

Abstract
Intracellular Salmonella enterica serovar Typhimurium (serovar Typhimurium) occupies a Salmonella-containing vacuole (SCV) where bacterial effector proteins are secreted into the host cell using type III secretion systems (T3SS). Cytoskeletal motor proteins and T3SS-delivered effector proteins facilitate SCV positioning to juxtanuclear positions where bacterial replication occurs. Here, we show that this characteristic SCV positioning is not maintained by all SCVs during infection of HeLa cells. Notably, juxtanuclear SCV localization that occurs by 8 to 14 h postinfection is followed by significant centrifugal displacement of a subset of SCVs toward the host cell periphery by 24 h postinfection. This novel phenotype requires bacterial protein synthesis, a functional Salmonella pathogenicity island 2 (SPI-2)-encoded T3SS, intact microtubules, and kinesin-1 motor protein. Bacteria lacking PipB2, a kinesin-recruiting T3SS effector, did not exhibit centrifugal displacement and remained at juxtanuclear positions throughout 24 h of infection. While levels of the SPI-2 effectors PipB2 and SifA increased during 24 h postinfection, a corresponding decrease in levels of the SPI-1 T3SS effectors SipA and SopB, both known to mediate juxtanuclear SCV positioning, was observed. A fluorescence-based assay indicated that wild-type serovar Typhimurium transferred from infected to uninfected epithelial cells while strains deficient in SPI-2 T3SS secretion or PipB2 did not. Our results reveal a novel SCV phenotype implicated in the cell-to-cell spread of serovar Typhimurium during infection.
Salmonella enterica serovar Typhimurium (serovar Typhimurium) is a cause of gastroenteritis in humans and typhoid-like disease in certain strains of mice (55). Serovar Typhimurium is a facultative intracellular pathogen that can actively invade nonphagocytic cells through the delivery of bacterial proteins, termed effectors, into the host cell cytosol using the type III secretion system (T3SS) encoded by Salmonella pathogenicity island 1 (SPI-1) (19). Following entry, serovar Typhimurium typically resides in a membrane-bound compartment termed the Salmonella-containing vacuole (SCV) (16, 50, 54). From here, additional effector proteins required for intracellular replication and virulence are delivered into the host cytosol using a second T3SS encoded by another pathogenicity island, SPI-2 (10, 28, 40, 48, 58). These effectors modulate various host cell activities, including endosomal trafficking, actin assembly dynamics, and microtubule-based transport (5, 8, 24, 33, 50, 51, 58).
One characteristic trait of SCVs is their localization to a juxtanuclear, Golgi apparatus-associated region of the host cell several hours postinfection (1, 5, 41, 45). In general, with some variation likely due to differing experimental methods, this distinct localization is observed as early as 4 h postinfection (hpi) and is maintained at 8 to 16 hpi (2, 5, 13, 24, 26, 41, 42, 45). A recent study has shown that serovar Typhimurium can redirect host secretory traffic, resulting in an accumulation of post-Golgi vesicles around the SCV (36). It has been proposed that serovar Typhimurium targets the Golgi apparatus to acquire nutrients and/or membrane materials for maintenance of a replicative niche within the SCV (23, 45).
Several different effectors secreted by the SPI-2 T3SS appear to mediate this centralized positioning of SCVs, including SseF, SseG, and SifA (2, 5, 13, 41, 42, 45). Deletion of sseF or sseG results in SCVs that are displaced from their usual juxtanuclear, Golgi compartment-associated position (1, 13, 45). SseF and SseG have been shown to interact with each other (13) and appear to promote the recruitment of the minus-end-directed microtubule motor dynein to SCVs to permit their juxtanuclear localization (2). SseG has also been proposed to act by tethering SCVs to the Golgi region (42).
Deletion of sifA also results in SCVs that are displaced from the nucleus and located toward the host cell periphery (5). SifA was shown to interact with a host SifA- and kinesin-interacting protein that negatively regulates the recruitment of plus-end-directed kinesin-1 motors to the SCV, thus favoring the inward migration and maintenance of the SCV around the nucleus (5). In apparent opposition to SifA, the SPI-2 effector PipB2 has been shown to recruit kinesin-1 to the SCV (26). However, the characteristic positioning of SCVs to juxtanuclear regions suggests that the kinesin-inhibitory action of SifA may be dominant over the effects of PipB2 (26), at least at 8 to 14 hpi.
Interestingly, some effectors secreted by the SPI-1 T3SS that is traditionally associated with Salmonella invasion appear to persist in host cells (6, 14) and are also implicated in modulating intracellular SCV positioning (6, 57). We recently demonstrated a role for the SPI-1 T3SS effector SopB in maintaining the juxtanuclear positioning of SCVs through the action of nonmuscle myosin II actin motors (57). Another SPI-1 effector, SipA, has also been shown to persist in host cells after bacterial entry and appears to act with SifA to ensure perinuclear positioning of SCVs (6). Hence, it appears that stringent control of microtubule and actin motor activity on the SCV by both SPI-1 and SPI-2 T3SS effectors is an important facet of SCV intracellular positioning (26).
Overall, much remains to be resolved regarding the mediators and implications of intracellular SCV positioning. By remaining at the juxtanuclear region, the bacteria likely modify their compartments into a replicative niche where nutrient acquisition and SCV maintenance can occur (23, 36, 45). As a result of replication, high numbers of intracellular bacteria would presumably lead to host cell lysis, resulting in bacterial release; however, little is known about any mechanism(s) of Salmonella escape from host cells.
The present study was conducted to examine the intracellular positioning of SCVs over the course of a 24-h infection. We show that at later stages of epithelial cell infection, the positioning of a significant proportion of SCVs is not maintained at a juxtanuclear region but is situated closer to the host cell periphery. This outward displacement of SCVs was dependent upon the SPI-2 T3SS, host microtubules and kinesin, and the SPI-2 effector PipB2. Furthermore, the dynamic positioning of SCVs is associated with a decrease in protein levels of SPI-1 effectors previously shown to mediate juxtanuclear positioning. Results from a cell-to-cell infection assay indicate that serovar Typhimurium strains that did not exhibit peripheral displacement at later stages of infection were also impaired in their ability to infect newly introduced host cells. Our results provide new insight into the nature of SCV positioning and demonstrate that intracellular SCV positioning is a dynamic process, with implications for bacterial cell-to-cell transfer.
MATERIALS AND METHODS
Bacterial strains and plasmids.
S. enterica serovar Typhimurium strains used in this work were wild-type (WT) SL1344 (29), WT CS401 (39), and their derivatives as follows. Strains derived from WT strain SL1344 were the ΔsifA (51) and ΔssaR (SPI-2 secretion deficient) (9) strains. Isogenic mutant strains of serovar Typhimurium CS401 include the ΔssaT (SPI-2 secretion deficient) (39) and ΔpipB2 (strain MBO207) strains. The ΔpipB2 strain was constructed using allelic exchange as outlined previously (12). To study promoter activity, we integrated single-copy transcriptional fusions of the sifA or pipB2 promoter to lacZ into serovar Typhimurium strain SL1344 by homologous recombination as described previously (11). Strain BKC11-13 encodes PsifA::lacZ, and strain BKC12-66 encodes PpipB2::lacZ.
Plasmid psifA-2HA encoding a SifA fusion to double hemagglutinin (HA) tags (SifA-2HA) was described previously (8). Plasmid pPipB2-2HA encoding a PipB2 fusion to double HA tags at the C terminus of the effector protein was constructed by amplifying pipB2 and its promoter region using primers pipB2-WSK1 (5′-GCGCGAATTCACGGCTCTACTACTCGATAG-3′; the EcoRI site is underlined) and pipB2-WSK2 (5′-GCGCGAGCTCTTACGCATAATCCGGCACATCATACGGATACGCATAATCCGGCACATCATACGGATAAATATTTTCACTATAAAATTCGTT-3′; the SacI site is underlined). PCR amplicons were ligated into the low-copy-number vector pWSK29 (56) restricted previously with EcoRI/SacI. Primer pipB2-WSK1 was designed to anneal upstream of the pipB2 promoter as described in a previous study (35). Plasmid pCB6-GFP-KLC-TPR encoding a kinesin light chain tetratricopeptide repeat (KLC-TPR) fusion to green fluorescent protein (GFP) was generously provided by M. Way, London Research Institute, London, United Kingdom.
Cell culture, transfection, and bacterial invasion.
