

Abstract
The RgpA-Kgp proteinase-adhesin complexes of Porphyromonas gingivalis were observed, using immunostaining, in human gingival tissue associated with periodontitis but not in healthy tissue. The staining pattern suggested a concentration gradient from the subgingival plaque into the subjacent gingival connective tissue. Intense immunostaining was observed in areas displaying gross disturbance of tissue architecture. P. gingivalis cells and the RgpA-Kgp complexes at low concentrations were shown to stimulate secretory intercellular adhesion molecule 1, interleukin-8 (IL-8), IL-6, and macrophage chemoattractant protein secretion from cultured human epithelial (KB) and fibroblast (MRC-5) cells. However, at high concentrations a reduction in the level of these mediators was observed. In contrast, macrophage inflammatory protein 1α and IL-1α were stimulated only at high P. gingivalis cell concentrations. P. gingivalis cells and the RgpA-Kgp complexes were shown to induce apoptosis in KB and MRC-5 cells in a time- and dose-dependent manner. These data suggest that the RgpA-Kgp complexes penetrate the gingival connective tissue; at low concentrations distal from the plaque the complexes stimulate the secretion of proinflammatory mediators, while at high concentrations proximal to the plaque they induce apoptosis and attenuate the secretion of proinflammatory mediators.
Chronic periodontitis is an inflammatory disease associated with specific bacteria in subgingival plaque that results in the destruction of the tooth's supporting tissues. The presence of the three bacterial species Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia as a consortium in subgingival plaque has been associated with chronic periodontitis, and of these bacteria, P. gingivalis is reported to be most closely associated with the severity of disease (51, 79). P. gingivalis produces extracellular complexes of proteinases and adhesins, designated the RgpA-Kgp complexes (or high-molecular-weight gingipains). Isogenic mutants of P. gingivalis lacking the RgpA-Kgp complexes are avirulent in an animal periodontitis model, and therefore the complexes have been proposed to be a major virulence factor for this bacterium (51, 56). The chronic interaction of the host immune system with subgingival plaque containing P. gingivalis and the RgpA-Kgp complexes in the subjacent tissue is believed to be a major factor in tissue destruction in chronic periodontitis (6, 21, 51, 83). Compared with healthy subjects, gingival tissues and gingival crevicular fluid of patients with chronic periodontitis are reported to have significantly increased amounts of proinflammatory cytokines such as interleukin-1 (IL-1), IL-6, and tumor necrosis factor alpha (TNF-α); chemokines such as IL-8, macrophage chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein 1α (MIP-1α); and adhesion molecules such as intracellular adhesion molecule 1 (ICAM-1) (12, 25, 27, 28, 32, 33, 37, 44, 46, 62, 84, 87). These cytokines and chemokines play a significant role in mediating the recruitment of a dense mononuclear infiltrate, consisting mainly of T cells and macrophages (21, 22, 39, 55, 81). Furthermore, a concentration gradient of secretory ICAM-1 (sICAM-1) across the junctional epithelium is reported to be an important mechanism leading to leukocyte recruitment into the gingival sulcus (85). Assuma et al. (2) have reported that blocking IL-1 and TNF-α activity in a nonhuman primate model significantly reduced tissue destruction and alveolar bone loss. These studies suggest that the chronic presence of specific pathogens such as P. gingivalis in subgingival plaque results in the secretion of inflammatory mediators which, in turn, may cause inappropriate accumulation and activation of circulating and resident leukocytes at the site of infection, producing chronic inflammation and tissue destruction. Another contributing factor for periodontal tissue destruction is reported to be the induction of host cell apoptosis by specific subgingival plaque pathogens (5). In gingival biopsies from patients with chronic periodontitis, apoptotic cells have been reported to constitute about 10% of the total cell population and included epithelial and fibroblast cells (31, 36). Tonetti et al. (86) have reported that exposure of clinically healthy gingival tissues to plaque bacteria induces apoptosis-associated DNA damage and the expression of proapoptotic p53 protein. Furthermore, in gingival biopsies from patients with periodontitis, epithelial cell apoptosis was reported to be more prevalent in the most apical part of the sulcus closest to the subgingival plaque (31, 86). These observations suggest that interactions with certain bacterial products may play an important role in inducing apoptosis in gingival tissue cells. Furthermore, a high prevalence of apoptotic cells expressing the p53 protein was detected in the subgingival inflammatory infiltrate, suggesting that apoptotic cell death may be important in the regulation of the inflammatory response to chronic bacterial challenge (86).
A number of prior studies have investigated the ability of P. gingivalis to induce secretion of proinflammatory mediators from oral epithelial and fibroblast cells. It has been reported that P. gingivalis induces expression of proinflammatory mediators such as IL-1β, IL-8, IL-6, and sICAM-1 from gingival epithelial or fibroblast cells (1, 70, 80, 93). However, in contrast, P. gingivalis cells have also been reported to degrade existing inflammatory cytokines and antagonize IL-1β, IL-8, IL-6, and sICAM-1 production by gingival epithelial or fibroblast cells (13, 16-18, 43). These apparent contradictions may be partially explained by the assay conditions related to factors such as bacterial strain variability and inclusion of human serum (38, 74, 80). However, we have postulated that this paradoxical situation where P. gingivalis both suppresses and induces the host immune response is a concentration-dependent phenomenon that is related to the level of P. gingivalis cells and the P. gingivalis major virulence factor, the RgpA-Kgp proteinase-adhesin complexes (51).
In the present study, we demonstrate the presence of RgpA-Kgp in gingival tissue associated with periodontitis, and we investigate the hypothesis that P. gingivalis W50 whole cells and RgpA-Kgp complexes differentially regulate the expression of proinflammatory mediators and the induction of apoptosis in human epithelial and fibroblast cells in a concentration-dependent fashion.
MATERIALS AND METHODS
Human subjects and gingival specimens.
The study was approved by the University of Melbourne Human Research Ethics Committee, and informed written consent was obtained from all participants. Subjects were chosen from adult patients, 18 years of age and over, presenting for treatment at the Periodontics Department of The Royal Dental Hospital of Melbourne. Specimens were collected from 10 control subjects (22 to 66 years old) with gingivitis or periodontal health (five males and five females) and 14 diseased subjects (25 to 59 yrs), 11 with chronic periodontitis (six males and five females), and 3 with generalized-aggressive periodontitis (GAP; all male). Of the 24 subjects, 21 had no relevant medical history, one subject with chronic periodontitis had non-insulin-dependent diabetes mellitus, one GAP subject was a light smoker, and another GAP subject had taken systemic tetracycline 10 weeks prior to tissue collection.
Control specimens were collected from sites with probing depths less than 4 mm and were harvested during crown-lengthening procedures. All diseased specimens were collected from sites with probing depths greater than 5 mm with bleeding and/or suppuration on deep probing. Specimens from all diseased subjects, except one with GAP, were harvested from the pocket wall including associated granulation tissue during periodontal flap reflection from sites that had not responded to previous nonsurgical treatment. Three specimens were harvested from sites adjacent to two teeth extracted for periodontal reasons from one GAP subject. Subjects in the control group were age matched and gender matched as closely as possible with those in the periodontitis group.
Immunohistochemical localization of RgpA-Kgp in gingival tissue specimens.
Gingival specimens were kept moist in phosphate-buffered saline (PBS) before being quickly frozen in liquid nitrogen. The specimens were stored at −70°C until required. Transverse 5-μm-thick sections were cut using a Frigocut E cryotome (Reichert-Jung, Sweden) and placed onto silane-coated slides that were then stored with desiccant at −70°C until required. The sections were fixed in ice-cold acetone for 2 min and air dried for 2 h. The slides were then immersed in 0.6% (wt/wt) H2O2 in methanol for 10 min at room temperature to quench endogenous peroxidase activity and then placed in a Sequenza slide rack (Shandon Inc., United Kingdom). The sections were kept moist with PBS.
Nonspecific binding sites were blocked with 10% (vol/vol) normal swine serum in PBS for 20 min at room temperature. The sections were then incubated with normal rabbit serum or specific anti-RgpA-Kgp antiserum diluted 1:4,000 in antibody diluting buffer (ADB) (PBS containing 500 mM NaCl, 1% [vol/vol] normal swine serum, and 0.05% [vol/vol] Tween 20) for 18 h at 4°C. Sections were then washed with ADB three times for 5 min each. The anti-RgpA-Kgp antiserum was generated in New Zealand White rabbits using standard procedures. Western blot analysis of the RgpA-Kgp antigen separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using the rabbit anti-RgpA-Kgp antiserum indicated reactivity predominately with the A1 adhesins of RgpA and Kgp, as described previously (52, 57). Sections were then incubated with biotin-conjugated swine anti-rabbit immunoglobulin G diluted 1:200 in ADB for 20 min at room temperature and washed again with ADB. For 20 min the sections were then incubated at room temperature with avidin-biotin complexes. The complexes were prepared by adding horseradish peroxidase-conjugated biotin to avidin, diluted 1:100 in ADB, and then left on ice for 30 min. After a washing step with ADB, the sections were then incubated in diaminobenzidine (DAB)-H2O2 for 15 min at room temperature, and the reaction was stopped with water. The sections were then washed in water for 5 min and then counterstained in Mayer's hematoxylin for 30 s, differentiated in HCl, and stained with Scotts tap water. After the sections were subjected to a final dehydration in alcohol and xylol and mounting in di-n-butylphthalate in xylene (DPX) with a coverslip, they were photographed with an Olympus OM2 camera attached to an Olympus BH2 microscope.
Bacterial growth conditions.
P. gingivalis W50 was grown in an anaerobic chamber (MK3 anaerobic workstation; Don Whitley Scientific Ltd., Shipley, England) at 37°C on horse blood agar plates supplemented with 10% (vol/vol) lysed horse blood. Bacterial colonies were used to inoculate 20 ml of brain heart infusion medium containing 5 μg/ml of hemin and 0.5 μg/ml of cysteine and grown overnight at 37°C. Typically, 2 ml of bacterial culture was then transferred to 200 ml of fresh brain heart infusion medium, and the batch culture growth was monitored at 650 nm using a spectrophotometer (Perkin-Elmer model 295E). The bacterial culture was harvested during the exponential phase of growth. Culture purity was routinely checked by Gram staining. Escherichia coli JM109 (Promega Corporation) was grown using LB medium following standard procedures.
Epithelial and fibroblast cell culture.
