all common colds (9). In fact, this may explain the false-positive result we obtained. Since cross-reactivity with the N1 polypeptide has been identified, it may be attributable to a highly conserved N111-118 motif described by Vlasova et al. and present in the N proteins of all CoVs (23). Although only one false-positive serum specimen was obtained from unexposed individuals (1%), the potential for cross-reactivity between SARS-CoV and other human CoVs was not directly assessed in this study and remains a concern.
Analysis with our ELISA showed that the recombinant polypeptides had different levels of immunogenicity with the SARS-CoV-positive sera, and one specific mixture of all four recombinant proteins demonstrated higher sensitivity and specificity in detecting antibodies in sera from patients. Hence, we can report a new ELISA-based IgG-plus-IgM antibody detection test for the diagnosis of SARS. Due to the high degrees of pathogenicity and infectivity of SARS-CoV for humans, detection of IgM and IgG antibodies may be the method of choice for this serodiagnosis. We have increased the sensitivity and specificity of the assay by using a cocktail of SARS-CoV N and S proteins. Furthermore, the use of protein fragments or polypeptides instead of whole recombinant proteins for antibody detection may in part resolve the issue of potential cross-reactivity with proteins of other human CoVs.
Acknowledgments
We are grateful to H. W. Doerr and Matthias Niedrig for kindly providing the RNA sample and a panel of sera from SARS patients. We thank Laura Altuzarra for technical assistance with illustrations.
Footnotes
Published ahead of print on 26 November 2008.
REFERENCES
- 1.Brinkmann, U., R. E. Mattes, and P. Buckel. 1989. High-level expression of recombinant genes in Escherichia coli is dependent on the availability of the dnaY gene product. Gene 85109-114. [DOI] [PubMed] [Google Scholar]
- 2.Callebaut, P., L. Enjuanes, and M. Pensaert. 1996. An adenovirus recombinant expressing the spike glycoprotein of porcine respiratory coronavirus is immunogenic in swine. J. Gen. Virol. 77309-313. [DOI] [PubMed] [Google Scholar]
- 3.Drosten, C., S. Gunther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L. Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguiere, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Muller, V. Rickerts, M. Sturmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz, and H. W. Doerr. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 3481967-1976. [DOI] [PubMed] [Google Scholar]
- 4.Freifelder, D. 1983. Physical biochemistry: applications to biochemistry and molecular biology, p.494-536. W. H. Freeman and Company, San Francisco, CA.
- 5.Gill, S. C., and P. H. von Hippel. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182319-326. [DOI] [PubMed] [Google Scholar]
- 6.Gómez, N., C. Carrillo, J. Salinas, F. Parra, M. V. Borca, and J. M. Escribano. 1998. Expression of immunogenic glycoprotein S polypeptides from transmissible gastroenteritis coronavirus in transgenic plants. Virology 249352-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han, D. P., H. G. Kim, Y. B. Kim, L. L. Poon, and M. W. Cho. 2004. Development of a safe neutralization assay for SARS-CoV and characterization of S-glycoprotein. Virology 326140-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haynes, L. M., C. Miao, J. L. Harcourt, J. M. Montgomery, M. Q. Le, S. A. Dryga, K. I. Kamrud, B. Rivers, G. J. Babcock, J. B. Oliver, J. A. Comer, M. Reynolds, T. M. Uyeki, D. Bausch, T. Ksiazek, W. Thomas, H. Alterson, J. Smith, D. M. Ambrosino, and L. J. Anderson. 2007. Recombinant protein-based assays for detection of antibodies to severe acute respiratory syndrome coronavirus spike and nucleocapsid proteins. Clin. Vaccine Immunol. 14331-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes, K. V. 2001. Coronaviruses, p. 1187-1203. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 10.Homberger, F. R. 1994. Nucleotide sequence comparison of the membrane protein genes of three enterotopic strains of mouse hepatitis virus. Virus Res. 3149-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackwood, M. W., and D. A. Hilt. 1995. Production and immunogenicity of multiple antigenic peptide (MAP) constructs derived from the S1 glycoprotein of infectious bronchitis virus (IBV). Adv. Exp. Med. Biol. 380213-219. [DOI] [PubMed] [Google Scholar]