HeLa (human epithelial cell line) cells were obtained from the ATCC. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; HyClone) supplemented with 10% fetal bovine serum (Wisent) at 37°C in 5% CO2. HeLa epithelial cells were seeded at 5 × 104 cells per well in 24-well tissue culture plates 16 to 24 h before use. Late-log-phase bacterial cultures were used for infecting HeLa cells, as outlined previously (7). The bacteria used for infections were logarithmic phase serovar Typhimurium grown for 3 h in LB medium. Bacteria were pelleted at 10,000 × g for 2 min and resuspended in phosphate-buffered saline (PBS). The inoculum was diluted and added to HeLa cells at a multiplicity of infection of 100:1 at 37°C for 10 min. The cells were then washed extensively with PBS, and growth medium containing 100 μg/ml of gentamicin was added until 2 h postinfection, at which point the cells were washed, and growth medium containing 10 μg ml−1 of gentamicin was added for the duration of each experiment. Where indicated (see the legend of Fig. 1), nocodazole was used at 2 μg/ml, and tetracycline was used at 20 μg/ml. For experiments involving CellTracker Blue CMAC (Molecular Probes) (see below), HeLa cells between passages 3 to 10 from thawed stocks were used. For all other experiments, HeLa cells between passages 3 to 30 were used. For transfection of HeLa cells, Fugene 6 (Roche) or Genejuice (Novagen) transfection reagents were used according to the manufacturer's instructions. Cells were transfected with plasmids encoding GFP or KLC-TPR at 2 hpi with serovar Typhimurium.
FIG. 1.

SCVs exhibit dynamic intracellular positioning over the course of infection. (A to D) HeLa cells were infected with WT serovar Typhimurium and fixed at the indicated times postinfection. Cells were immunostained for bacteria (red) and LAMP1 (green), and the nucleus was stained using DAPI (blue). The left panel shows immunofluorescence images, with dotted lines indicating cell borders; the right panel shows corresponding differential interference contrast (DIC) images. Arrows indicate LAMP1+ SCVs. Bar, 10 μm. (E) Distribution of SCVs in HeLa cells relative to the host cell nucleus over time. Cells were fixed at the selected time points postinfection and immunostained as above. The distances of LAMP1+ SCVs to the nearest edge of the host cell nucleus were measured and grouped according to the specific distances shown in the histogram. Numbers above the square brackets indicate total percentage of SCVs that are located >6 μm from the host cell nucleus. Shown at the bottom of the histogram are the average distances ± standard deviations for three separate experiments, calculated as outlined in the Materials and Methods. Where indicated, nocodazole (2 μg/ml) or tetracycline (20 μg/ml) was added at 8 hpi. At least 100 LAMP1+ SCVs were measured from the nucleus for each time point in a given experiment. *, P < 0.001) in comparison to the corresponding 8-hpi measurement; ** P < 0.001 in the average distances of SCVs to the nucleus between the drug-treated cells and the corresponding WT control time points, as determined by Kruskal-Wallis one-way ANOVA with Dunn's multiple comparison posttest.
Immunofluorescence microscopy.
Cells were fixed with 2.5% paraformaldehyde, and immunofluorescence microscopy was carried out as described previously (32). Coverslips were mounted onto glass slides using DakoCytomation fluorescent mounting medium. Images were acquired using an inverted Leica DMIRE2 fluorescent microscope equipped with a Hamamatsu ORCA-ER camera and OpenLab, version 3.1.7, software. Images were imported into Adobe Photoshop and assembled in Adobe Illustrator.
Antibodies and reagents.
Rabbit polyclonal antibodies to serovar Typhimurium O antiserum group B were obtained from Difco Laboratories (Kansas City, MO) and used at a dilution of 1:200 for immunofluorescence studies. Mouse anti-human lysosome-associated membrane protein 1 (LAMP1) antibody (clone H4A3), used at a 1:50 dilution, was developed by J. Thomas August and obtained from the Developmental Studies Hybridoma Bank under the auspices of the National Institute of Child Health and Human Development, National Institutes of Health, and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA. Rabbit anti-kinesin heavy chain (PCP42) was used at 1:1,000 for immunofluorescence studies and was obtained from R. Vale, University of California. Secondary antibodies used were Alexa 488-conjugated goat anti-mouse immunoglobulin G and Alexa 568-conjugated goat anti-rabbit immunoglobulin G (Molecular Probes). To stain host cell nuclei in fixed cells, coverslips were immersed in 2 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI) for 10 min prior to being mounted on glass slides. For Western blotting, mouse monoclonal anti-HA (Covance) was used at 1:2,000, and rabbit anti-SipA (generously provided by D. Zhou, Purdue University) was used at 1:1,000.
Documenting intracellular positions of SCVs.
The intracellular position of SCVs was determined by measuring the distance of LAMP1-positive (LAMP1+) SCVs to the nearest edge of the host cell nucleus (labeled by DAPI staining) in fixed HeLa cells. Images of infected cells stained for serovar Typhimurium, LAMP1, and nuclear DNA were acquired using epifluorescence microscopy, and measurements were determined using Openlab 3.1.7 software.
An average SCV-to-nucleus distance was calculated for each time point in each experiment. The distances of at least 100 LAMP1+ SCVs from the nuclei of at least 20 infected HeLa cells were measured for each time point in a given experiment. As experiments were done in triplicate, the resulting finalized averages were calculated from three independent average values. The average distances ± standard deviations for three separate experiments are reported in each case. Statistical analyses of data representing average distances of SCVs were conducted in the following manner. All data were tested for normality using the D'Agostino and Pearson normality test. Where required, data sets were subjected to log transformation, and normality testing was repeated. Any non-Gaussian data sets were analyzed using Kruskal-Wallis one-way analysis of variance (ANOVA) with Dunn's multiple comparison posttest. Depending on the nature of the experiment, statistical analyses for data that did not represent measurements of average distances to host cell nuclei were performed using either a two-tailed unpaired t test, Mann-Whitney test, or Tukey one-way ANOVA (Prism 4 software). In all cases P values of <0.05 were considered significant (see figure legends for details).
Promoter activity studies.
Transcriptional activity was monitored using a chemiluminescent β-galactosidase detection method (11). HeLa cells were seeded into six-well tissue culture plates (2 × 105 cells/well; two wells per time point) and infected with serovar Typhimurium carrying either PsifA::lacZ or PpipB2::lacZ, as described above. Cells were also infected with WT serovar Typhimurium (without any lacZ fusion) as a control. At selected times postinfection, cells were washed twice with PBS and harvested using a total of 1 ml of ice-cold lysis buffer per well (1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS], 1 mM phenylmethylsulfonyl fluoride, 5 mM NaF, 5 mM NaV, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μM pepstatin). One hundred microliters of cell lysate from each time point was used to enumerate intracellular bacteria by dilution plating. The remaining 900 μl of cell lysate was centrifuged at 10,000 × g for 3 min, and the pellet was resuspended in 50 μl of PBS and mixed with 13 μl of chloroform. Ten microliters of cell lysates was transferred to white microtiter plate wells and mixed with 100 μl of Galacto-Star substrate mix (Applied Biosystems). This volume of cell lysate was determined to produce β-galactosidase activity values that lie within the linear range of the assay (volumes of 5, 10, 15, and 20 μl were tested in each trial). After a 70-min incubation at room temperature, the relative light units of β-galactosidase activity were measured using a Microlumat Plus luminometer. Light signal data were normalized to the number of CFU, based on dilution plating done earlier. From this, the relative increase in β-galactosidase activity compared to the control infection was determined.
Effector protein level studies.
To study the levels of specific effector proteins expressed by intracellular serovar Typhimurium during infection, HeLa cells were infected with ΔpipB2 (pPipB2-2HA) and ΔsifA (psifA-2HA) strains of bacteria. HeLa cells were seeded into six-well tissue culture plates (2 × 105 cells/well; two wells per time point) and infected with bacteria as described above. At selected times postinfection, cells were washed twice with PBS and harvested using a total of 400 μl of ice-cold lysis buffer for every two wells (1% Triton X-100, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride, 5 mM NaF, 5 mM NaV, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μM pepstatin). One hundred microliters of cell lysate from each time point was used to enumerate intracellular bacteria by dilution plating. The remaining cell lysate samples were loaded onto 12% polyacrylamide gels according to equal numbers of bacteria, resolved by SDS-polyacrylamide gel electrophoresis, and transferred to Immobilin-PSQ membranes for immunoblotting.
Cell-to-cell infection assays of serovar Typhimurium.
To investigate cell-to-cell infection of serovar Typhimurium, fluorescently labeled, uninfected HeLa cells were seeded over a layer of unlabeled, infected cells in 24-well tissue culture plates. This permitted examination of whether intracellular bacteria could migrate into the newly introduced uninfected cells. HeLa cells (5 × 104 cells/well) were infected with isogenic WT, ΔssaT (SPI-2 secretion deficient), and ΔpipB2 serovar Typhimurium strains as outlined previously (7). To fluorescently label HeLa cells with CellTracker Blue CMAC (Molecular Probes), a flask of HeLa cells (80 to 90% confluence) was washed three times in PBS and incubated in serum-free DMEM containing 25 μM CellTracker Blue for 45 min. Cells were thoroughly washed three times with PBS and allowed to recover in DMEM supplemented with 10% fetal bovine serum for 30 min. At 2 hpi, these uninfected HeLa cells labeled with CellTracker Blue were seeded over the previously infected cells at two times the original density (10 × 104 cells/well). At various time points (upwards to 24 h), cells were fixed and immunostained for Salmonella and LAMP1 and visualized by fluorescent microscopy. LAMP1+ SCVs found within CellTracker Blue-labeled cells were regarded as evidence for cell-to-cell infection. All experiments were conducted in the presence of gentamicin (10 μg/ml) to inhibit invasion of HeLa cells by any extracellular bacteria. In addition, fresh medium containing gentamicin (10 μg/ml) was added to the wells 14 hpi to ensure that extracellular bacterial inhibition was maintained throughout the experiment.