The human epithelial cell line KB (ATCC CCL-17) and human oral fibroblast cell line MRC-5 (JRH Biosciences, Australia) were maintained as frozen stocks and cultured in Earl's minimum essential medium (EMEM) supplemented with 2 mM l-glutamine, 10% (vol/vol) heat-inactivated fetal calf serum, 100 IU/ml penicillin-streptomycin, and 30 μg/ml gentamicin in a 5% CO2 incubator at 37°C. Cell passage was performed twice a week, and the cells used in experiments were never older than 12 passages. Epithelial and fibroblast cells were seeded into 24-well tissue culture plates in a volume of 1 ml per well and grown to 95% confluence (106 cells/well) before being used for cytokine and apoptosis studies.
Detection of cytokines in the culture fluid of the epithelial and fibroblast cell cultures.
P. gingivalis W50 grown to late exponential phase was washed twice with PBS and resuspended in EMEM supplemented with 25 mM glutamine and 20 mM HEPES (serum-free culture medium). KB and MRC-5 cell monolayers (1 × 106 cells/well) were incubated with P. gingivalis cells added in 200 μl/well at a bacterial cell-to-host cell ratio (bacteria cell ratio, or BCR) of 10:1, 50:1, 100:1, 500:1, or 1,000:1. The RgpA-Kgp complexes were purified from P. gingivalis W50 as described previously (57) and dissolved in serum-free culture medium to a final concentration of either 10, 5, 2.5, 1.25, or 0.625 μg per 200 μl and added onto the KB and MRC-5 cell monolayers. These amounts of RgpA-Kgp complexes were equivalent to a BCR of 200:1, 100:1, 50:1, 25:1, and 12.5:1, respectively (51, 57). The cell culture plates were then centrifuged at 800 × g for 5 min at room temperature and then incubated at 37°C for 90 min in a 5% CO2 incubator. After incubation, the culture fluid was collected, the cell monolayers were washed twice with sterile PBS (500 μl), and the washings were pooled with the collected culture fluid. KB or MRC-5 cells detached during the incubation or washings were collected by centrifugation at 800 × g for 5 min at room temperature and washed twice with sterile PBS. Each cell pellet was then resuspended in 200 μl of fresh EMEM supplemented with 25 mM glutamine, 100 IU/ml penicillin-streptomycin, 30 μg/ml gentamicin, 20 mM HEPES, and 10% (vol/vol) heat-inactivated fetal calf serum; the pellet suspensions were then added back to the corresponding cell culture wells and incubated for a further 16 h at 37°C in a 5% CO2 incubator. After the incubation period, the supernatants were collected and centrifuged at 800 × g for 5 min at room temperature. The collected supernatants were then stored at −70°C until analyzed by enzyme-linked immunosorbent assay (ELISA). The concentrations of IL-1α, IL-1β, IL-6, IL-8, TNF-α, MIP-1α, MCP-1, and sICAM-1 in the supernatants of KB and MRC-5 cell monolayers incubated with either P. gingivalis or RgpA-Kgp were measured using CytoScreen ELISA systems (Biosource International, Camarillo, CA) according to manufacturer's protocols. Data are expressed as pg/ml of three separate values from three separate experiments. The data were analyzed using a one-way analysis of variance with SPSS software, and the effect sizes represented as Cohen's d were calculated using the effect size calculator provided online by the Evidence-Based Education UK website at http://www.cemcenter.org/ebeuk/research/effectsize/default.htm. According to Cohen (10) a small effect size is a d of ≥0.2 and <0.5, a moderate d is ≥0.5 and <0.8, and a large d is ≥0.8.
Measurement of epithelial cell and fibroblast apoptosis by flow cytometry.
An annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) apoptosis/necrosis detection kit (Beckton Dickinson Pharmingen) was used to analyze epithelial cell and fibroblast apoptosis/necrosis following incubation with P. gingivalis W50 and the RgpA-Kgp complexes. P. gingivalis W50 was grown to late exponential phase as described above, washed twice in PBS, and resuspended in serum-free culture medium. KB and MCR-5 cells grown in 24-well cell culture plates as described above were washed twice in sterile PBS and incubated with P. gingivalis at a BCR of 10:1, 50:1, 100:1, 500:1, 1,000:1, and 10,000:1. RgpA-Kgp was dissolved in serum-free culture medium to a final concentration of either 0.625, 1.25, 2.5, 5, or 10 μg per 200 μl and added onto the KB and MCR-5 cell monolayers. The cell culture plates were centrifuged (800 × g for 5 min at room temperature) and incubated for 90 min at 37°C. Following the incubation, supernatants containing detached KB or MRC-5 cells were collected, and the remaining epithelial or fibroblast cells were detached from the culture plate wells by incubating with a trypsin-EDTA mixture (500 μl/well) (JRH Biosciences, Victoria, Australia) for 5 min at 37°C and then pooled with the corresponding supernatants. KB or MRC-5 cells were collected by centrifugation at 400 × g for 5 min at 4°C and washed twice with cold PBS (400 × g for 5 min at 4°C). Following the washing steps, the KB and MRC-5 cell pellets were resuspended in binding buffer (Beckton Dickinson Pharmingen) and stained simultaneously with FITC-labeled annexin V and PI, according to the manufacturer's protocol; pellets were assessed by bivariate flow cytometry using a FACSCalibur flow cytometer (Beckton Dickinson) equipped with lasers operating at 488 and 610 nm. The flow cytometer was set for annexin V-FITC (FL1) versus PI (FL2) bivariate analysis. Forward and side scatter properties were used to acquire data from 10,000 cells and to gate out small particles such as cell debris and bacterial cells. The quadrants on the two-dimensional dot plots were set based upon healthy unstimulated samples, and the percentage of cells in the respective quadrants was analyzed by CellQuest software (Beckton Dickinson).
RESULTS
Immunolocalization of RgpA-Kgp in gingival tissue. (i) Healthy tissue incubated with RgpA-Kgp.
One of the healthy specimens was incubated with RgpA-Kgp for 30 min to 24 h and then probed with the nonimmune or specific antiserum diluted 1:50 to 1:4,000. Sections probed with the specific serum exhibited a diffuse, relatively homogeneous and moderately intense brown stain (peroxidase-DAB reaction product) extending to various extents from the cut surface into the connective tissue. This was clearly more intense and well demarcated from the less intense background stain (data not shown). This more intense stain was not observed in sections probed with nonimmune serum.
Time-dependent penetration of RgpA-Kgp into the connective tissue of these specimens was also observed. A band of specific immunostaining confined to the edge of the connective tissue was observed in specimens incubated with the antigen for 30 min and extended farther into specimens incubated with the antigen for 1 h. It was also interesting that after incubation for 24 h the macroscopic structure of the tissue was clearly altered, with the edges of the specimen appearing degraded.
(ii) Tissue associated with health, gingivitis, and periodontitis.
Gingival tissue biopsies from 21 diseased sites from 14 patients and 11 healthy sites from 10 patients were probed with the specific antibody to determine whether the RgpA-Kgp antigen was present. Tissue from 7 of the 21 diseased sites contained RgpA-Kgp (Fig. 1) whereas none of the healthy sites was positive for the antigen. Results were analyzed using Fisher's exact probability test using SPSS, version 15 (50). There was a significant association (P = 0.035) between the disease status of the gingival tissue and the presence of the RgpA-Kgp antigen in that tissue as determined by immunolocalization.
FIG. 1.

Photomicrographs of frozen sections of gingival tissue associated with chronic periodontitis, GAP, and periodontal health/gingivitis stained for the P. gingivalis RgpA-Kgp proteinase-adhesin complexes. Panels A to D show sections of diseased tissue from a previously untreated GAP patient that demonstrate discrete staining, observed as dark brown spots often in aggregates (especially panels B and D), as well as less intense diffuse staining, observed as a light brown “smear” around the discrete staining. The diffuse staining is also seen in areas devoid of the discrete staining. Note the large amount of discrete staining scattered among inflammatory cells in panels C and D. Panels E to H show sections of diseased tissue from a previously treated chronic periodontitis patient. Note the comparatively high proportions of diffuse staining in these panels. Panel E contains mostly diffuse staining, whereas panels F to H show both discrete and diffuse staining with relatively large amounts of diffuse staining also observed independent of the discrete staining. The arrow in panel F points to diffuse staining associated with a small blood vessel extending around its entire circumference. The pink amorphous material observed in panels G and H represents plaque bacteria. Panel I shows a section of diseased tissue from another previously treated chronic periodontitis patient, showing mostly diffuse staining associated with a moderately dense inflammatory cell infiltrate. Spots indicating discrete staining, some in small aggregates, are also evident. Panels J and K show sections of diseased tissue from another previously treated chronic periodontitis patient that contain discrete stains of various sizes, sometimes in aggregates, along with moderate amounts of surrounding diffuse staining. Panel L shows a section of diseased tissue from another previously treated chronic periodontitis patient that contains discrete stains, mostly in aggregates, along with surrounding diffuse staining. Panels M to P show sections of control tissue (healthy or gingivitis only) from four different patients that demonstrate the absence of either discrete or diffuse staining in areas with various amounts of inflammatory cell infiltrate. Scale bars: 30 μm (A to E, G, H, J, L to P), 20 μm =(F), and 50 μm (I and K).
An intense peroxidase-DAB reaction product for RgpA-Kgp was observed in localized areas of connective tissue from subjects with chronic periodontitis and GAP. Specimens from four chronic periodontitis subjects are shown in Fig. 1 (panels E to H, I, J and K, and L), and a specimen from one GAP subject is also shown (Fig. 1A to D). The proportional area of specimens containing this reaction product was generally limited and varied between specimens, even specimens collected from the same site. The intense reaction product was observed as discrete dark brown to black spots of various sizes. A diffuse lighter brown stain generally surrounded the discrete spots but was also observed in areas without the discrete spots. These two patterns of staining were observed in sections probed with a high dilution of the specific anti-RgpA-Kgp antiserum (1:4,000) in which nonspecific background staining was not observed. This intense reaction product was not observed in connective tissue of any diseased or control specimen probed with nonimmune serum or in any control specimen probed with the immune serum (Fig. 1M to P).
