

Abstract
The Sm16/SmSLP/SmSPO-1 (Sm16) protein is secreted by the parasite Schistosoma mansoni during skin penetration and has been ascribed immunosuppressive activities. Here we describe the strategy behind the design of a modified Sm16 protein with a decreased aggregation propensity, thus facilitating the expression and purification of an Sm16 protein that is soluble in physiological buffers. The Stokes radii and sedimentation coefficients of recombinant and native proteins indicate that Sm16 is an approximately nine-subunit oligomer. Analysis of truncated Sm16 derivatives showed that both oligomerization and binding to the plasma membrane of human cells depend on multiple C-terminal regions. For analysis of immunomodulatory activities, Sm16 was expressed in Pichia pastoris to facilitate the preparation of a pyrogen/endotoxin-free purified protein. Recombinant Sm16 was found to have no effect on T-lymphocyte activation, cell proliferation, or the basal level of cytokine production by whole human blood or monocytic cells. However, Sm16 exerts potent inhibition of the cytokine response to the Toll-like receptor (TLR) ligands lipopolysaccharide (LPS) and poly(I:C) while being less efficient at inhibiting the response to the TLR ligand peptidoglycan or a synthetic lipopeptide. Since Sm16 specifically inhibits the degradation of the IRAK1 signaling protein in LPS-stimulated monocytes, our findings indicate that inhibition is exerted proximal to the TLR complex.
Innate immunity provides a first line of defense toward invading microorganisms and depends on the expression of “pattern recognition receptors,” which recognize a restricted repertoire of molecules common to many pathogens (reviewed in reference 1). The Toll-like receptor (TLR) family is the archetypal pattern recognition receptor type and triggers signaling pathways leading to inflammatory responses through the production of an array of cytokines/chemokines. TLR signaling involves a variety of adaptor proteins, e.g., the bridging adaptor MAL and MyD88, which recruit members of the interleukin-1 receptor-associated kinase (IRAK) family to specific TLRs (reviewed in reference 1). While most of the identified TLR ligands are of viral, bacterial, or fungal origin, recent reports suggest that components of parasitic helminths may also act as TLR ligands (reviewed in reference 11).
The life cycle of the helminth Schistosoma mansoni, the parasite that causes schistosomiasis, involves human and snail hosts, as well as two free-living waterborne larval stages (reviewed in reference 4). The developmental stages include the miracidium that is infectious to fresh water snails, the sporocyst that is the parasitic form in the snail, and the cercaria that is released by snails and penetrates the skin of the human host. During entry into the dermis, the parasite transforms into a schistosomulum. The parasite then enters the blood system and develops into the adult worm, which is the long-term parasitic form.
Contact with free fatty acids in lipids on human skin stimulates holocrine release of protease secretions from the acetabular glands of the cercariae, which facilitate entry into the dermis (reviewed in reference 13). Acetabular gland secretions also contain proteins with a likely role in controlling the host immune defense (reviewed in reference 11). These include a glutathione S-transferase that has prostaglandin D2 synthase activity (8) and a protein termed Sm16 (3, 16, 18). Sm16 has also been identified as a developmental-stage-specific protein and has been termed in these studies either SmSPO-1 (15) or SmSLP (23). A recent proteomic study showed that Sm16 constitutes 3 to 4% of the proteins present in cercarial secretions (5).
Immunomodulation by Sm16 has been studied by two distinct approaches, namely, by addition of the partially purified protein to cultured cells (16) and by intradermal gene delivery of an Sm16 cDNA in mice (3). While these studies do not provide any mechanistic clues, it was reported that Sm16 suppresses both expression of proinflammatory cytokines by keratinocytes and antigen-specific lymphocyte proliferation, which implies a broad immunosuppressive action.