RESULTS
SCVs display dynamic intracellular positioning over the course of infection.
It has been established that, following invasion, serovar Typhimurium localizes to a juxtanuclear position within the host cell (5, 45). Maximal rates of bacterial replication occur at this location, where it is proposed that the acquisition of nutrients and membrane materials for SCV maintenance is optimal (15, 45). In general, studies examining this characteristic intracellular SCV positioning have focused on factors that are required for the maintenance of this phenotype (5, 13, 26, 42, 45). We sought to determine whether the typical juxtanuclear positioning of SCVs persists or whether their intracellular localization changes over a 24-h course of infection.
HeLa cells were infected with WT serovar Typhimurium CS401 and fixed at 4, 8, 14, and 24 hpi. Cells were immunostained for serovar Typhimurium and LAMP1 (a marker of SCVs) and DAPI stained to visualize the host cell nucleus. At 4 hpi, SCVs were situated near the host cell nucleus (Fig. 1A). By 8 hpi, the majority of SCVs were clustered near the nucleus of infected cells (Fig. 1B) in accordance with other studies (5, 45). These cells also presented LAMP1+ Salmonella-induced filaments (SIFs) that are typical at this time point (4, 20). Interestingly, at 14 hpi, we noted that many infected cells presented a more dispersed SCV positioning phenotype (Fig. 1C). Specifically, SCVs were localized more toward the host cell periphery and not necessarily in tight juxtanuclear clusters. SCV positioning was also similar at 24 hpi (Fig. 1D), with SCVs often appearing much farther from the nucleus than those at 8 hpi. As these experiments were carried out in the presence of gentamicin (100 μg/ml in the first 2 hpi, followed by 10 μg/ml for the remainder of each experiment), the appearance of SCVs near the peripheries of host cells at later stages postinfection was not due to reinfection of HeLa cells with extracellular bacteria.
To quantify SCV positioning, the distances of LAMP1+ SCVs to the nearest edge of the host cell nucleus were measured for each time point (see Materials and Methods). We, along with several other groups, have used the host cell nucleus as a reference point for documenting intracellular positioning of SCVs (1, 5, 6, 24, 26, 57). We categorized the intracellular distribution of LAMP1+ SCVs by grouping measurements according to their distances from the nucleus (Fig. 1E). A higher percentage of SCVs were situated >6 μm from the nucleus at both 14 hpi (32.3%) and 24 hpi (32.5%) than at 8 hpi (4.4%) (Fig. 1E). In addition, there was a significant increase in the proportion of SCVs located >10 μm away from the nucleus at 14 hpi and 24 hpi compared to 8 hpi (Fig. 1E). Figure 1E also shows that the average distance of WT SCVs to the nucleus was 2.3 ± 0.1 μm at 8 hpi. At later stages postinfection, average distances were significantly greater and were determined to be 5.6 ± 0.1 μm at 14 hpi and 5.4 ± 0.2 μm at 24 hpi. Hence, there is a change in the distribution of SCVs during infection, with an increased proportion localizing farther away from the nucleus after 8 hpi.
Addition of tetracycline at 8 hpi to inhibit bacterial protein synthesis abrogated the centrifugal displacement of WT SCVs at 14 and 24 hpi (Fig. 1E). In addition, disruption of the microtubule network using nocodazole affected centrifugal SCV displacement (Fig. 1E). The addition of nocodazole did not adversely affect the morphology or the surface area covered by the infected HeLa cells (data not shown). We also tested the effects of the actin-disrupting drug cytochalasin D; however, the toxic effects of the drug over the lengthy course of study precluded measurements of SCV positioning. The displacement of SCVs away from the nucleus at later stages of infection was also observed in WT strain SL1344, represented by histograms showing average distances from the nucleus (Fig. 2B) and the percentage of SCVs >6 μm from the nucleus (Fig. 2C). Hence, the centrifugal displacement phenotype is not unique to strain CS401. However, there was a difference in the dynamics of positioning between the two WT strains as the average distances to the nucleus for WT CS401 SCVs were greater at 14 hpi than for WT SL1344 (Fig. 1E and 2B). Importantly, SCVs from both strains were juxtaposed to the nucleus at 8 hpi, and approximately one-third of SCVs were displaced significantly farther away at 24 hpi. Thus, two different strains of serovar Typhimurium exhibited similar dynamic intracellular positioning within host cells. Overall, these results indicate that WT SCVs undergo dynamic intracellular positioning during infection, with localization toward the host cell nucleus by the majority of SCVs followed by a novel phenotype of displacement toward the host cell periphery by a subset of SCVs.
FIG. 2.

SPI-2 is needed for dynamic intracellular positioning over the course of a 24-h infection. (A) HeLa cells were infected with WT or isogenic SPI-2 deficient (ΔssaR) serovar Typhimurium strains for 24 h and then fixed. Cells were immunostained for bacteria (red) and LAMP1 (green), and the nucleus was stained using DAPI (white). Dotted lines indicate cell borders. Arrows indicate LAMP1+ SCVs. Bar, 10 μm. (B) Comparison of intracellular positioning of WT, isogenic SPI-2 deficient (ΔssaR), ΔsifA, and ΔsifA (psifA-2HA) SCVs in HeLa cells during infection. The average distance of SCVs ± standard deviations for three separate experiments is shown. At least 150 LAMP1+ SCVs were measured for each time point in a given experiment. Triple asterisks indicate a significant difference between the indicated mutant strain measurement and the corresponding WT control measurement (P < 0.001) as determined by Kruskal-Wallis one-way ANOVA with Dunn's multiple comparison posttest. (C) Percentage of SCVs examined in panel B that were located >6 μm from their host cell nucleus at the indicated times postinfection. Averages ± standard deviations for three separate experiments are shown. The asterisk indicates a significant difference between the indicated mutant strain measurement and the corresponding WT control measurement (P < 0.05), as determined by one-way ANOVA and Tukey posthoc analysis.
Centrifugal displacement of SCVs is SPI-2 dependent.
Several studies have shown that effector proteins secreted by the SPI-2 T3SS mediate the close association of SCVs with the host cell nucleus and Golgi apparatus (5, 13, 42, 45). We therefore examined whether the SPI-2 T3SS was required for the displacement of SCVs away from the host cell nucleus at later stages postinfection (up to 24 hpi). In contrast to WT SL1344 SCVs, an isogenic ΔssaR (SPI-2 T3SS deficient) strain remained close to the nucleus throughout the course of infection, including at 24 hpi (Fig. 2A to C). The localization of the ΔssaR strain was generally perinuclear, in accordance with previous studies (57), and not concentrated on any particular side of the nucleus. Similar results were observed with a ΔssaT (SPI-2 T3SS deficient) strain in the CS401 background (data not shown). Hence, the late-onset outward displacement that is evident by 24 hpi in WT serovar Typhimurium SCVs is dependent on a functional SPI-2 T3SS.
SifA is an SPI-2 T3SS effector involved in inhibiting kinesin recruitment to the SCV, thus helping to maintain the SCV near the nucleus. As such, ΔsifA mutant SCVs tend to localize at the host cell periphery by 14 hpi (5). We determined the intracellular positioning of ΔsifA mutant SCVs over the course of 24 h. Interestingly, ΔsifA SCVs were close to the nucleus at 4 hpi and indistinguishable from isogenic WT SL1344 SCVs, suggesting that there exists an initial inward movement of SCVs following invasion that is independent of SifA (Fig. 2B and C). However, by 14 hpi, ΔsifA SCVs were significantly farther away from the nucleus than WT SCVs, consistent with previous observations (5). The ability of ΔsifA SCVs to localize near the nucleus at 14 hpi was complemented by a plasmid-encoded HA-tagged SifA (SifA-2HA) (Fig. 2B and C). By 24 hpi, both WT and ΔsifA SCVs had similar displacements from the host cell nucleus, indicating that sifA is not required for centrifugal movement of SCVs at this later stage of infection.
Kinesin is involved in the centrifugal displacement of SCVs at later stages of infection.