The largest amount of discrete specific immunostaining was observed in the GAP subjects as shown in Fig. 1A to D, whereas in specimens from chronic periodontitis subjects (Fig. 1E to H, I, J and K, and L), proportionally larger amounts of specific diffuse immunostaining were observed. The sizes of the discrete spots within each specimen varied, and the spots were often observed in close proximity to each other or in aggregates that also varied in size both between and within specimens. Both patterns of specific immunostaining were observed in areas with moderate to heavy infiltrates of chronic inflammatory cells, as shown in Fig. 1C and D. In the presence of specific immunostaining, inflammatory cells appeared to be somewhat dispersed compared with the cells in infiltrates devoid of specific immunostaining. However, specific immunostaining was also observed in areas of some specimens with relatively few inflammatory cells and also in some areas of tissue exhibiting marked architectural changes (degradation). In one chronic periodontitis specimen (Fig. 1F), specific diffuse immunostaining was observed in the wall of a small blood vessel extending around its entire circumference.
Secretion of sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, IL-1α, IL-1β, and TNF-α from oral epithelial (KB) and fibroblast (MRC-5) cells following incubation with P. gingivalis W50.
The levels of sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, IL-1α, IL-1β, and TNF-α in culture supernatants of KB and MRC-5 cells stimulated with P. gingivalis W50 were analyzed by ELISA (Biosource International, Camarillo, CA). KB and MRC-5 cell monolayers were incubated with increasing cell numbers of P. gingivalis W50 at BCRs ranging from 10:1 to 1,000:1 for 90 min at 37°C. After incubation, P. gingivalis cells were removed, and cell monolayers were washed and reincubated in fresh cell culture medium for a further 16 h at 37°C. As controls, KB and MRC-5 cell monolayers were incubated with cell culture medium alone or with an equivalent cell number of E. coli JM109 cells.
Figure 2 shows the levels of sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, and IL-α in KB and MRC-5 cell supernatants following incubation with P. gingivalis W50. For both cell types, incubation with increasing numbers of P. gingivalis cells resulted in a corresponding increase in sICAM-1, IL-8, and IL-6 secretion up to a BCR of 100:1 that was significantly (P < 0.01) higher than the basal levels of secretion (Fig. 2A, B, and C). However, at higher BCRs (500:1 and 1,000:1), there was a significant (P < 0.01) decrease in sICAM-1 and IL-8 secretion from KB and MRC-5 cells compared with the basal levels of secretion. Although there was a reduction in IL-6 secretion from both KB and MRC-5 cells at high BCRs (500:1 and 1,000:1), it was only significantly less than the basal level of secretion (P < 0.01) in KB cells incubated with P. gingivalis at a BCR of 1,000:1. For KB cells, P. gingivalis was more effective (as indicated by effect sizes) at inducing the secretion of sICAM-1 (d = 22; 99% confidence interval [CI], 3.17 to 23.31) compared with MRC-5 cells (d = 11; 99% CI, 1.15 to 11.35), with KB cells secreting a maximum of 1,442 pg/ml of sICAM-1 above the basal level of secretion (BCR of 100:1) compared with 353 pg/ml from MRC-5 cells (BCR of 50:1). P. gingivalis was also more effective in inducing IL-8 secretion from KB cells (d = 106; 99% CI, 30.48 to 207.47) than MRC-5 cells (d = 24; 99% CI, 6.90 to 47.46) with significantly (P < 0.01) more IL-8 secreted above the basal level from KB cells (1,594 pg/ml; BCR of 100:1) than from MRC-5 cells (1,300 pg/ml; BCR of 100:1). In contrast to sICAM-1 and IL-8 secretion, P. gingivalis was more effective at inducing IL-6 secretion in MRC-5 cells (d = 14.00; 99% CI, 3.54 to 25.65) than in KB cells (d = 7; 99% CI, 1.49 to 13.17); MRC-5 cells secreted a maximum of 3,380 pg/ml, above the basal level of secretion (BCR of 100:1) compared with 107 pg/ml from KB cells (BCR of 100:1).
FIG. 2.

Secretion of sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, and IL-1α by KB and MRC-5 cells following incubation with viable P. gingivalis W50 whole cells. Confluent KB and MRC-5 cell monolayers were incubated with P. gingivalis W50 at various BCRs (10:1. 50:1, 100:1 500:1, 1,000:1, and 10,000:1 for 90 min at 37°C. After incubation, P. gingivalis was removed, and the cell monolayers were incubated with fresh culture medium for a further 16 h at 37°C. Following incubation, the culture supernatants were collected and assayed for sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, and IL-1α by ELISA. The values represent the means and standard deviations of triplicate determinations from three representative experiments. The dotted line represents basal cytokine levels as determined by unstimulated cells (control). Data were analyzed by a t test and effect size (Cohen's d) compared to the control.
P. gingivalis induced a significant (P < 0.01) increase in the secretion of MCP-1 compared to controls from both KB and MRC-5 cells, with a maximum secretion reached at a BCR of 10:1 for both host cells (Fig. 2D). At higher BCRs, the amount of MCP-1 secreted from KB cells decreased to basal levels, and for MRC-5 cells, the reduction was significantly (P < 0.01) less than basal levels at a BCR ≥100:1. P. gingivalis was more effective at inducing MCP-1 from MRC-5 cells (d = 38; 99% CI, 2.58 to 19.58) than from KB cells (d = 20; 99% CI, 3.43 to 34.97); MCR-5 cells secreted a maximum of 121 pg/ml of MCP-1 above the basal level (BCR of 10:1), whereas epithelial cells secreted a maximum of 15 pg/ml of MCP-1 (BCR of 10:1) above basal level.
P. gingivalis was also found to stimulate MIP-1α secretion from KB and MRC-5 cells but only at high BCRs of 500:1 and 1,000:1 (Fig. 2E). Effect size values indicate that P. gingivalis was more effective in inducing MIP-1α from MRC-5 cells (d = 365; 99% CI, 105.71 to 718.83) than from KB cells (d = 306; 99% CI, 5.76 to 40.18); MRC-5 cells secreted a maximum of 75 μg/ml MIP-1α above basal levels (BCR of 1,000:1), whereas epithelial cells secreted a maximum of 31 pg/ml of MIP-1α above basal levels (BCR of 1,000:1). P. gingivalis was also found to induce a significant (P < 0.01) increase in IL-1α secretion but only from KB cells at BCRs of 100:1, 500:1, and 1,000:1, with maximum secretion above basal level (11.6 pg/ml) reached at 500:1 (Fig. 2F). However, at a BCR of 1,000:1, there was a threefold reduction in the amount of IL-1α compared with the amount induced at a BCR of 500:1. P. gingivalis was found not to induce detectable levels of IL-1β or TNF-α from KB or MRC-5 cells.
The incubation of E. coli JM109 cells with KB or MRC-5 cells at the BCRs used for P. gingivalis under the same experimental conditions did not result in significant cell binding, induction of cytokines, or the pattern of suppression of the cytokines at high BCR values as observed with P. gingivalis.
Secretion of sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, IL-1α, IL-1β, and TNF-α from KB and MRC-5 cells following incubation with RgpA-Kgp proteinase-adhesin complexes.
The amounts of sICAM-1, IL-8, IL-6, MCP-1, MIP-1α, IL-1α, IL-1β, and TNF-α in supernatants of KB and MRC-5 cells following incubation with RgpA-Kgp were determined by ELISA. Overall, incubation with increasing amounts of RgpA-Kgp resulted in corresponding and significant (P < 0.01) increases in IL-8, IL-6, sICAM-1, and MCP-1 secretion from both KB and MRC-5 cells, with a maximum secretion level reached at an RgpA-Kgp concentration of between 1.25 to 5 μg/well for each cytokine (Fig. 3A-D). Incubation with higher concentrations of RgpA-Kgp resulted in a reduction in the amounts of IL-8, IL-6, sICAM-1, and MCP-1 for both KB and MRC-5 cells (Fig. 3). The RgpA-Kgp complexes did not induce secretion of MIP-1α, IL-1α, or IL-1β from either KB or MRC-5 cells (data not shown).
FIG. 3.

Secretion of sICAM-1, IL-8, MCP-1, and IL-6 by KB and MRC-5 cells following incubation with P. gingivalis RgpA-Kgp complexes. Confluent KB and MRC-5 cell monolayers were incubated with RgpA-Kgp complexes at various concentrations (0.625, 1.25, 2.5, 5, and 10 μg) equivalent to BCRs of 12.5:1, 25:1, 50:1, 100:1, and 200:1, respectively, for 90 min at 37°C. After incubation, the cells were washed and incubated with fresh culture medium for another 16 h at 37°C. Following incubation, the culture supernatants were assayed for sICAM-1, IL-8, IL-6, MCP-1, and IL-6 by ELISA. The values represent the mean and standard deviation of triplicate determinations from three representative experiments. The dotted line represents basal cytokine levels as determined by unstimulated cells (control). Data were analyzed by a t test and effect size (Cohen's d) compared to control.
Induction of apoptosis in KB epithelial and MRC-5 fibroblast cells by P. gingivalis.
Apoptosis was quantified by flow cytometry after staining KB and MRC-5 cells with annexin V-FITC and PI following incubation with increasing BCRs of P. gingivalis. KB and MRC-5 cells incubated with medium alone or E. coli JM109 were used as controls. Viable cells were defined as FITC negative/PI negative, early apoptotic cells were defined as FITC positive/PI negative, late apoptotic/necrotic cells were defined as FITC positive/PI positive, and necrotic cells with a permeabilized membrane only were defined as FITC negative/PI positive.
The induction of apoptosis in KB and MRC-5 cells after incubation with various concentrations of P. gingivalis for 90 min is shown in Fig. 4. For KB cells, incubation with P. gingivalis resulted in a significant (P < 0.01) reduction in the number of viable cells from 97.9% in the control to 80.9% at a BCR of 1,000:1, and at a BCR of 10,000:1 only 5.3% of KB cells remained viable. The percentage of KB cells undergoing early apoptosis was shown to significantly (P < 0.01) increase from 0.4% in the control to 2.2% at a BCR of 50:1 and continued to increase, reaching 19.7% at a BCR of 10,000:1. P. gingivalis was shown to induce late apoptosis/necrosis in KB cells only at a high BCR of 10,000:1, where 71.8% of KB cells were in the late apoptotic/necrotic stage.
FIG. 4.