We have recently expressed Sm16-encoding DNA constructs in human cells (9), which revealed that the secreted protein associates with the plasma membrane. Interestingly, Sm16 binds protein-free lipids, as well as the plasma membrane of diverse cell types, which results in internalization by endocytosis. Moreover, expression of Sm16 derivatives lacking the signal peptide, which introduce the otherwise secreted Sm16 into the cytosol, causes rapid apoptotic death of transfected human cells. However, given that addition of Sm16 to the culture medium failed to induce apoptosis of human cell lines, it seems clear that the secreted Sm16 protein does not enter the cytosol by penetration of lipid bilayers. It follows that the physiological function of the cell surface-binding Sm16 protein seems likely to involve interactions on the extracellular and/or luminal side of membranes of host cells.
In the present study, we performed structural and functional studies of the secreted form of Sm16 engineered to allow recombinant expression of a soluble protein in either Escherichia coli or, to circumvent pyrogen/endotoxin contamination, the methylotrophic yeast Pichia pastoris. We show that Sm16 forms an approximately nine-subunit oligomer that has the potential to inhibit TLR signaling of monocytes without detectable inhibitory/toxic effects on T-lymphocyte activation or proliferation of human cells. Our findings suggest a TLR-dependent mechanism whereby Sm16 secretion from the acetabular glands may inhibit activation of the innate immune response during skin penetration.
MATERIALS AND METHODS
DNA constructs.
Low expression of Sm16 from the native cDNA (18), which in part can be attributed to many rare codons for mammals and E. coli, prompted us to construct a codon-optimized synthetic Sm16-encoding gene by ligation of a set of overlapping oligonucleotides. This synthetic sequence encodes amino acid 23 to 117 of Sm16 [termed Sm16(23-117)], which corresponds to the secreted form of Sm16 (9). An NcoI site was introduced at the ATG initiation codon, and a SacI site was introduced by silent mutations within the codons for the last three C-terminal amino acids (Glu-Ser-Ser) (9). To construct the engineered version of Sm16 that contains Ala substitutions at the codons for Ile-92 and Leu-93, termed Sm16(23-117)AA, we employed a PCR strategy using the synthetic Sm16-encoding gene as a template and 3′ primers specifying the substitutions, as well as a C-terminal DYKDDDDK Flag epitope tag (primer sequences are available on request). To construct stepwise C-terminal deletions (outlined in Fig. 1A; primer sequences are available on request), we employed a PCR-based strategy using 3′ primers defining each one of the deletions. These primers all contained a SacI site within the sequence that corresponds to the last three codons of the Sm16 sequence. The PCR products were then used to replace the NcoI-SacI fragment of Flag epitope-tagged Sm16(23-117). For expression in E. coli, the coding sequences of Flag-tagged Sm16 derivatives were subcloned as NcoI-BamHI fragments into E. coli T7 expression plasmid pET3d (Novagen), which introduced an N-terminal six-His tag. For the expression of Flag epitope-tagged Sm16 derivatives in the eukaryotic organism P. pastoris, PCR-amplified fragments were subcloned into the EcoRI and XbaI sites of the pPICZ B vector, which introduced an N-terminal six-His tag (primer sequences are available on request). The coding sequences of PCR-generated fragments were confirmed by nucleotide sequence analysis with an ABI PRISM dye terminator cycle sequencing kit from Perkin-Elmer.
FIG. 1.