Since the centrifugal displacement of ΔsifA SCVs is attributed to the recruitment of kinesin-1 to the bacterial compartment (5), we next explored the involvement of this motor protein in the peripheral positioning of WT SCVs. We transfected serovar Typhimurium-infected HeLa cells with GFP-KLC-TPR to exert a dominant-negative effect on host cell kinesin (44). In HeLa cells infected with either the WT or ΔsifA strain, expression of GFP-KLC-TPR resulted in bacteria that were significantly closer to the nucleus than infected control cells expressing GFP alone at 24 hpi (Fig. 3A to C). Hence, host cell kinesin contributes to the peripheral displacement of SCVs from the nucleus at late time points postinfection.
FIG. 3.

Kinesin is required for the centrifugal displacement of intracellular serovar Typhimurium at late time points postinfection. (A) HeLa cells were infected with WT or ΔsifA serovar Typhimurium, and subsequently transfected (2 h postinfection) with a plasmid encoding GFP alone or GFP-KLC-TPR that has dominant-negative activity against kinesin. Cells were fixed at 24 hpi and immunostained for serovar Typhimurium. The distances of bacteria to the nearest edge of the host cell nucleus were measured in transfected cells. The averages ± standard deviations for three separate experiments are shown. A triple asterisk indicates a significant difference from the respective GFP transfection control (P < 0.0.001), as determined by Kruskal-Wallis one-way ANOVA with Dunn's multiple comparison posttest. (B) Percentage of SCVs examined in panel A that were located >6 μm from their host cell nucleus. Averages ± standard deviations for three separate experiments are shown. The asterisk indicates a significant difference from the respective GFP transfection control (P < 0.05), as determined by a two-tailed, unpaired t test. (C) Positioning of WT and ΔsifA serovar Typhimurium bacteria (red) 24 hpi in HeLa cells transfected with GFP or GFP-KLC-TPR. Arrowheads indicate bacteria positioned toward the host cell periphery in GFP-transfected cells. Arrows indicate bacteria positioned next to the host cell nucleus in GFP-KLC-TPR-transfected cells. Size bar, 10 μm. (D) Typical kinesin labeling of WT (top panel) and ΔsifA (bottom panels) LAMP1+ SCVs in infected HeLa cells at 14 hpi. Arrows indicate distinct recruitment of kinesin to LAMP1+ SCVs of the ΔsifA strain. Arrowheads indicate colocalization of LAMP1 with WT bacteria, which do not display significant kinesin recruitment regardless of their position in the host cell. Cells were also DAPI stained. Size bar, 10 μm. (E) HeLa cells were infected with WT, ΔsifA, ΔssaR, or ΔsifA (psifA-2HA) strains of serovar Typhimurium for the times indicated. Cells were fixed and immunostained for LAMP1, bacteria, and kinesin. The percentage of LAMP1+ SCVs colocalizing with kinesin was determined for each strain. Values shown are the averages ± standard deviations of three separate experiments. The presence of kinesin on 100 LAMP1+ SCVs was determined for each strain at each time point in a given experiment. The asterisk indicates a significant difference between the indicated mutant strain and the corresponding WT control (P < 0.05), as determined by a two-tailed Mann-Whitney test.
To determine whether displacement of SCVs toward the host cell periphery is coupled with the accumulation of kinesin on SCVs, we immunostained for kinesin heavy chain in infected HeLa cells (Fig. 3D). The ΔsifA mutant served as a positive control for kinesin labeling as it was previously demonstrated that ΔsifA SCVs accumulate kinesin over the course of 14 h (5). At 14 hpi, we observed that LAMP1+ ΔsifA SCVs displayed noticeable recruitment of kinesin whether they were situated far from or close to the nucleus (Fig. 3D, lower panels). In contrast, WT SCVs did not tend to accumulate significant amounts of kinesin at either 14 or 24 hpi (Fig. 3D and E). This included the subset of SCVs that were situated significantly farther away from the nucleus at 24 hpi (Fig. 2). The complemented ΔsifA strain harboring plasmid psifA-2HA did not recruit noticeable amounts of kinesin (Fig. 3E). Interestingly, kinesin recruitment on ΔsifA SCVs decreased to a level similar to WT vacuoles at 24 hpi (Fig. 3E). No significant recruitment of kinesin was observed on the ΔssaR SPI-2 secretion mutant (Fig. 3E), in accordance with previous observations using another SPI-2 mutant (5). Together, these results show that increased accumulation of kinesin on the SCV is not an absolute requirement for maintaining the peripheral positioning of WT and ΔsifA SCVs at 24 hpi. Hence, a basal level of kinesin on the SCV or levels that are below the limit of detection by fluorescence microscopy are sufficient to drive centrifugal displacement of bacteria.
The SPI-2 effector PipB2 is required for centrifugal displacement of SCVs at later stages of infection.
The SPI-2 effector PipB2 has recently been shown to bind to kinesin-1 and to act as a linker of this motor protein to the SCV (26). To determine whether this effector is involved in mediating centrifugal SCV migration, the intracellular SCV positioning of WT and isogenic ΔpipB2 strains of serovar Typhimurium were determined during a 24-h infection. As expected, WT SCVs were localized near the nucleus at 8 hpi and displayed a more scattered distribution toward the host cell periphery at later time points postinfection (Fig. 4A to C). In contrast, ΔpipB2 SCVs were situated at relatively short distances from the nucleus at all time points postinfection. The peripheral displacement of ΔpipB2 SCVs at later time points postinfection was restored in bacteria bearing a plasmid encoding HA-tagged PipB2 (pPipB2-2HA) under the control of its native promoter (Fig. 4B and C). These results indicate that the kinesin-binding effector protein PipB2 is required for serovar Typhimurium migration toward the host cell periphery at later time points postinfection.
FIG. 4.

The centrifugal displacement of SCVs is dependent on the effector PipB2. (A) Comparison of intracellular positioning of WT and isogenic ΔpipB2 SCVs in HeLa cells after 8 and 24 hpi. Cells were immunostained for bacteria (red) and LAMP1 (green), and the nucleus was stained using DAPI (blue). Dotted lines indicate boundaries of indicated cells. Arrows indicate SCVs that are adjacent to the nucleus. Arrowheads indicate SCVs that are located toward the host cell periphery. Bar, 10 μm. (B) HeLa cells were infected with WT, ΔpipB2, or ΔpipB2 (pPipB2-2HA) strains of serovar Typhimurium. Cells were fixed at the indicated time points, immunostained, and analyzed as described in the legend of Fig. 1. The triple asterisk indicates a significant difference (P < 0.001) between the indicated mutant strain measurement and the corresponding WT control measurement, as determined by Kruskal-Wallis one-way ANOVA with Dunn's multiple comparison posttest. (C) Percentage of SCVs examined in panel B that were located >6 μm from their host cell nucleus at the indicated times postinfection. Averages ± standard deviations for three separate experiments are shown. The asterisk indicates a significant difference between the indicated mutant strain measurement and the corresponding WT control measurement (P < 0.05), as determined by one-way ANOVA and Tukey post hoc analysis.
SifA participates in retaining SCVs at a juxtanuclear position by acting as a kinesin uncoupler, resulting in ΔsifA mutant SCVs that show a scattered distribution away from the host cell nucleus by 10 to 14 hpi (5). Since SifA is necessary for maintaining juxtanuclear SCV localization up to 14 hpi and since PipB2 is necessary for outward SCV movement after 14 hpi, we tested whether the dynamic localization of SCVs during the course of infection may be due to changes in expression of these antagonistic effector proteins over time. HeLa cells were infected with serovar Typhimurium strains containing a single-copy chromosomal lacZ reporter gene under the control of sifA or pipB2 promoters, designated PsifA::lacZ and PpipB2::lacZ, respectively. A similar strategy has been used previously to examine the expression of other serovar Typhimurium genes (11, 17). The relative increase in β-galactosidase activity compared to control infections using the parental WT strain was determined by normalizing numbers of intracellular bacteria. As seen in Fig. 5A, both sifA and pipB2 promoters are induced by 4 hpi, have maximal activity at 14 hpi, and show a decline in activity by 24 hpi. Hence, sifA and pipB2 follow a similar pattern of gene expression over the course of a 24-h serovar Typhimurium infection.
FIG. 5.