Induction of apoptosis in KB and MRC-5 cells after incubation with P. gingivalis W50 at various BCRs. Confluent monolayers of KB and MRC-5 cells were incubated with P. gingivalis W50 at various BCRs (10:1, 50:1, 100:1, 500:1, 1,000:1, and 10,000:1) for 90 min at 37°C. The cells were stained simultaneously with annexin V-FITC and PI and then analyzed on a FACSCalibur flow cytometer to determine the proportion of apoptotic and viable cells. The quadrants on the two-dimensional dot plots were set based upon healthy untreated cells (Control). The lower-left quadrant represents viable cells negative for both annexin V-FITC and PI. The lower-right quadrant represents a PI-negative, annexin V-FITC-positive early apoptotic cell population. The upper-left quadrant represents PI-positive, annexin V-FITC-negative necrotic cells with a permeabilized membrane only. The upper-right quadrant represents an annexin V-FITC- and PI-positive late apoptotic/necrotic cell population. Analysis is based on duplicate samples of 10,000 cells, and the data are representative of three experiments.
For MRC-5 cells, incubation with P. gingivalis resulted in a significant (P < 0.05) reduction in the number of viable cells from 99.0% in the control to 93.2% at a BCR of 1,000:1, and at a BCR of 10,000:1, only 4.2% of MRC-5 cells were viable (Fig. 4). Incubation with P. gingivalis was shown to significantly (P < 0.05) increase the percentage of MRC-5 cells undergoing early apoptosis from 0.5% in the control to 2.8% at a BCR of 50:1 and continued to increase, reaching 48.0% at a BCR of 10,000:1. P. gingivalis induced late apoptosis/necrosis in MRC-5 cells only at a high BCR of 10,000:1, where 47.6% of MRC-5 cells were in the late apoptotic/necrotic stage. KB cells were found to be more susceptible to P. gingivalis-induced apoptosis as a higher percentage of KB cells than MRC-5 cells were shown to undergo early apoptosis (BCRs of 500:1 and 1000:1) and late apoptosis/necrosis (BCR 10,000:1).
For time course experiments, KB and MRC-5 cells were incubated with P. gingivalis at a BCR of 10,000:1 for 30, 60, and 90 min (Fig. 5). The percentage of viable KB cells was shown to significantly (P < 0.01) decrease from 97.8% in the control to 10.2% following a 30-min incubation with P. gingivalis, and at 90 min, only 2.5% of the KB cells were viable. KB cells undergoing early apoptosis significantly (P < 0.01) increased from 0.0% in the control to 73.7% following a 30-min incubation with P. gingivalis, reached a maximum of 80.4% at 60 min of incubation, and then decreasing to 71.8% at 90 min of incubation. The percentage of late apoptotic/necrotic KB cells was shown to significantly (P < 0.05) increase with a corresponding increase in incubation time from 0.1% in the control to 12.5% following a 30-min incubation with P. gingivalis and reached a maximum of 18.1% at 90 min.
FIG. 5.

Time course analysis of KB and MRC-5 cell apoptosis induction by P. gingivalis W50. Confluent monolayers of KB and MRC-5 cells were incubated for 30, 60, and 90 min with P. gingivalis at a BCR of 10,000:1. The cells were stained simultaneously with annexin V-FITC and PI and then analyzed on a FACSCalibur flow cytometer to determine the proportion of apoptotic and viable cells. The quadrants on the two-dimensional dot plots were set based upon healthy untreated cells (Control). The lower-left quadrant represents viable cells negative for both annexin V-FITC and PI. The lower-right quadrant represents the PI-negative, annexin V-FITC-positive early apoptotic cell population. The upper-left quadrant represents PI-positive, annexin V-FITC-negative necrotic cells with a permeabilized membrane only. The upper-right quadrant represents the annexin V-FITC- and PI-positive late apoptotic/necrotic cell population. Analysis is based on duplicate samples of 10,000 cells, and the data are representative of three experiments.
For MRC-5 cells, the percentage of viable cells was shown to significantly (P < 0.01) decrease from 99.1% in the control to 36.9% following a 30-min incubation with P. gingivalis, and only 4.1% of MRC-5 cells were viable after a 90-min incubation (Fig. 5). MRC-5 cells undergoing early apoptosis increased from 0.6% in the control to 44.0% following a 30-min incubation with P. gingivalis, reaching a maximum of 71.5% at 60 min of incubation and then decreasing to 61.9% at 90 min of incubation. At 90 min of incubation with P. gingivalis, two distinct populations of MRC-5 cells undergoing either early apoptosis (61.9%) or late apoptosis/necrosis (22.6%) were observed. In general, KB cells were more susceptible to P. gingivalis-induced apoptosis as a greater percentage of KB cells than MRC-5 cells was shown to be apoptotic or necrotic at each time point. E. coli JM109 at the same BCR values did not induce apoptosis of the KB or MRC-5 cells under the same experimental conditions.
Induction of KB epithelial and MRC-5 fibroblast cell apoptosis by the RgpA-Kgp proteinase-adhesin complexes.
To investigate the effect of RgpA-Kgp on KB and MRC-5 cell apoptosis, cells were incubated with the complexes at various concentrations and times and then stained with annexin V-FITC and PI and analyzed by flow cytometry. The induction of apoptosis in KB and MRC-5 cells after incubation with either 10 or 50 μg of RgpA-Kgp for 90 min is shown in Fig. 6. The percentage of viable KB cells was shown to significantly (P < 0.05) decrease from 90.0% in the control to 87.0% and 75.5% following incubation with 10 and 50 μg of RgpA-Kgp, respectively. An increase in RgpA-Kgp concentration resulted in an increase in KB cells undergoing both early apoptosis and late apoptosis/necrosis. KB cells in the early apoptotic stage increased from 2% in the control to 16.9% following incubation with 50 μg of RgpA-Kgp complex. Late apoptosis/necrosis in KB cells was shown to significantly (P < 0.05) increase from 2.3% in the control to 6.6% following incubation with 50 μg of RgpA-Kgp.
FIG. 6.

KB and MRC-5 cell apoptosis induced after incubation with RgpA-Kgp complexes at various concentrations. Confluent monolayers of KB and MRC-5 cells were incubated with 10 μg and 50 μg of RgpA-Kgp complexes (equivalent to BCRs of 200:1 and 1,000:1, respectively) for 90 min at 37°C. The KB and MRC-5 ells were stained simultaneously with annexin V-FITC and PI and then analyzed on a FACSCalibur flow cytometer to determine the proportion of apoptotic and viable cells. The quadrants on the two-dimensional dot plots were set based upon healthy untreated cells (Control). The lower-left quadrant represents viable cells negative for both annexin V-FITC and PI. The lower-right quadrant represents the PI-negative, annexin V-FITC-positive early apoptotic cell population. The upper-left quadrant represents PI-positive, annexin V-FITC-negative necrotic cells with a permeabilized membrane only. The upper-right quadrant represents the annexin V-FITC- and PI-positive late apoptotic/necrotic cell population. Analysis is based on duplicate samples of 10,000 cells, and the data are representative of three experiments.
For MRC-5 cells, the number of viable cells significantly (P < 0.05) decreased from 91.1% in the control to 90.6% and 57.3% following incubation with 10 and 50 μg of RgpA-Kgp, respectively (Fig. 6). The percentage of MRC-5 cells in the early apoptotic stage was shown to significantly (P < 0.01) increase from 2.6% in the control to 33.2% following incubation with 50 μg of RgpA-Kgp. The late apoptotic/necrotic MRC-5 cell population increased from 1.0% to 4.1% after incubation with 50 μg of RgpA-Kgp.
For the time course experiments, KB and MRC-5 cells were incubated with 50 μg of RgpA-Kgp for 30, 60, and 90 min (Fig. 7). The percentage of viable KB cells was shown to significantly (P < 0.05) decrease from 88.2% in the control to 65.9% after 30 min of incubation with RgpA-Kgp and continued to decrease; at 90 min only 50.1% of the cells remained viable. The early apoptotic cell population was shown to increase from 1.0% in the control to 10.3%, 29.6%, and 34.4% after 30, 60, and 90 min of incubation with RgpA-Kgp, respectively. There was also a significant (P < 0.05) increase in the percentage of KB cells undergoing late apoptosis/necrosis from 3.5% in the control to 11.3% after 90 min of incubation with RgpA-Kgp.
FIG. 7.

Time course analysis of KB and MRC-5 cell apoptosis induction by RgpA-Kgp complexes. Confluent monolayers of KB and MRC-5 cells were incubated with 50 μg of RgpA-Kgp complexes (equivalent to a BCR of 1,000:1) for 30, 60, and 90 min. The KB and MRC-5 cells were stained with annexin V-FITC and PI and then analyzed on a FACSCalibur flow cytometer to determine the proportion of apoptotic/necrotic and viable cells. The quadrants on the two-dimensional dot plots were set based upon healthy untreated cells (Control). The lower-left quadrant represents viable cells negative for both annexin V-FITC and PI. The lower-right quadrant represents the PI-negative, annexin V-FITC-positive early apoptotic cell population. The upper-left quadrant represents PI-positive, annexin V-FITC-negative necrotic cells with a permeabilized membrane only. The upper-right quadrant represents the annexin V-FITC- and PI-positive late apoptotic/necrotic cell population. Analysis is based on duplicate samples of 10,000 cells, and the data are representative of three experiments.
The percentage of viable MRC-5 cells was shown to significantly (P < 0.05) decrease from 91.1% in the control to 83.5% after 30 min of incubation with RgpA-Kgp, and at 90 min only 57.3% of the fibroblast cells remained viable. The cell population in the early apoptotic stage was shown to increase from 2.6% in the control to 6.4%, 15.2%, and 33.2% after 30, 60, and 90 min of incubation with RgpA-Kgp, respectively. Late apoptosis/necrosis in the fibroblast cell population increased from 1.0% in the control to 4.1% after 90 min of incubation with RgpA-Kgp.
DISCUSSION
The P. gingivalis RgpA-Kgp complex was immunolocalized in connective tissue of gingival tissue specimens associated with periodontitis using an immunoperoxidase staining technique. The immunolocalization was evident from the presence of discrete and diffuse peroxidase-DAB reaction products (specific immunostaining) in localized areas of these specimens probed with specific antiserum and from the absence of the discrete and diffuse staining in serial sections from these specimens probed with nonimmune serum. None of the control specimens probed with either nonimmune or immune serum contained either discrete or diffuse specific immunostaining.