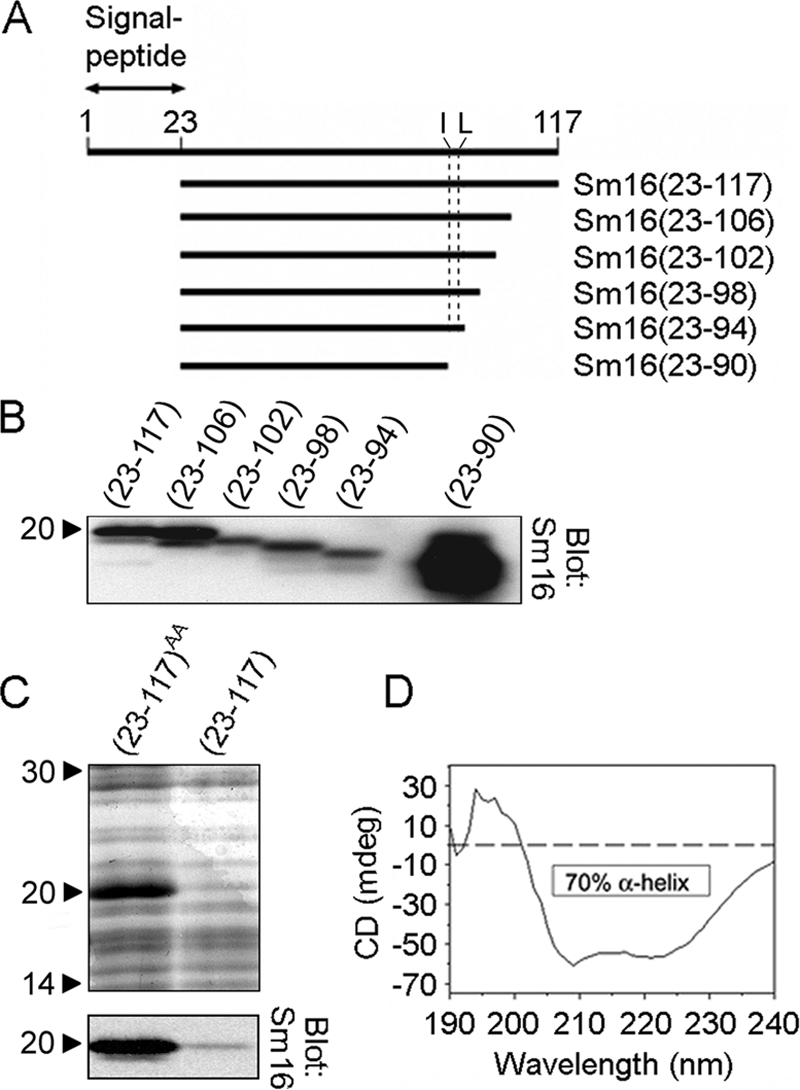
Construction of the engineered version of Sm16, Sm16(23-117)AA, with improved solubility and protein expression. (A) Schematic representation of recombinant Sm16 derivatives with successive truncations at the C terminus. Each truncated derivative is defined according to the amino acid sequence deduced from the cDNA sequence (numbers in parentheses). The location of hydrophobic residues Ile-92 and Leu-93 is indicated by a dashed line. The signal peptide of Sm16 consists of the first 22 residues, which are absent in the secreted form of the protein termed Sm16(23-117). All Sm16 derivatives contain an N-terminal His tag and an eight-residue C-terminal Flag tag. (B) Anti-Sm16 antibodies were used to detect recombinant Sm16 by immunoblot analysis of crude lysates of E. coli transformed with pET3d plasmid derivatives directing the expression of the indicated Sm16 proteins. (C) Coomassie brilliant blue-stained SDS-PAGE (upper panel) and immunoblot analysis with anti-Sm16 (lower panel) of crude lysates of E. coli transformed with pET3d derivatives directing the expression of the indicated Sm16 proteins. The abundantly expressed Sm16(23-117)AA protein was modified for improved solubility by replacing Ile-92 and Leu-93 with Ala, which also greatly improved expression. (D) Far-UV circular-dichroism (CD) spectra of Sm16(23-117)AA expressed in E. coli as a His-tagged protein and purified by metal ion affinity. Proteins were dissolved at 0.2 mg/ml in 2 mM sodium phosphate buffer and scanned at 20°C. The data indicate that ∼70% of the protein has an α-helical conformation. The values to the left of panels B and C are molecular sizes in kilodaltons.
Expression of Sm16 in E. coli and P. pastoris, purification of recombinant proteins, and detection of endotoxins and pyrogenic activities.