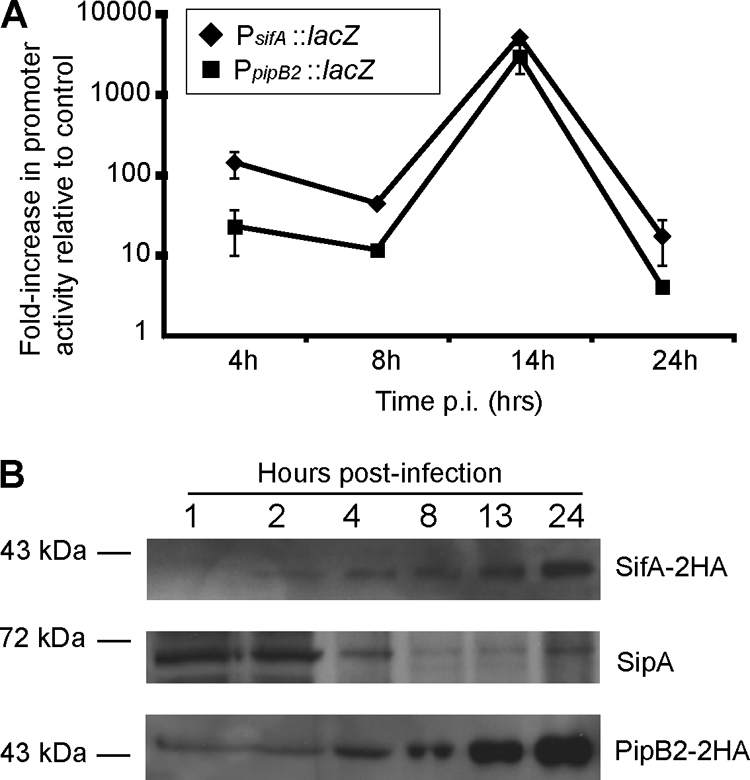
Analysis of promoter activity and protein expression levels of SifA, PipB2, and SipA. (A) HeLa cells were infected with serovar Typhimurium carrying chromosomal transcriptional lacZ fusions to either the sifA or pipB2 promoters (PsifA::lacZ or PpipB2::lacZ) or with the WT parental strain. At selected times, cell lysates were collected, and β-galactosidase activity was determined using a chemiluminescence assay. Light signal data were normalized to the numbers of CFU, based on dilution plating of lysates. Shown are the relative increases in β-galactosidase activity resulting from PsifA::lacZ or PpipB2::lacZ activity compared to the WT control infection. Values are the averages from two independent experiments and average deviations. (B) Immunoblot analysis of effector protein levels in infected HeLa cells. Cells were infected with ΔsifA (psifA-2HA) (top and middle blots) or ΔpipB2 (pPipB2-2HA) (bottom blot) strains of serovar Typhimurium. Whole-cell lysates were harvested at the indicated times and analyzed by immunoblotting samples that were loaded according to equal bacterial numbers. Top and middle blots show Western blotting to detect SifA-2HA and SipA, as indicated, from the same blot. At the bottom is a Western blot to detect PipB2-2HA.
To determine whether gross differences in intracellular bacterial protein levels could account for dynamic SCV positioning, HeLa cells infected with ΔsifA bacteria carrying plasmid psifA-2HA or with ΔpipB2 bacteria carrying pPipB2-2HA were analyzed by immunoblotting with anti-HA antibody to determine temporal changes in levels of SifA-2HA or PipB2-2HA (Fig. 5B). In each case, samples were normalized to the number of intracellular bacteria. Expression of both HA-tagged effectors was under the control of their own native promoters, and the dynamic intracellular positioning of these strains was similar to WT SCVs due to complementation by their respective plasmid-encoded genes (Fig. 2B and 4B). As seen in Fig. 5B, levels of the SPI-2 T3SS effectors SifA-2HA and PipB2-2HA continuously increased over the course of the 24-h infection. Hence, the centrifugal displacement of SCVs was not attributed to gross differences in promoter activity between pipB2 and sifA nor to loss of kinesin-inhibiting SifA.
The SPI-1 T3SS effector SipA is a cofactor for SifA function in regulating SCV position. In the absence of sipA, SCVs are situated away from the host cell nucleus at 8 hpi (6). We investigated the temporal changes in SipA levels during serovar Typhimurium infection of HeLA cells and found that SipA levels decreased over time (Fig. 5B). We also examined the levels of the SPI-1 T3SS effector SopB, which promotes SCV localization near the nucleus at 8 hpi by activating myosin II (57), and found it was maintained up to 12 hpi in accordance with a previous report (14) but was undetectable after 12 hpi (data not shown). Hence, the decline of SPI-1 effector proteins SipA and SopB, coupled with the increase of SPI-2 effector proteins PipB2 and SifA, is associated with the centrifugal displacement of SCVs at 24 hpi.
Centrifugal displacement of SCVs from the host cell nucleus is required for infection of newly introduced HeLa cells.
We investigated whether centrifugal displacement of SCVs toward the host cell periphery might facilitate the infection of neighboring cells. Using an assay similar to one used to study cell-to-cell transfer by the intracellular pathogen Listeria monocytogenes (3), we determined the ability of intracellular serovar Typhimurium to infect a newly introduced population of fluorescently labeled host cells. Isogenic WT, ΔpipB2, and ΔssaT (SPI-2 secretion deficient) strains of serovar Typhimurium were used to infect HeLa cells. At 2 hpi, an excess of uninfected HeLa cells fluorescently labeled with CellTracker Blue CMAC was seeded onto the previously infected cells in a 2:1 ratio. Cells were subsequently fixed at various time points postinfection and examined for bacteria residing within CellTracker Blue-labeled cells as evidence of cell-to-cell infection. We used LAMP1 staining of the SCV as an indicator of intracellular bacteria within CellTracker Blue-labeled cells.
After 14 hpi, no strain of bacteria exhibited evidence of cell-to-cell transfer to newly introduced CellTracker Blue HeLa cells (Fig. 6A and E). However, by 24 hpi, approximately 12% of CellTracker Blue-labeled HeLa cells contained WT LAMP1+ SCVs (Fig. 6B and E). We observed that these infected CellTracker Blue-labeled HeLa cells were often in close apposition to infected unlabeled cells (Fig. 6A, second row), suggesting that the bacteria had exhibited cell-to-cell transfer. In most infected CellTracker Blue cells, the number of LAMP1+ SCVs within ranged from one to two SCVs. However, in some instances two or more SCVs were observed in CellTracker Blue cells (Fig. 6B).
FIG. 6.
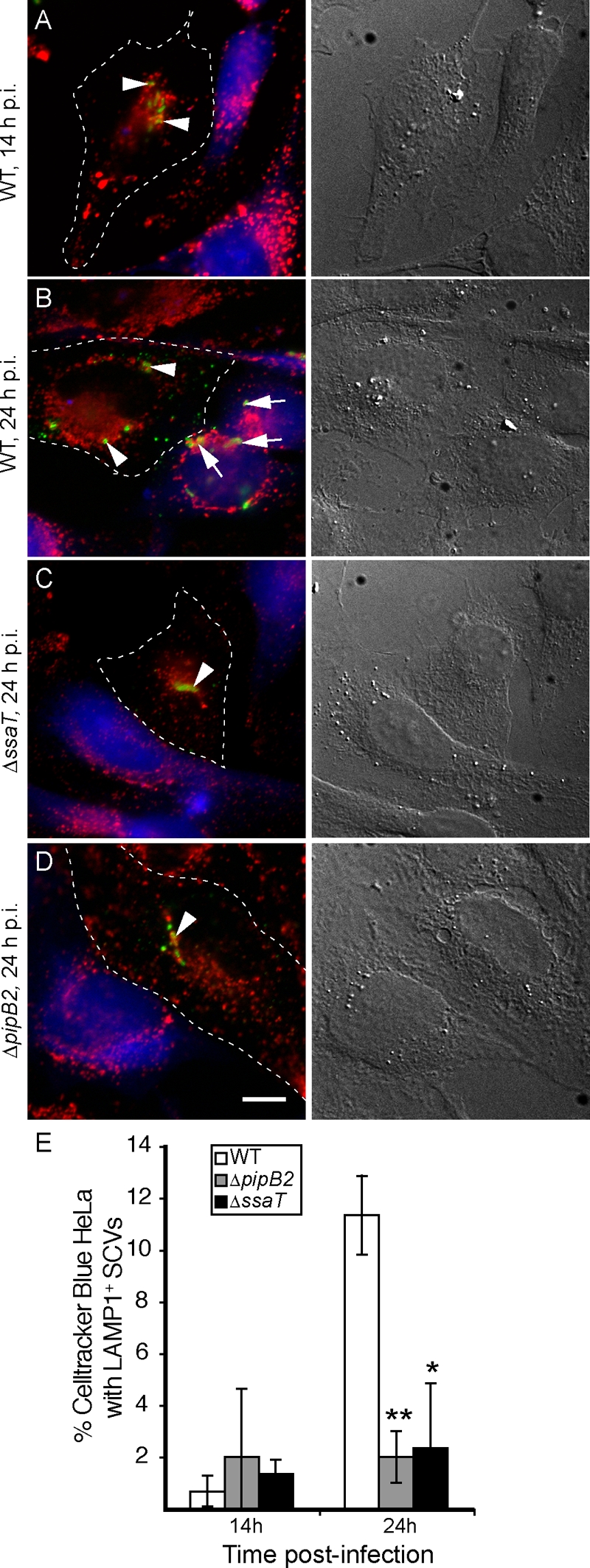
Intracellular wild-type serovar Typhimurium can transfer into newly introduced HeLa cells, while SPI-2-deficient and ΔpipB2 mutants cannot. (A to D) HeLa cells were infected with WT, isogenic SPI-2 deficient (ΔssaT), or ΔpipB2 strains of serovar Typhimurium. Two hours after bacterial internalization, new uninfected HeLa cells labeled with CellTracker Blue CMAC (blue; left panels) were seeded over the previously infected cells at twice the original density. Cells were fixed at various time points and immunostained for Salmonella (green) and LAMP1 (red) and visualized by fluorescence microscopy (left panels). LAMP+ SCVs appearing in CellTracker Blue-labeled HeLa cells were used as indicators of cell-to-cell transfer. Corresponding differential interference contrast images are in the right panels. Dotted lines indicate boundaries of originally infected HeLa cells. Blue cells represent newly introduced HeLa cells. All experiments were conducted in the presence of gentamicin (100 μg/ml for the first 2 hpi, followed by 10 μg/ml for the remainder of each experiment). (E) Percent CellTracker Blue-labeled HeLa cells from the above experiment that contained LAMP+ SCVs with WT, SPI-2 deficient (ΔssaT), or ΔpipB2 strains of serovar Typhimurium. The averages ± standard deviations for three separate experiments are shown. One hundred CellTracker Blue-labeled HeLa cells were counted for each time point in each experiment. Asterisks indicate significant differences from the WT strain at 24 hpi as determined by a two-tailed Mann-Whitney test: **, P < 0.005; *, P < 0.05.