The precise physical nature (e.g., discrete antigen, outer membrane vesicles, or whole bacterial cells) of the antigens stained with the specific antiserum could not be determined. The combination of both staining patterns (discrete and diffuse) resembled that reported previously in epithelial and connective tissues of gingival specimens from sites associated with experimental gingivitis, chronic periodontitis, and localized aggressive periodontitis probed with antiserum raised against whole P. gingivalis, Actinobacillus actinomycetemcomitans, and Capnocytophaga sputigena cells and stained with peroxidase-DAB (64-68). The discrete immunostaining observed in the localized aggressive periodontitis and chronic periodontitis specimens probed with antisera raised against these species was subsequently shown to represent bacterial cells (64, 65, 68). Discrete immunostaining has also been identified in periodontitis tissue using immunofluorescence after probing with antiserum to P. gingivalis (11, 58), Actinomyces, Prevotella intermedia (58), and A. actinomycetemcomitans (9). Similar to our results, previous studies also found discrete immunostaining interspersed with varied amounts of diffuse immunostaining (65, 66, 68). The diffuse immunostaining may have represented released bacterial antigens (68).
The discrete immunostaining was typical of the insoluble and homogeneous peroxidase-DAB reaction product observed with DAB immunocytochemistry. While it is possible that the spots represented P. gingivalis cells, evidence from histological investigations suggests that active invasion of chronic periodontitis tissue by whole bacterial cells is limited and difficult to confirm in naturally occurring disease (40, 41, 69, 78).
While the size of some discrete spots was consistent with that of P. gingivalis cells or outer membrane vesicles (45), the identity of the discretely stained material could not be ascertained from size since this reflects the duration of the oxidation reaction. Therefore, the discrete spots may have represented only released antigen at higher concentrations. In this context, E. coli lipopolysaccharide stained with avidin-biotin complexes appears as discrete structures and often in clusters at the transmission electron microscopy level (42). In our study, the intensity of the discrete spots was generally higher than that of the diffuse specific stain, which likely reflected differences in antigen concentration. (92) Watanabe et al. have reported that the intensity of DAB-peroxidase staining of alpha-fetoprotein was proportional to the levels of this antigen in the tissue. Therefore, the intense immunostaining in our study is likely to represent higher concentrations of antigen that were located predominantly at the edge of the tissue samples (Fig. 1) which were proximal to the subgingival plaque.
Diffuse specific staining was found near discrete spots and in areas devoid of these spots. A similar pattern of diffuse immunostaining was also observed interspersed among discrete spots in gingivitis and periodontitis tissue probed with antiserum to P. gingivalis and to other species using an immunoperoxidase technique (65-68). It is likely that the diffuse immunostaining in our diseased specimens represented RgpA-Kgp at lower concentrations in the tissues (92). Hence, the staining pattern is indicative of a concentration gradient of the RgpA-Kgp antigen in the gingival connective tissue, as would be expected from diffusion of the antigen from subgingival plaque into the subjacent tissue. It is significant that the A1 adhesins associated with the RgpA-Kgp proteinase/adhesin complexes have been shown to bind to various connective tissue proteins and proteoglycans such as fibronectin, laminin, and collagen type V (53, 59, 63). Therefore, it is likely that these adhesins were responsible for binding RgpA-Kgp to tissue. Specific immunostaining was also observed in the wall of a small blood vessel. This is consistent with binding of RgpA-Kgp adhesin domains to collagen type V and laminin associated with vascular endothelium (90) and binding to heparan sulfate found in the extracellular matrix of blood vessels. The result is also consistent with binding of RgpA-Kgp to endothelial cells (51). Targeting of the RgpA-Kgp complexes to blood vessels and subsequent vascular disruption may be highly significant in the virulence of P. gingivalis in periodontitis (51).
Specific immunostaining was often localized between chronic inflammatory cell infiltrates. It is possible therefore that these cells included antibody-producing B cells specific for the P. gingivalis RgpA-Kgp (15, 19, 54). Specific immunostaining was occasionally observed in areas containing few chronic inflammatory cells, possibly suggesting suppression or dysregulation of cellular infiltration. Specific immunostaining was also identified in areas with significant loss of tissue architecture, which is consistent with the proteolytic enzyme activity of RgpA-Kgp and cellular apoptosis/necrosis. This may have been due to the capacity of RgpA-Kgp at high concentrations to directly and indirectly participate in the degradation of collagens type I, III, IV, and V and fibronectin (7, 59, 61, 76, 77) and to induce apoptosis/necrosis. The relatively low numbers of fibroblasts in many stained areas may also be partly due to apoptotic/necrotic effects displayed by RgpA-Kgp at high concentrations (73).
The presence of RgpA-Kgp complexes in contact with pocket epithelial and fibroblast cells led us to investigate the effect of these complexes on the secretion of inflammatory mediators by these cells. There is increasing evidence to suggest that both epithelial cells and fibroblasts are integral parts of the innate immune system as they actively participate in the inflammatory response by secreting cytokines in response to the presence of pathogens (26, 49). Hence, we investigated the secretion of inflammatory mediators IL-1α, IL-1β, IL-6, IL-8, MCP-1, MIP-1α, TNF-α, and sICAM-1 from KB epithelial and MRC-5 fibroblast cells and the degree of apoptosis in these cells following incubation with various concentrations of P. gingivalis and the RgpA-Kgp proteinase-adhesin complexes.
Results presented show that incubation with low concentrations of P. gingivalis whole cells (BCR of ≤500:1) and RgpA-Kgp (1.25 to 5 μg) stimulated secretion of sICAM-1, IL-8, IL-6, and MCP-1 from KB and MRC-5 fibroblast cells. Secretion of proinflammatory cytokines in response to P. gingivalis and RgpA-Kgp is likely to have a significant impact on the pathogenesis of chronic periodontitis. IL-6 is known to have pleiotropic effects including the maturation of B cells into immunoglobulin-producing plasma cells; hence, excessive secretion of IL-6 by epithelial and fibroblast cells may contribute to the production of P. gingivalis-specific antibody that was observed in serum of chronic periodontitis patients (52). In addition to the effects on B cells, IL-6 has been reported to induce bone resorption (30); hence, excessive secretion of IL-6 in response to P. gingivalis may play a role in inducing alveolar bone loss, a clinical hallmark of chronic periodontitis. Enhanced secretion of IL-8, MCP-1, and sICAM-1 by epithelial and fibroblast cells in response to P. gingivalis and the RgpA-Kgp complexes would induce the migration of cells such as monocytes, neutrophils, and T cells to the periodontal site (22, 88). This cellular infiltrate is considered to play a major role in the destructive processes including periodontal bone resorption that lead to the loss of tooth attachment (5, 83). However, the ability of P. gingivalis cells and RgpA-Kgp to induce secretion of these cytokines from epithelial and fibroblast cells was found to be concentration dependent. While P. gingivalis cells and RgpA-Kgp at low concentrations stimulated secretion of sICAM-1, IL-8, IL-6, and MCP-1, at higher concentrations the levels of these cytokines were substantially reduced.
In previous studies where epithelial cells were incubated with P. gingivalis strains ATCC 33277 or 381 at a single BCR of <100:1, there was in a moderate increase in IL-8 secretion; however, in other studies using higher BCRs of >150:1 of the same P. gingivalis strains, there was a decrease in the secretion of IL-8 and surface expression of ICAM-1 (24, 29, 43). These previous papers are consistent with the findings of the current study. The reduction in the levels of IL-6, IL-8, MCP-1, and sICAM-1 secretion at high P. gingivalis cell numbers and high concentrations of RgpA-Kgp may in part be attributed to degradation of these cytokines by the Arg- and Lys-specific proteinases of RgpA-Kgp. Previous studies have shown that P. gingivalis Arg- and Lys-specific proteinases degrade a variety of cytokines such as IL-8 (95), IL-6 (4), TNF-α (8), and surface ICAM-1 on gingival epithelial cells (82, 91). Furthermore, mutant strains of P. gingivalis deficient in Arg- and Lys-specific proteinase activity have been reported to stimulate significantly more IL-8, ICAM-1, and IL-6 from gingival epithelial cells and fibroblasts than wild-type P. gingivalis (29, 80). The reduction in sICAM-1, IL-8, IL-6, and MCP-1 secretion could also be attributed to epithelial and fibroblast cell apoptosis induced by high concentrations of P. gingivalis cells and RgpA-Kgp. Although P. gingivalis cells at high concentrations (BCR of ≥500:1) attenuated secretion of sICAM-1, IL-8, IL-6, and MCP-1, the secretion of MIP-1α in both epithelial and fibroblast cells and IL-1α in epithelial cells was enhanced following incubation with high P. gingivalis cell numbers. Both IL-1α and MIP-1α have been associated with the induction of antiapoptotic pathways (34, 35). Epithelial cells pretreated with IL-1 have been reported to be resistant to apoptosis induced by TNF-associated ligands such as APO-2 (34, 35). Members of the CCR3 family of chemokines, to which MIP-1α belongs, have been shown to inhibit apoptosis of cells such as lymphocytes (60) and eosinophils (75). Hence, the secretion of IL-1α and MIP-1α by epithelial and fibroblast cells in response to high concentrations of P. gingivalis cells may be an immunological defensive/survival strategy that protects surrounding and infiltrating cells against P. gingivalis-induced apoptosis.
Gingival cell apoptosis is reported to be a common feature in diseased periodontal sites (36, 71). We assessed apoptosis of cultured human epithelial and fibroblast cells following incubation with P. gingivalis W50 whole cells or RgpA-Kgp on the basis of labeling with annexin V-FITC and PI as described previously (3, 89). For both cell types, it was shown that incubation with P. gingivalis induced early apoptosis at a BCR of ≥50:1, while a mixture of early apoptosis and late apoptosis/necrosis was observed at a BCR of 10,000:1. A BCR of 10,000:1 was based on 104 epithelial cells lining a 6-mm periodontal pocket (14, 72) harboring 108 bacterial cells in subgingival plaque (20). Time course experiments showed an increase in epithelial cells undergoing early apoptosis and late apoptosis/necrosis after 30 min of incubation with P. gingivalis, while for fibroblast cells only early apoptosis was observed at this time point. An increase in fibroblast cells undergoing late apoptosis/necrosis was observed only after a 90-min incubation with P. gingivalis, suggesting that epithelial cells are more susceptible than fibroblasts to programmed cell death induced by P. gingivalis. RgpA-Kgp also induced apoptosis of the epithelial and fibroblast cells, with the epithelial cells being more susceptible. In contrast to results presented here, a number of studies have reported that prolonged incubation (24 h) with P. gingivalis strain ATCC 33277 did not induce epithelial cell apoptosis (47, 94). Furthermore, Nakhjiri et al. (47) reported that P. gingivalis ATCC 33277 prevented apoptosis of epithelial cells following incubation with camptothecin, a chemical inducer of apoptosis. These conflicting results may be due to the different human epithelial cells or P. gingivalis strains used in this and other studies. P. gingivalis strain W50 used here has been reported to be more virulent than strain ATCC 33277 based on invasiveness in animal models and prevalence in patients with chronic periodontitis (23, 48).