- 12.Krokhin, O., Y. Li, A. Andonov, H. Feldmann, R. Flick, S. Jones, U. Stroeher, N. Bastien, K. V. Dasuri, K. Cheng, J. N. Simonsen, H. Perreault, J. Wilkins, W. Ens, F. Plummer, and K. G. Standing. 2003. Mass spectrometric characterization of proteins from SARS virus: a preliminary report. Mol. Cell. Proteomics 2346-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li, S., L. Lin, H. Wang, J. Yin, Y. Ren, Z. Zhao, J. Wen, C. Zhou, X. Zhang, X. Li, J. Wang, Z. Zhou, J. Liu, J. Shao, T. Lei, J. Fang, N. Xu, and S. Liu. 2003. The epitope study on the SARS-CoV nucleocapsid protein. Genomics Proteomics Bioinformatics 1198-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, L., I. Manopo, B. P. Leung, H. H. Chng, A. E. Ling, L. L. Chee, E. E. Ooi, S. W. Chan, and J. Kwang. 2004. Immunological characterization of the spike protein of the severe acute respiratory syndrome coronavirus. J. Clin. Microbiol. 421570-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahony, J. B., and S. Richardson. 2005. Molecular diagnosis of severe acute respiratory syndrome: the state of the art. J. Mol. Diagn. 7551-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McIntosh, K. 1996. Coronaviruses, p. 1095-1103. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 1. Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 17.Méchin, M. C., M. Der Vartanian, and C. Martín. 1996. The major subunit ClpG of Escherichia coli CS31A fibrillae as an expression vector for different combinations of two TGEV coronavirus epitopes. Gene 179211-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ndifuna, A., A. K. Waters, M. Zhou, and E. W. Collison. 1998. Recombinant nucleocapsid protein is potentially an inexpensive, effective serodiagnostic reagent for infectious bronchitis virus. J. Virol. Methods 7037-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice, P., I. Longden, and A. Bleasby. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16276-277. [DOI] [PubMed] [Google Scholar]
- 20.Rota, P. A., M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli, J. P. Icenogle, S. Penaranda, B. Bankamp, K. Maher, M. H. Chen, S. Tong, A. Tamin, L. Lowe, M. Frace, J. L. DeRisi, Q. Chen, D. Wang, D. D. Erdman, T. C. Peret, C. Burns, T. G. Ksiazek, P. E. Rollin, A. Sanchez, S. Liffick, B. Holloway, J. Limor, K. McCaustland, M. Olsen-Rasmussen, R. Fouchier, S. Gunther, A. D. Osterhaus, C. Drosten, M. A. Pallansch, L. J. Anderson, and W. J. Bellini. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 3001394-1399. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 22.Studier, F. W. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 18560-89. [DOI] [PubMed] [Google Scholar]
- 23.Vlasova, A. N., X. Zhang, M. Hasoksuz, H. S. Nagesha, L. M. Haynes, Y. Fang, S. Lu, and L. J. Saif. 2007. Two-way antigenic cross-reactivity between severe acute respiratory syndrome coronavirus (SARS-CoV) and group 1 animal CoV is mediated through an antigenic site in the N-terminal region of the SARS-CoV nucleoprotein. J. Virol. 8113365-13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo, P. C., S. K. Lau, B. H. Wong, H. W. Tsoi, A. M. Fung, R. Y. Kao, K. H. Chan, J. S. Peiris, and K. Y. Yuen. 2005. Differential sensitivities of severe acute respiratory syndrome (SARS) coronavirus spike polypeptide enzyme-linked immunosorbent assay (ELISA) and SARS coronavirus nucleocapsid protein ELISA for serodiagnosis of SARS coronavirus pneumonia. J. Clin. Microbiol. 433054-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu, F., M. Q. Le, S. Inoue, F. Hasebe, C. Parquet Mdel, S. Morikawa, and K. Morita. 2007. Recombinant truncated nucleocapsid protein as antigen in a novel immunoglobulin M capture enzyme-linked immunosorbent assay for diagnosis of severe acute respiratory syndrome coronavirus infection. Clin. Vaccine Immunol. 14146-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao, J., Q. Huang, W. Wang, Y. Zhang, P. Lv, and X.-M. Gao. 2007. Identification and characterization of dominant helper T-cell epitopes in the nucleocapsid protein of severe acute respiratory syndrome coronavirus. J. Virol. 816079-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou, T., H. Wang, D. Luo, T. Rowe, Z. Wang, R. J. Hogan, S. Qiu, R. J. Bunzel, G. Huang, V. Mishra, T. G. Voss, R. Kimberly, and M. Luo. 2004. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J. Virol. 787217-7226. [DOI] [PMC free article] [PubMed] [Google Scholar]