In contrast to WT bacteria, there was no significant increase at 24 hpi in the appearance of either ΔpipB2 or ΔssaT SCVs in CellTracker Blue-labeled HeLa cells (Fig. 6C to E). Intracellular replication assays were also conducted to compare the growth rates of WT, ΔpipB2, and ΔssaT strains in HeLa cells. Both WT and ΔpipB2 strains had similar growth rates (data not shown), in agreement with previous observations in both epithelial cells and macrophages (35). Therefore, the lack of cell-to-cell infection of the ΔpipB2 mutant was not due to decreased intracellular growth. Overall, these results show an association between the ability of serovar Typhimurium to undergo SPI-2- and PipB2-dependent centrifugal displacement, with its ability to achieve cell-to-cell transfer.
DISCUSSION
Studies to date have examined the characteristic intracellular localization of SCVs at a juxtanuclear, Golgi apparatus-associated region (5, 13, 26, 42, 45). In the present study, we report the novel observation that a subset of LAMP+ SCVs displace away from the host cell nucleus and localize toward the host cell periphery at later stages of infection. This displacement was not strain specific and was shown to occur 14 to 24 hpi.
From our studies and those of others, we propose a model for dynamic intracellular SCV positioning during infection of host cells (Fig. 7). During the first few hours postinfection, SCV movement toward the host cell nucleus occurs (Fig. 7A). This characteristic inward migration does not require a functional SPI-2 T3SS, as we (this study) and others have observed (2, 42). The GTPase Rab7 recruits Rab7-interacting lysosomal protein that brings the dynein/dynactin complex to the SCV (21, 24, 37), contributing to SCV movement toward the host cell nucleus. Despite their association with invasion, specific SPI-1 effectors persist in the host cell after secretion to further modulate host cell activities well after invasion. The SPI-1 effector SopB persists for up to 12 h after Salmonella entry (14), and we have demonstrated its involvement in mediating the juxtanuclear positioning of SCVs through activation of the nonmuscle myosin II actin motor via Rho GTPases (Fig. 7A) (57). The SPI-1 effector SipA has also been shown to persist on the SCV for up to 8 hpi. This persistence allows SipA to cooperate with the SPI-2 T3SS effector SifA, which is secreted later during infection, to mediate perinuclear SCV positioning (6) (Fig. 7B). Hence, SPI-1 effectors have important roles in positioning the SCV.
FIG. 7.

Model of dynamic SCV positioning during the course of host cell infection. (A) Following invasion, SCVs move centripetally toward the host cell nucleus in an SPI-2-independent manner. Rab7 GTPase recruits Rab7-interacting lysosomal protein (RILP) that brings the dynein/dynactin complex to the SCV (21, 24, 37), mediating SCV movement toward the host cell nucleus. The SPI-1 effector SopB is involved in mediating the juxtanuclear positioning of SCVs through activation of the nonmuscle myosin II actin motor via Rho GTPases (57) and persists for up to 12 h after invasion (14). The SPI-1 effector SipA also persists on the SCV for up to 8 hpi and cooperates with the SPI-2 T3SS effector SifA to mediate perinuclear SCV positioning (6). (B) By 8 to 14 hpi, SCVs have localized to their characteristic juxtanuclear position. SPI-2 effectors including the Golgi apparatus-tethering and/or dynein-recruiting effectors SseG (2, 42) and SseF (2) and the kinesin-inhibiting effector SifA (5) maintain the juxtanuclear positioning of SCVs (27, 41). Bacterial replication and SIF formation occur. Although the kinesin-linking SPI-2 effector PipB2 is also localized to SCVs, its effect on SCV positioning is masked by the combined actions of SipA, SopB, SifA, SseG, and SseF, which retain SCVs at juxtanuclear positions. PipB2 is involved in the extension of SIFs at this stage of infection (26, 34, 41). (C) At later times postinfection (≥14 h), SCVs move centrifugally toward the host cell periphery in a manner dependent upon the host microtubule network, active bacterial protein synthesis, an intact SPI-2 T3SS, the kinesin-recruiting effector PipB2, and host kinesin. Movement toward the host cell periphery may facilitate escape from the host cell and/or cell-to-cell transfer. (D) During the course of a 24-h infection, levels of SPI-2 effector proteins such as SifA and PipB2 increase (dotted line) while levels of SPI-1 effector proteins such as SipA and SopB decrease (solid line). These changes in effector protein levels are associated with centrifugal displacement of SCVs.
As infection progresses, expression of the SPI-2 T3SS and associated effector genes is upregulated, resulting in increased SPI-2 effector protein levels (Fig. 7D) (18, 25). By 8 to 14 hpi, SCVs are localized near the nucleus (Fig. 7B). SPI-2 effectors including the Golgi apparatus-tethering and/or dynein-recruiting effectors SseG (2, 42) and SseF (2) and the kinesin-inhibiting effector SifA (5) maintain the juxtanuclear positioning of SCVs (27, 41) and facilitate bacterial replication and SIF formation (4, 20). Persisting SPI-1 effectors, including the SifA cofactor SipA, continue to exert their effects on SCV positioning (6). While also localized to SCVs, the effects of the kinesin-linking SPI-2 effector PipB2 on SCV positioning appear to be masked at this stage by a combination of effects from SipA, SopB, SifA, SseG, and SseF. However, PipB2 is involved in the extension of SIFs at this stage of infection (26, 34, 41).
At later times postinfection (≥14 h), a subset of SCVs undergoes centrifugal displacement toward the host cell periphery (Fig. 7C). This is dependent upon host microtubules, active bacterial protein synthesis, a functional SPI-2 T3SS, PipB2, and host kinesin. A significant accumulation of kinesin on WT SCVs was not observed in our studies, including at 24 hpi when SCVs were centrifugally displaced from the nucleus. It is possible that PipB2 helps recruit and/or control basal levels of kinesin-1 that are below the level of detection used in this study but which are sufficient to mediate the outward displacement of SCVs at later times postinfection. Indeed, kinesin is not detected on SIFs though they are formed in a kinesin-dependent manner. By this late stage, levels of SPI-1 effectors such as SipA and SopB have decreased significantly while those of SPI-2 effectors continue to increase (Fig. 7D). Since SipA works in conjunction with SifA to ensure perinuclear positioning of SCVs (6), it is conceivable that there is insufficient SipA to cooperate with SifA to help maintain perinuclear SCV positioning at later stages of infection. Hence, the noticeable decrease in global SipA levels in a WT background at later stages of infection likely contributes to peripheral displacement of SCVs, similar to that observed with a ΔsipA mutant (6). This suggests that simple cell lysis is likely not the sole means of escape from a host cell used by serovar Typhimurium. The modulation of other factors implicated in SCV positioning such as SseF and SseG is still not clear.
Approximately one-third of SCVs were no longer within 6 μm of the nucleus at 24 hpi, as measured under our study conditions. Why only a fraction of SCVs exhibit centrifugal displacement away from a centralized perinuclear region and can localize as far as the host cell periphery is not clear. It is possible that only a subset of intracellular bacteria acquires the necessary nutrients or expresses the appropriate combination of virulence factors to mediate centrifugal displacement. It is also possible that higher proportions of SCVs displace to the host cell periphery but are not observed due to exit from the host cell.