In summary, we show that the ability to stimulate the secretion of inflammatory mediators and the induction of apoptosis in epithelial and fibroblast cells is dependent on the concentration of P. gingivalis cells and the RgpA-Kgp complexes. These results have led us to propose a new pathogenic mechanism for P. gingivalis-induced periodontitis that helps explain the bacterial specificity of disease related to the presence of the bacterium's major virulence factor, the RgpA-Kgp proteinase-adhesin complexes that are released from subgingival plaque into the subjacent epithelium and connective tissue (Fig. 8). In the subjacent tissue distal from the biofilm, the concentration of the RgpA-Kgp complexes will be low due to the natural concentration gradient into the tissue of bacterial products produced in the biofilm. At these low concentrations RgpA-Kgp will induce, in a susceptible host, the secretion of proinflammatory mediators, resulting in cellular infiltration, inflammation, and bone resorption, the clinical hallmarks of chronic periodontitis. However, at sites proximal to the biofilm where the concentration of RgpA-Kgp will be high, the proinflammatory mediators will be rapidly degraded, and the plaque biofilm will be protected from the host's defenses. Furthermore, RgpA-Kgp at high concentrations at sites proximal to the biofilm may also induce host cell apoptosis, further leading to tissue destruction and protection of the plaque biofilm. The attenuation of host proinflammatory mediators close to the plaque biofilm by high RgpA-Kgp concentrations would result in the dysregulation of the host immune system, thereby ineffectively clearing the bacterial toxic products, which allows them to penetrate deep into the connective tissue and stimulate chronic inflammation and disease.
FIG. 8.
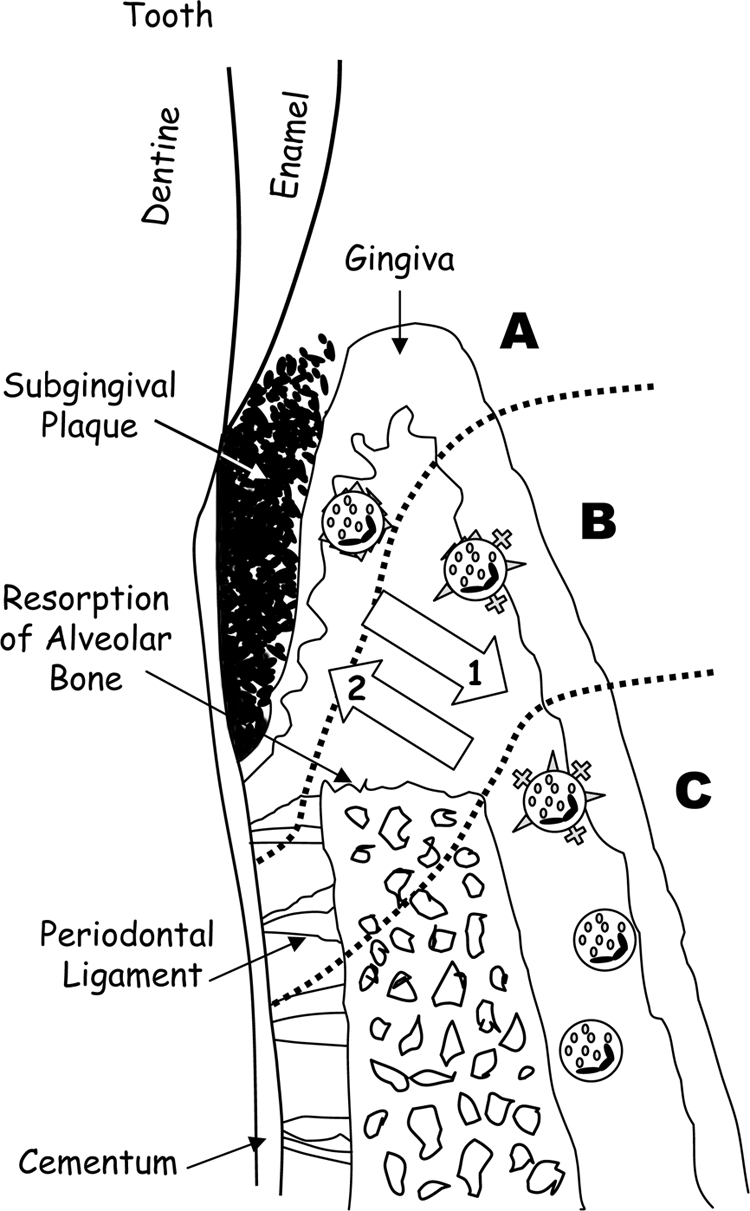
Schematic representation of a progressive periodontal lesion. (A) The area shows a high concentration of the RgpA-Kgp complexes, high level of host cell surface receptor, cytokine/chemokine degradation, apoptosis, and inflammation. (B) Decreasing concentrations of the RgpA-Kgp complexes result in decreased host cell surface receptor, cytokine/chemokine degradation, and apoptosis, but there is still a strong inflammatory response leading to bone resorption and host cell recruitment and activation. (C) Low concentrations of the RgpA-Kgp complexes and bacterial products result in host cell stimulation and recruitment and activation of host immune cells. Arrow 1 indicates the decreasing concentration of the RgpA-Kgp complexes and bacterial products in host tissue; arrow 2 indicates increasing dysregulation of the immune response.
Acknowledgments
This project is supported by the Australian National Health and Medical Research Council (project no. 454475) and the National Institutes of Health (grant no. 1R01DE1419801).
Editor: B. A. McCormick
Footnotes
Published ahead of print on 29 December 2008.
REFERENCES
- 1.Ansai, T., E. Yamamoto, S. Awano, W. Yu, A. J. Turner, and T. Takehara. 2002. Effects of periodontopathic bacteria on the expression of endothelin-1 in gingival epithelial cells in adult periodontitis. Clin. Sci. (London) 103327S-331S. [DOI] [PubMed] [Google Scholar]
- 2.Assuma, R., T. Oates, D. Cochran, S. Amar, and D. T. Graves. 1998. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J. Immunol. 160403-409. [PubMed] [Google Scholar]
- 3.Aubry, J. P., A. Blaecke, S. Lecoanet-Henchoz, P. Jeannin, N. Herbault, G. Caron, V. Moine, and J. Y. Bonnefoy. 1999. Annexin V used for measuring apoptosis in the early events of cellular cytotoxicity. Cytometry 37197-204. [DOI] [PubMed] [Google Scholar]
- 4.Banbula, A., M. Bugno, A. Kuster, P. C. Heinrich, J. Travis, and J. Potempa. 1999. Rapid and efficient inactivation of IL-6 gingipains, lysine- and arginine-specific proteinases from Porphyromonas gingivalis. Biochem. Biophys. Res. Commun. 261598-602. [DOI] [PubMed] [Google Scholar]
- 5.Bascones, A., J. Gamonal, M. Gomez, A. Silva, and M. A. Gonzalez. 2004. New knowledge of the pathogenesis of periodontal disease. Quintessence Int. 35706-716. [PubMed] [Google Scholar]
- 6.Bascones, A., S. Noronha, M. Gomez, P. Mota, M. A. Gonzalez Moles, and M. V. Dorrego. 2005. Tissue destruction in periodontitis: bacteria or cytokines fault? Quintessence Int. 36299-306. [PubMed] [Google Scholar]
- 7.Birkedal-Hansen, H., R. E. Taylor, J. J. Zambon, P. K. Barwa, and M. E. Neiders. 1988. Characterization of collagenolytic activity from strains of Bacteroides gingivalis. J. Periodontal Res. 23258-264. [DOI] [PubMed] [Google Scholar]
- 8.Calkins, C. C., K. Platt, J. Potempa, and J. Travis. 1998. Inactivation of tumor necrosis factor-alpha by proteinases (gingipains) from the periodontal pathogen, Porphyromonas gingivalis. Implications of immune evasion. J. Biol. Chem. 2736611-6614. [DOI] [PubMed] [Google Scholar]
- 9.Christersson, L. A., B. Albini, J. J. Zambon, U. M. Wikesjo, and R. J. Genco. 1987. Tissue localization of Actinobacillus actinomycetemcomitans in human periodontitis. I. Light, immunofluorescence and electron microscopic studies. J. Periodontol. 58529-539. [DOI] [PubMed] [Google Scholar]
- 10.Cohen, J. 1969. Statistical power analysis for the behavioral sciences. Academic Press, New York, NY.
- 11.Courant, P. R., and H. Bader. 1966. Bacteroides melaninogenicus and its products in the gingiva of man. Periodontics 4131-136. [PubMed] [Google Scholar]
- 12.Crawford, J. M. 1992. Distribution of ICAM-1, LFA-3 and HLA-DR in healthy and diseased gingival tissues. J. Periodontal Res. 27291-298. [DOI] [PubMed] [Google Scholar]
- 13.Darveau, R. P., C. M. Belton, R. A. Reife, and R. J. Lamont. 1998. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect. Immun. 661660-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawes, C. 2003. Estimates, from salivary analyses, of the turnover time of the oral mucosal epithelium in humans and the number of bacteria in an edentulous mouth. Arch. Oral Biol. 48329-336. [DOI] [PubMed] [Google Scholar]
- 15.Ebersole, J. L., D. E. Frey, M. A. Taubman, and D. J. Smith. 1980. An ELISA for measuring serum antibodies to Actinobacillus actinomycetemcomitans. J. Periodontal Res. 15621-632. [DOI] [PubMed] [Google Scholar]
- 16.Eick, S., J. Rodel, J. W. Einax, and W. Pfister. 2002. Interaction of Porphyromonas gingivalis with KB cells: comparison of different clinical isolates. Oral Microbiol. Immunol. 17201-208. [DOI] [PubMed] [Google Scholar]
- 17.Fletcher, J., S. Nair, S. Poole, B. Henderson, and M. Wilson. 1998. Cytokine degradation by biofilms of Porphyromonas gingivalis. Curr. Microbiol. 36216-219. [DOI] [PubMed] [Google Scholar]
- 18.Fletcher, J., K. Reddi, S. Poole, S. Nair, B. Henderson, P. Tabona, and M. Wilson. 1997. Interactions between periodontopathogenic bacteria and cytokines. J. Periodontal Res. 32200-205. [DOI] [PubMed] [Google Scholar]
- 19.Gemmell, E., and G. J. Seymour. 1992. Different responses in B cells induced by Porphyromonas gingivalis and Fusobacterium nucleatum. Arch. Oral Biol. 37565-573. [DOI] [PubMed] [Google Scholar]
- 20.Gmur, R. 1988. Applicability of monoclonal antibodies to quantitatively monitor subgingival plaque for specific bacteria. Oral Microbiol. Immunol. 3187-191. [DOI] [PubMed] [Google Scholar]
- 21.Graves, D. T., and D. Cochran. 2003. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J. Periodontol. 74391-401. [DOI] [PubMed] [Google Scholar]
- 22.Graves, D. T., and Y. Jiang. 1995. Chemokines, a family of chemotactic cytokines. Crit. Rev. Oral Biol. Med. 6109-118. [DOI] [PubMed] [Google Scholar]
- 23.Griffen, A. L., S. R. Lyons, M. R. Becker, M. L. Moeschberger, and E. J. Leys. 1999. Porphyromonas gingivalis strain variability and periodontitis. J. Clin. Microbiol. 374028-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han, Y. W., W. Shi, G. T. Huang, S. Kinder Haake, N. H. Park, H. Kuramitsu, and R. J. Genco. 2000. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect. Immun. 683140-3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanazawa, S., Y. Kawata, A. Takeshita, H. Kumada, M. Okithu, S. Tanaka, Y. Yamamoto, T. Masuda, T. Umemoto, and S. Kitano. 1993. Expression of monocyte chemoattractant protein 1 (MCP-1) in adult periodontal disease: increased monocyte chemotactic activity in crevicular fluids and induction of MCP-1 expression in gingival tissues. Infect. Immun. 615219-5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hedges, S. R., W. W. Agace, and C. Svanborg. 1995. Epithelial cytokine responses and mucosal cytokine networks. Trends Microbiol. 3266-270. [DOI] [PubMed] [Google Scholar]
- 27.Hirose, M., K. Ishihara, A. Saito, T. Nakagawa, S. Yamada, and K. Okuda. 2001. Expression of cytokines and inducible nitric oxide synthase in inflamed gingival tissue. J. Periodontol. 72590-597. [DOI] [PubMed] [Google Scholar]
- 28.Hou, L. T., C. M. Liu, B. Y. Liu, S. J. Lin, C. S. Liao, and E. F. Rossomando. 2003. Interleukin-1beta, clinical parameters and matched cellular-histopathologic changes of biopsied gingival tissue from periodontitis patients. J. Periodontal Res. 38247-254. [DOI] [PubMed] [Google Scholar]
- 29.Huang, G. T., D. Kim, J. K. Lee, H. K. Kuramitsu, and S. K. Haake. 2001. Interleukin-8 and intercellular adhesion molecule 1 regulation in oral epithelial cells by selected periodontal bacteria: multiple effects of Porphyromonas gingivalis via antagonistic mechanisms. Infect. Immun. 691364-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishimi, Y., C. Miyaura, C. H. Jin, T. Akatsu, E. Abe, Y. Nakamura, A. Yamaguchi, S. Yoshiki, T. Matsuda, and T. Hirano. 1990. IL-6 is produced by osteoblasts and induces bone resorption. J. Immunol. 1453297-3303. [PubMed] [Google Scholar]
- 31.Jarnbring, F., E. Somogyi, J. Dalton, A. Gustafsson, and B. Klinge. 2002. Quantitative assessment of apoptotic and proliferative gingival keratinocytes in oral and sulcular epithelium in patients with gingivitis and periodontitis. J. Clin. Periodontol. 291065-1071. [DOI] [PubMed] [Google Scholar]
- 32.Kinane, D. F., F. P. Winstanley, E. Adonogianaki, and N. A. Moughal. 1992. Bioassay of interleukin 1 (IL-1) in human gingival crevicular fluid during experimental gingivitis. Arch. Oral Biol. 37153-156. [DOI] [PubMed] [Google Scholar]
- 33.Kirby, A. C., G. Griffiths, A. Gokbuget, H. N. Newman, S. R. Porter, and I. Olsen. 1999. Localized adhesion molecule expression and circulating LFA-3 levels in adult and early onset forms of periodontitis. J. Clin. Periodontol. 26793-801. [DOI] [PubMed] [Google Scholar]
- 34.Kothny-Wilkes, G., D. Kulms, T. A. Luger, M. Kubin, and T. Schwarz. 1999. Interleukin-1 protects transformed keratinocytes from tumor necrosis factor-related apoptosis-inducing ligand- and CD95-induced apoptosis but not from ultraviolet radiation-induced apoptosis. J. Biol. Chem. 27428916-28921. [DOI] [PubMed] [Google Scholar]
- 35.Kothny-Wilkes, G., D. Kulms, B. Poppelmann, T. A. Luger, M. Kubin, and T. Schwarz. 1998. Interleukin-1 protects transformed keratinocytes from tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 27329247-29253. [DOI] [PubMed] [Google Scholar]
- 36.Koulouri, O., D. F. Lappin, M. Radvar, and D. F. Kinane. 1999. Cell division, synthetic capacity and apoptosis in periodontal lesions analysed by in situ hybridisation and immunohistochemistry. J. Clin. Periodontol. 26552-559. [DOI] [PubMed] [Google Scholar]
- 37.Kurtis, B., H. Develioglu, I. L. Taner, K. Balos, and I. O. Tekin. 1999. IL-6 levels in gingival crevicular fluid (GCF) from patients with non-insulin dependent diabetes mellitus (NIDDM), adult periodontitis and healthy subjects. J. Oral Sci. 41163-167. [DOI] [PubMed] [Google Scholar]
- 38.Lamont, R. J., and H. F. Jenkinson. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 621244-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lappin, D. F., M. Kjeldsen, L. Sander, and D. F. Kinane. 2000. Inducible nitric oxide synthase expression in periodontitis. J. Periodontal Res. 35369-373. [DOI] [PubMed] [Google Scholar]
- 40.Listgarten, M. A. 1988. Letter. J. Periodontol 59412. [DOI] [PubMed] [Google Scholar]
- 41.Listgarten, M. A. 1986. Pathogenesis of periodontitis. J. Clin. Periodontol. 13418-430. [DOI] [PubMed] [Google Scholar]
- 42.Lucas, R. M., A. Subramoniam, and J. J. Aleo. 1985. Intracellular localization of bacterial lipopolysaccharide using the avidin biotin complex method at the electron microscopic level. J. Periodontol. 56553-557. [DOI] [PubMed] [Google Scholar]
- 43.Madianos, P. N., P. N. Papapanou, and J. Sandros. 1997. Porphyromonas gingivalis infection of oral epithelium inhibits neutrophil transepithelial migration. Infect. Immun. 653983-3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathur, A., B. Michalowicz, M. Castillo, and D. Aeppli. 1996. Interleukin-1α, interleukin-8 and interferon-α levels in gingival crevicular fluid. J. Periodontal Res. 31489-495. [DOI] [PubMed] [Google Scholar]
- 45.Mayrand, D., and S. C. Holt. 1988. Biology of asaccharolytic black-pigmented Bacteroides species. Microbiol. Rev. 52134-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mogi, M., J. Otogoto, N. Ota, H. Inagaki, M. Minami, and K. Kojima. 1999. Interleukin 1β, interleukin 6, β2-microglobulin, and transforming growth factor α in gingival crevicular fluid from human periodontal disease. Arch. Oral Biol. 44535-539. [DOI] [PubMed] [Google Scholar]
- 47.Nakhjiri, S. F., Y. Park, O. Yilmaz, W. O. Chung, K. Watanabe, A. El-Sabaeny, K. Park, and R. J. Lamont. 2001. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol. Lett. 200145-149. [DOI] [PubMed] [Google Scholar]
- 48.Neiders, M. E., P. B. Chen, H. Suido, H. S. Reynolds, J. J. Zambon, M. Shlossman, and R. J. Genco. 1989. Heterogeneity of virulence among strains of Bacteroides gingivalis. J. Periodontal Res. 24192-198. [DOI] [PubMed] [Google Scholar]
- 49.Nixon, C. S., M. J. Steffen, and J. L. Ebersole. 2000. Cytokine responses to Treponema pectinovorum and Treponema denticola in human gingival fibroblasts. Infect. Immun. 685284-5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Norusis, M. 1993. SPSS for Windows: base system user's guide, release 6.0. SPSS Inc., Chicago, IL.