Using a cell-to-cell transfer assay, we show that bacteria capable of movement to the host cell periphery at later stages of infection in our SCV positioning experiments are associated with an increased ability to infect new host cells. The exit of intracellular pathogens from host cells is important for the spread of infection and transmission to new hosts and can occur through multiple pathways (30). Intracellular bacteria such as L. monocytogenes and Shigella flexneri use actin-mediated propulsion to drive infection into neighboring host cells (52). Chlamydia species employ protease-dependent host cell lysis, active extrusion of the chlamydial inclusion (31), and the transfer of reticulate bodies to secondary inclusions as methods of dissemination (53). The induction of host cell apoptosis may also be exploited by various pathogens, including serovar Typhimurium, to facilitate dissemination (22, 30).
Serovar Typhimurium has been shown to escape from oncotic macrophages using a nonlytic, flagella-dependent mechanism (47). While it is possible that serovar Typhimurium exits epithelial cells in a similar manner, we believe the oncotic route is distinct from the dynamic SCV route observed in our studies. Oncotic macrophages are filled with extreme numbers of bacteria and display extensive morphological changes (47). In our in vitro studies, infected epithelial cells were still intact after 24 hpi, and individual LAMP1+ SCVs were visualized near the host cell periphery without crowding. Containment within an SCV would also likely prevent bacteria from using flagella to mediate outward movement. It is possible that serovar Typhimurium uses different mechanisms of escape, depending on the infected host cell type. Using live-cell imaging, we have thus far been unable to directly visualize the exit of serovar Typhimurium from epithelial cells at late stages of infection; hence, the possible mechanism of exit is not known. It is clear from time-lapse imaging that many late-stage SCVs at the periphery of the host epithelial cell do not tend to migrate back toward the host cell center even after several hours (data not shown). Hence, there is a tendency for centrifugally displaced cells to remain at the periphery once they are there.
From in vivo studies in mice, it has been shown that systemic typhoid-like disease is characterized by infected phagocytes clustered within pathological lesions, or foci (16, 43, 46). Interestingly, there are typically only one to two bacteria in each phagocyte within a lesion (38, 49). Serovar Typhimurium growth in mice appears to result from an increase in the number of infected cells and not from an increase in bacterial numbers per cell (38, 49). This suggests that bacteria may escape from the host cell to infect other cells once a low threshold of intracellular bacteria is achieved (38). It is possible that intracellular serovar Typhimurium use centrifugal displacement toward host cell peripheries as one strategy to facilitate cell-to-cell transfer in vivo.
Our results reveal a novel phenotype associated with SCV positioning in host cells. We demonstrate that the intracellular localization of SCVs is dynamic during the course of infection and that SCVs do not simply remain at juxtanuclear positions following invasion. Centrifugal SCV displacement is orchestrated by both bacterial and host factors. The characteristic displacement of WT SCVs toward the host cell periphery at later stages of infection might be one pathway exploited by the pathogen to escape from the host cell to infect other cells.
Acknowledgments
We thank members of the Brumell laboratory for helpful discussions and critical reading of the manuscript. We are grateful to L. Knodler (NIAID/NIH), S. I. Miller (University of Washington), M. B. Ohlson (Stanford University), and O. Steele-Mortimer (NIAID/NIH) for providing ΔpipB2 serovar Typhimurium strains and for helpful discussions regarding the manuscript. We thank R. Vale (University of California) for providing anti-kinesin heavy chain antibody, D. Zhou (Purdue University) for providing anti-SipA antibody, and M. Way (London Research Institute) for providing plasmid pCB6-GFP-KLC-TPR.
J.S. was supported by fellowships from the Canadian Association of Gastroenterology (CAG)/Canadian Institutes of Health Research (CIHR)/Solvay Pharma and the Natural Sciences and Engineering Research Council. A.N. was supported by a studentship from CAG/CIHR/Crohn's and Colitis Foundation of Canada. J.H.B. holds an Investigators in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. This work was supported by a grant from the Canadian Institutes of Health Research given to J.H.B. (grant no. 62890).
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 22 December 2008.
REFERENCES
- 1.Abrahams, G. L., and M. Hensel. 2006. Manipulating cellular transport and immune responses: dynamic interactions between intracellular Salmonella enterica and its host cells. Cell Microbiol. 8728-737. [DOI] [PubMed] [Google Scholar]
- 2.Abrahams, G. L., P. Muller, and M. Hensel. 2006. Functional dissection of SseF, a type III effector protein involved in positioning the Salmonella-containing vacuole. Traffic 7950-965. [DOI] [PubMed] [Google Scholar]
- 3.Alberti-Segui, C., K. R. Goeden, and D. E. Higgins. 2007. Differential function of Listeria monocytogenes listeriolysin O and phospholipases C in vacuolar dissolution following cell-to-cell spread. Cell Microbiol. 9179-195. [DOI] [PubMed] [Google Scholar]
- 4.Birmingham, C. L., X. Jiang, M. B. Ohlson, S. I. Miller, and J. H. Brumell. 2005. Salmonella-induced filament formation is a dynamic phenotype induced by rapidly replicating Salmonella enterica serovar Typhimurium in epithelial cells. Infect. Immun. 731204-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucrot, E., T. Henry, J. P. Borg, J. P. Gorvel, and S. Meresse. 2005. The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 3081174-1178. [DOI] [PubMed] [Google Scholar]
- 6.Brawn, L. C., R. D. Hayward, and V. Koronakis. 2007. Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 163-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown, N. F., J. Szeto, X. Jiang, B. K. Coombes, B. B. Finlay, and J. H. Brumell. 2006. Mutational analysis of Salmonella translocated effector members SifA and SopD2 reveals domains implicated in translocation, subcellular localization and function. Microbiology 1522323-2343. [DOI] [PubMed] [Google Scholar]
- 8.Brumell, J. H., D. L. Goosney, and B. B. Finlay. 2002. SifA, a type III secreted effector of Salmonella Typhimurium, directs Salmonella-induced filament (Sif) formation along microtubules. Traffic 3407-415. [DOI] [PubMed] [Google Scholar]
- 9.Brumell, J. H., C. M. Rosenberger, G. T. Gotto, S. L. Marcus, and B. B. Finlay. 2001. SifA permits survival and replication of Salmonella typhimurium in murine macrophages. Cell Microbiol. 375-84. [DOI] [PubMed] [Google Scholar]
- 10.Cirillo, D. M., R. H. Valdivia, D. M. Monack, and S. Falkow. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30175-188. [DOI] [PubMed] [Google Scholar]
- 11.Coombes, B. K., N. F. Brown, Y. Valdez, J. H. Brumell, and B. B. Finlay. 2004. Expression and secretion of Salmonella pathogenicity island-2 virulence genes in response to acidification exhibit differential requirements of a functional type III secretion apparatus and SsaL. J. Biol. Chem. 27949804-49815. [DOI] [PubMed] [Google Scholar]
- 12.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 976640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deiwick, J., S. P. Salcedo, E. Boucrot, S. M. Gilliland, T. Henry, N. Petermann, S. Waterman, J. P. Gorvel, D. W. Holden, and S. Meresse. 2006. The translocated Salmonella effector proteins SseF and SseG interact and are required to establish an intracellular replication niche. Infect. Immun. 746965-6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drecktrah, D., L. A. Knodler, K. Galbraith, and O. Steele-Mortimer. 2005. The Salmonella SPI1 effector SopB stimulates nitric oxide production long after invasion. Cell Microbiol. 7105-113. [DOI] [PubMed] [Google Scholar]
- 15.Drecktrah, D., L. A. Knodler, D. Howe, and O. Steele-Mortimer. 2007. Salmonella trafficking is defined by continuous dynamic interactions with the endolysosomal system. Traffic 8212-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunlap, N. E., W. H. Benjamin, Jr., R. D. McCall, Jr., A. B. Tilden, and D. E. Briles. 1991. A “safe-site: for Salmonella typhimurium is within splenic cells during the early phase of infection in mice. Microb. Pathog. 10297-310. [DOI] [PubMed] [Google Scholar]
- 17.Duong, N., S. Osborne, V. H. Bustamante, A. M. Tomljenovic, J. L. Puente, and B. K. Coombes. 2007. Thermosensing coordinates a cis-regulatory module for transcriptional activation of the intracellular virulence system in Salmonella enterica serovar Typhimurium. J. Biol. Chem. 28234077-34084. [DOI] [PubMed] [Google Scholar]