- 51.O'Brien-Simpson, N., P. D. Veith, S. G. Dashper, and E. C. Reynolds. 2003. Porphyromonas gingivalis gingipains: the molecular teeth of a microbial vampire. Curr. Protein Pept. Sci. 4409-426. [DOI] [PubMed] [Google Scholar]
- 52.O'Brien-Simpson, N. M., C. L. Black, P. S. Bhogal, S. M. Cleal, N. Slakeski, T. J. Higgins, and E. C. Reynolds. 2000. Serum immunoglobulin G (IgG) and IgG subclass responses to the RgpA-Kgp proteinase-adhesin complex of Porphyromonas gingivalis in adult periodontitis. Infect. Immun. 682704-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Brien-Simpson, N. M., R. D. Pathirana, R. A. Paolini, Y. Y. Chen, P. D. Veith, V. Tam, N. Ally, R. N. Pike, and E. C. Reynolds. 2005. An immune response directed to proteinase and adhesin functional epitopes protects against Porphyromonas gingivalis-induced periodontal bone loss. J. Immunol. 1753980-3989. [DOI] [PubMed] [Google Scholar]
- 54.Ogawa, T., M. L. McGhee, Z. Moldoveanu, S. Hamada, J. Mestecky, J. R. McGhee, and H. Kiyono. 1989. Bacteroides-specific IgG and IgA subclass antibody-secreting cells isolated from chronically inflamed gingival tissues. Clin. Exp. Immunol. 76103-110. [PMC free article] [PubMed] [Google Scholar]
- 55.Page, R. C., and H. E. Schroeder. 1976. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab. Investig. 34235-249. [PubMed] [Google Scholar]
- 56.Pathirana, R. D., N. M. O'Brien-Simpson, G. C. Brammar, N. Slakeski, and E. C. Reynolds. 2007. Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect. Immun. 751436-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pathirana, R. D., N. M. O'Brien-Simpson, P. D. Veith, P. F. Riley, and E. C. Reynolds. 2006. Characterization of proteinase-adhesin complexes of Porphyromonas gingivalis. Microbiology 1522381-2394. [DOI] [PubMed] [Google Scholar]
- 58.Pekovic, D. D., and E. D. Fillery. 1984. Identification of bacteria in immunopathological mechanisms of human periodontal diseases. J. Periodontal Res. 19329-351. [DOI] [PubMed] [Google Scholar]
- 59.Pike, R. M., J. Potempa, W. McGraw, H. T. Coetzer, and J. Travis. 1996. Characterization of the binding activities of proteinase-adhesin complexes from Porphromonas gingivalis. J. Bacteriol. 1782876-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinto, L. A., M. S. Williams, M. J. Dolan, P. A. Henkart, and G. M. Shearer. 2000. Beta-chemokines inhibit activation-induced death of lymphocytes from HIV-infected individuals. Eur. J. Immunol. 302048-2055. [DOI] [PubMed] [Google Scholar]
- 61.Potempa, J., A. Banbula, and J. Travis. 2000. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000. 24153-192. [DOI] [PubMed] [Google Scholar]
- 62.Prabhu, A., B. S. Michalowicz, and A. Mathur. 1996. Detection of local and systemic cytokines in adult periodontitis. J. Periodontol. 67515-522. [DOI] [PubMed] [Google Scholar]
- 63.Reynolds, E. C., and N. M. O'Brien-Simpson. 2000. Cardiovascular disease and periodontal diseases: microbial direct effects on endothelial cells. Periodontology 21:24-31 [Google Scholar]
- 64.Saglie, F. R., F. A. Carranza, Jr., M. G. Newman, L. Cheng, and K. J. Lewin. 1982. Identification of tissue-invading bacteria in human periodontal disease. J. Periodontal Res. 17452-455. [DOI] [PubMed] [Google Scholar]
- 65.Saglie, F. R., A. Marfany, and P. Camargo. 1988. Intragingival occurrence of Actinobacillus actinomycetemcomitans and Bacteroides gingivalis in active destructive periodontal lesions. J. Periodontol. 59259-265. [DOI] [PubMed] [Google Scholar]
- 66.Saglie, F. R., J. Pertuiset, M. T. Rezende, M. Nestor, A. Marfany, and J. Cheng. 1988. In situ correlative immuno-identification of mononuclear infiltrates and invasive bacteria in diseased gingiva. J. Periodontol. 59688-696. [DOI] [PubMed] [Google Scholar]
- 67.Saglie, F. R., J. H. Pertuiset, M. T. Rezende, M. S. Sabet, D. Raoufi, and F. A. Carranza, Jr. 1987. Bacterial invasion in experimental gingivitis in man. J. Periodontol. 58837-846. [DOI] [PubMed] [Google Scholar]
- 68.Saglie, F. R., C. T. Smith, M. G. Newman, F. A. Carranza, Jr., J. H. Pertuiset, L. Cheng, E. Auil, and R. J. Nisengard. 1986. The presence of bacteria in the oral epithelium in periodontal disease. II. Immunohistochemical identification of bacteria. J. Periodontol. 57492-500. [DOI] [PubMed] [Google Scholar]
- 69.Sanavi, F., M. A. Listgarten, F. Boyd, K. Sallay, and A. Nowotny. 1985. The colonization and establishment of invading bacteria in periodontium of ligature-treated immunosuppressed rats. J. Periodontol. 56273-280. [DOI] [PubMed] [Google Scholar]
- 70.Sandros, J., C. Karlsson, D. F. Lappin, P. N. Madianos, D. F. Kinane, and P. N. Papapanou. 2000. Cytokine responses of oral epithelial cells to Porphyromonas gingivalis infection. J. Dent. Res. 791808-1814. [DOI] [PubMed] [Google Scholar]
- 71.Sawa, T., F. Nishimura, H. Ohyama, K. Takahashi, S. Takashiba, and Y. Murayama. 1999. In vitro induction of activation-induced cell death in lymphocytes from chronic periodontal lesions by exogenous Fas ligand. Infect. Immun. 671450-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schroeder, H. E., E. Preisig, and C. P. Marinello. 1985. Size of corneocytes and non-keratinized superficial cells of the human oral mucosa at different ages. J. Biol. Buccale 13237-249. [PubMed] [Google Scholar]
- 73.Shah, H. N., S. V. Seddon, and S. E. Gharbia. 1989. Studies on the virulence properties and metabolism of pleiotropic mutants of Porphyromonas gingivalis (Bacteroides gingivalis) W50. Oral Microbiol. Immunol. 419-23. [DOI] [PubMed] [Google Scholar]
- 74.Shapira, L., C. Champagne, T. E. Van Dyke, and S. Amar. 1998. Strain-dependent activation of monocytes and inflammatory macrophages by lipopolysaccharide of Porphyromonas gingivalis. Infect. Immun. 662736-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shinagawa, K., A. Trifilieff, and G. P. Anderson. 2003. Involvement of CCR3-reactive chemokines in eosinophil survival. Int. Arch. Allergy Immunol. 130150-157. [DOI] [PubMed] [Google Scholar]
- 76.Smalley, J. W., A. J. Birss, and C. A. Shuttleworth. 1988. The degradation of type I collagen and human plasma fibronectin by the trypsin-like enzyme and extracellular membrane vesicles of Bacteroides gingivalis W50. Arch. Oral Biol. 33323-329. [DOI] [PubMed] [Google Scholar]
- 77.Smalley, J. W., A. J. Birss, and C. A. Shuttleworth. 1988. The effect of the outer membrane fraction of Bacteroides gingivalis W50 on glycosaminoglycan metabolism by human gingival fibroblasts in culture. Arch. Oral Biol. 33547-553. [DOI] [PubMed] [Google Scholar]
- 78.Socransky, S. S., and A. D. Haffajee. 1992. The bacterial etiology of destructive periodontal disease: current concepts. J. Periodontol. 63322-331. [DOI] [PubMed] [Google Scholar]
- 79.Socransky, S. S., A. D. Haffajee, M. A. Cugini, C. Smith, and R. L. Kent, Jr. 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25134-144. [DOI] [PubMed] [Google Scholar]
- 80.Steffen, M. J., S. C. Holt, and J. L. Ebersole. 2000. Porphyromonas gingivalis induction of mediator and cytokine secretion by human gingival fibroblasts. Oral Microbiol. Immunol. 15172-180. [DOI] [PubMed] [Google Scholar]
- 81.Stoufi, E. D., M. A. Taubman, J. L. Ebersole, D. J. Smith, and P. P. Stashenko. 1987. Phenotypic analyses of mononuclear cells recovered from healthy and diseased human periodontal tissues. J. Clin. Immunol. 7235-245. [DOI] [PubMed] [Google Scholar]
- 82.Tada, H., S. Sugawara, E. Nemoto, T. Imamura, J. Potempa, J. Travis, H. Shimauchi, and H. Takada. 2003. Proteolysis of ICAM-1 on human oral epithelial cells by gingipains. J. Dent. Res. 82796-801. [DOI] [PubMed] [Google Scholar]
- 83.Taubman, M. A., P. Valverde, X. Han, and T. Kawai. 2005. Immune response: the key to bone resorption in periodontal disease. J. Periodontol. 762033-2041. [DOI] [PubMed] [Google Scholar]
- 84.Tokoro, Y., T. Yamamoto, and K. Hara. 1996. IL-1β mRNA as the predominant inflammatory cytokine transcript: correlation with inflammatory cell infiltration into human gingiva. J. Oral Pathol. Med. 25225-231. [DOI] [PubMed] [Google Scholar]
- 85.Tonetti, M. S. 1997. Molecular factors associated with compartmentalization of gingival immune responses and transepithelial neutrophil migration. J. Periodontal Res. 32104-109. [DOI] [PubMed] [Google Scholar]
- 86.Tonetti, M. S., D. Cortellini, and N. P. Lang. 1998. In situ detection of apoptosis at sites of chronic bacterially induced inflammation in human gingiva. Infect. Immun. 665190-5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tonetti, M. S., M. A. Imboden, L. Gerber, N. P. Lang, J. Laissue, and C. Mueller. 1994. Localized expression of mRNA for phagocyte-specific chemotactic cytokines in human periodontal infections. Infect. Immun. 624005-4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van de Stolpe, A., and P. T. van der Saag. 1996. Intercellular adhesion molecule-1. J. Mol. Med. 7413-33. [DOI] [PubMed] [Google Scholar]
- 89.Vermes, I., C. Haanen, H. Steffens-Nakken, and C. Reutelingsperger. 1995. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods 18439-51. [DOI] [PubMed] [Google Scholar]
- 90.Voss, B., and J. Rauterberg. 1986. Localization of collagen types I, III, IV and V, fibronectin and laminin in human arteries by the indirect immunofluorescence method. Pathol. Res. Pract. 181568-575. [DOI] [PubMed] [Google Scholar]
- 91.Wang, P. L., M. Shinohara, N. Murakawa, M. Endo, S. Sakata, M. Okamura, and K. Ohura. 1999. Effect of cysteine protease of Porphyromonas gingivalis on adhesion molecules in gingival epithelial cells. Jpn. J. Pharmacol. 8075-79. [DOI] [PubMed] [Google Scholar]
- 92.Watanabe, J., Y. Asaka, and S. Kanamura. 1996. Relationship between immunostaining intensity and antigen content in sections. J. Histochem. Cytochem. 441451-1458. [DOI] [PubMed] [Google Scholar]
- 93.Yang, L. C., C. H. Tsai, F. M. Huang, C. M. Liu, C. C. Lai, and Y. C. Chang. 2003. Induction of interleukin-6 gene expression by pro-inflammatory cytokines and black-pigmented Bacteroides in human pulp cell cultures. Int. Endod. J. 36352-357. [DOI] [PubMed] [Google Scholar]
- 94.Yilmaz, O., T. Jungas, P. Verbeke, and D. M. Ojcius. 2004. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 723743-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang, J., H. Dong, S. Kashket, and M. J. Duncan. 1999. IL-8 degradation by Porphyromonas gingivalis proteases. Microb. Pathog. 26275-280. [DOI] [PubMed] [Google Scholar]