- 18.Eriksson, S., S. Lucchini, A. Thompson, M. Rhen, and J. C. Hinton. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47103-118. [DOI] [PubMed] [Google Scholar]
- 19.Galan, J. E. 2001. Salmonella interactions with host cells: type III secretion at work. Annu. Rev. Cell Dev. Biol. 1753-86. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-del Portillo, F., M. B. Zwick, K. Y. Leung, and B. B. Finlay. 1993. Salmonella induces the formation of filamentous structures containing lysosomal membrane glycoproteins in epithelial cells. Proc. Natl. Acad. Sci. USA 9010544-10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guignot, J., E. Caron, C. Beuzon, C. Bucci, J. Kagan, C. Roy, and D. W. Holden. 2004. Microtubule motors control membrane dynamics of Salmonella-containing vacuoles. J. Cell Sci. 1171033-1045. [DOI] [PubMed] [Google Scholar]
- 22.Guiney, D. G. 2005. The role of host cell death in Salmonella infections. Curr. Top. Microbiol. Immunol. 289131-150. [DOI] [PubMed] [Google Scholar]
- 23.Haraga, A., M. B. Ohlson, and S. I. Miller. 2008. Salmonellae interplay with host cells. Nat. Rev. Microbiol. 653-66. [DOI] [PubMed] [Google Scholar]
- 24.Harrison, R. E., J. H. Brumell, A. Khandani, C. Bucci, C. C. Scott, X. Jiang, B. B. Finlay, and S. Grinstein. 2004. Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles. Mol. Biol. Cell. 153146-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hautefort, I., A. Thompson, S. Eriksson-Ygberg, M. L. Parker, S. Lucchini, V. Danino, R. J. Bongaerts, N. Ahmad, M. Rhen, and J. C. Hinton. 2008. During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell Microbiol. 10958-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry, T., C. Couillault, P. Rockenfeller, E. Boucrot, A. Dumont, N. Schroeder, A. Hermant, L. A. Knodler, P. Lecine, O. Steele-Mortimer, J. P. Borg, J. P. Gorvel, and S. Meresse. 2006. The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. USA 10313497-13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henry, T., J. P. Gorvel, and S. Meresse. 2006. Molecular motors hijacking by intracellular pathogens. Cell Microbiol. 823-32. [DOI] [PubMed] [Google Scholar]
- 28.Hensel, M., J. E. Shea, S. R. Waterman, R. Mundy, T. Nikolaus, G. Banks, A. Vazquez-Torres, C. Gleeson, F. C. Fang, and D. W. Holden. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30163-174. [DOI] [PubMed] [Google Scholar]
- 29.Hoiseth, S. K., and B. A. Stocker. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291238-239. [DOI] [PubMed] [Google Scholar]
- 30.Hybiske, K., and R. S. Stephens. 2008. Exit strategies of intracellular pathogens. Nat. Rev. Microbiol. 699-110. [DOI] [PubMed] [Google Scholar]
- 31.Hybiske, K., and R. S. Stephens. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 10411430-11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang, X., O. W. Rossanese, N. F. Brown, S. Kujat-Choy, J. E. Galan, B. B. Finlay, and J. H. Brumell. 2004. The related effector proteins SopD and SopD2 from Salmonella enterica serovar Typhimurium contribute to virulence during systemic infection of mice. Mol. Microbiol. 541186-1198. [DOI] [PubMed] [Google Scholar]
- 33.Knodler, L. A., and O. Steele-Mortimer. 2003. Taking possession: biogenesis of the Salmonella-containing vacuole. Traffic 4587-599. [DOI] [PubMed] [Google Scholar]
- 34.Knodler, L. A., and O. Steele-Mortimer. 2005. The Salmonella effector PipB2 affects late endosome/lysosome distribution to mediate Sif extension. Mol. Biol. Cell 164108-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knodler, L. A., B. A. Vallance, M. Hensel, D. Jackel, B. B. Finlay, and O. Steele-Mortimer. 2003. Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol. Microbiol. 49685-704. [DOI] [PubMed] [Google Scholar]
- 36.Kuhle, V., G. L. Abrahams, and M. Hensel. 2006. Intracellular Salmonella enterica redirect exocytic transport processes in a Salmonella pathogenicity island 2-dependent manner. Traffic 7716-730. [DOI] [PubMed] [Google Scholar]
- 37.Marsman, M., I. Jordens, C. Kuijl, L. Janssen, and J. Neefjes. 2004. Dynein-mediated vesicle transport controls intracellular Salmonella replication. Mol. Biol. Cell 152954-2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mastroeni, P., and M. Sheppard. 2004. Salmonella infections in the mouse model: host resistance factors and in vivo dynamics of bacterial spread and distribution in the tissues. Microbes Infect. 6398-405. [DOI] [PubMed] [Google Scholar]
- 39.Miao, E. A., C. A. Scherer, R. M. Tsolis, R. A. Kingsley, L. G. Adams, A. J. Baumler, and S. I. Miller. 1999. Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol. Microbiol. 34850-864. [DOI] [PubMed] [Google Scholar]
- 40.Ochman, H., F. C. Soncini, F. Solomon, and E. A. Groisman. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. USA 937800-7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramsden, A. E., D. W. Holden, and L. J. Mota. 2007. Membrane dynamics and spatial distribution of Salmonella-containing vacuoles. Trends Microbiol. 15516-524. [DOI] [PubMed] [Google Scholar]
- 42.Ramsden, A. E., L. J. Mota, S. Munter, S. L. Shorte, and D. W. Holden. 2007. The SPI-2 type III secretion system restricts motility of Salmonella-containing vacuoles. Cell Microbiol. 92517-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richter-Dahlfors, A., A. M. Buchan, and B. B. Finlay. 1997. Murine salmonellosis studied by confocal microscopy: Salmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J. Exp. Med. 186569-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rietdorf, J., A. Ploubidou, I. Reckmann, A. Holmstrom, F. Frischknecht, M. Zettl, T. Zimmermann, and M. Way. 2001. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 3992-1000. [DOI] [PubMed] [Google Scholar]
- 45.Salcedo, S. P., and D. W. Holden. 2003. SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J. 225003-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salcedo, S. P., M. Noursadeghi, J. Cohen, and D. W. Holden. 2001. Intracellular replication of Salmonella typhimurium strains in specific subsets of splenic macrophages in vivo. Cell Microbiol. 3587-597. [DOI] [PubMed] [Google Scholar]
- 47.Sano, G., Y. Takada, S. Goto, K. Maruyama, Y. Shindo, K. Oka, H. Matsui, and K. Matsuo. 2007. Flagella facilitate escape of Salmonella from oncotic macrophages. J. Bacteriol. 1898224-8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shea, J. E., M. Hensel, C. Gleeson, and D. W. Holden. 1996. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 932593-2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheppard, M., C. Webb, F. Heath, V. Mallows, R. Emilianus, D. Maskell, and P. Mastroeni. 2003. Dynamics of bacterial growth and distribution within the liver during Salmonella infection. Cell Microbiol. 5593-600. [DOI] [PubMed] [Google Scholar]
- 50.Steele-Mortimer, O., S. Méresse, J.-P. Gorvel, B.-H. Toh, and B. B. Finlay. 1999. Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway. Cell. Microbiol. 133-51. [DOI] [PubMed] [Google Scholar]
- 51.Stein, M. A., K. Y. Leung, M. Zwick, F. Garcia-del Portillo, and B. B. Finlay. 1996. Identification of a Salmonella virulence gene required for formation of filamentous structures containing lysosomal membrane glycoproteins within epithelial cells. Mol. Microbiol. 20151-164. [DOI] [PubMed] [Google Scholar]
- 52.Stevens, J. M., E. E. Galyov, and M. P. Stevens. 2006. Actin-dependent movement of bacterial pathogens. Nat. Rev. Microbiol. 491-101. [DOI] [PubMed] [Google Scholar]
- 53.Suchland, R. J., D. D. Rockey, S. K. Weeks, D. T. Alzhanov, and W. E. Stamm. 2005. Development of secondary inclusions in cells infected by Chlamydia trachomatis. Infect. Immun. 733954-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takeuchi, A. 1967. Electron microscope studies of experimental Salmonella infection. I. Penetration into the intestinal epithelium by Salmonella typhimurium. Am. J. Pathol. 50109-136. [PMC free article] [PubMed] [Google Scholar]
- 55.Tsolis, R. M., R. A. Kingsley, S. M. Townsend, T. A. Ficht, L. G. Adams, and A. J. Baumler. 1999. Of mice, calves, and men. Comparison of the mouse typhoid model with other Salmonella infections. Adv. Exp. Med. Biol. 473261-274. [PubMed] [Google Scholar]
- 56.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100195-199. [PubMed] [Google Scholar]
- 57.Wasylnka, J. A., M. A. Bakowski, J. Szeto, M. B. Ohlson, W. S. Trimble, S. I. Miller, and J. H. Brumell. 2008. Role for myosin II in regulating positioning of Salmonella-containing vacuoles and intracellular replication. Infect. Immun. 762722-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterman, S. R., and D. W. Holden. 2003. Functions and effectors of the Salmonella pathogenicity island 2 type III secretion system. Cell Microbiol. 5501-511. [DOI] [PubMed] [Google Scholar]