Sm16 proteins expressed in E. coli and P. pastoris contained an N-terminal six-His tag and an eight-residue C-terminal Flag tag. T7 expression plasmid pET3d was transformed into E. coli strain BL21(DE3)pLysS, and recombinant His-tagged Sm16 proteins were purified by metal ion affinity as described previously (19), with the modifications that all of the buffers contained 5 mM 2-mercaptoethanol and that HisTrap crude columns (Amersham Pharmacia Biotech) were subjected to an extensive washing protocol optimized to minimize contaminating endotoxins/pyrogens (10 ml Tris-HCl buffer [pH 8.0] containing 2% Triton X-100-0.5% deoxycholate, followed by 20 ml 20 mM Tris-HCl [pH 8.0] and 10 ml phosphate buffer [pH 8.0]-0.5 M NaCl). His-tagged Sm16 proteins were then eluted by 0.5 M imidazole-0.5 M NaCl-20 mM phosphate buffer, pH 8.0. The eluted proteins were diluted 10-fold in 20 mM phosphate buffer, pH 8.0, and further purified by a second round of binding to a HisTrap crude column, followed by the same stringent washing protocol outline above. The eluted Sm16 proteins were dialyzed against phosphate-buffered saline. The final preparations were essentially pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) criteria, and the yield of recombinant Sm16 derivatives was about 2 mg of pure protein per liter of E. coli culture grown to an optical density at 650 nm (OD650) of 0.6.
Sm16(23-117)AA and Sm16(23-70) were expressed in the eukaryotic organism P. pastoris by employing the pPICZ B expression vector as described by the supplier (www.invitrogen.com). In brief, the KM71 strain of P. pastoris containing the pPICZ B expression vector was grown in buffered glycerol-complex medium to stationary phase (an OD650 of around 12), and protein expression was then induced by shifting to a methanol-complex medium at an initial cell density corresponding to an OD650 of 30. Cells were harvested after 15 h of induction and disrupted with a bead beater. Lysates were cleared by centrifugation (100,000 × g for 1 h) and filtered through a 0.22-μm filter. The His-tagged Sm16 proteins were then purified by two rounds of metal ion affinity purification by the same extensive washing protocol described above for Sm16 expressed in E. coli. The final preparations were essentially pure by SDS-PAGE criteria, and the final yield of Sm16(23-117)AA was about 2 mg of pure protein per liter of P. pastoris culture induced at an OD650 of 30.
For the detection of endotoxins by the Limulus amoebocyte lysate test, a QCL-1000 turbidimetric chromogenic kit (Lonza) was used according to the manufacturer's instructions; the detection limit of the test was <0.1 endotoxin unit/ml. The Sm16(23-117)AA protein prepared from E. coli by two rounds of metal ion affinity purification contained around 150 endotoxin units/mg protein, while no endotoxins at all could be detected in the corresponding recombinant protein expressed in P. pastoris.
Sm16 antibodies, detection of cell surface binding of Sm16, flow cytometry, and immunoblotting.
Antibodies to Sm16 were raised by immunizing rabbits with a His-tagged Sm16(23-117)-nonrelated helix fusion protein expressed by and purified from E. coli (9). The resulting antibodies were affinity purified by absorption to Sm16(23-70) coupled to Sepharose, which resulted in antibodies that allowed equivalent detection of full-length and C-terminally truncated Sm16 proteins. For analysis of Sm16 binding to cell surfaces, human K562 erythroleukemia cells were incubated (1 × 106 in 0.5 ml RPMI 1640 medium-10% fetal calf serum [FCS]) with 2 μg/ml Sm16 protein for 30 min at 37°C. Cells were subsequently washed two times in phosphate-buffered saline, and surface-bound Sm16 was detected by staining of live cells with affinity-purified anti-Sm16 (1 μg/ml) and fluorescein-conjugated swine anti-rabbit immunoglobulin. Cell surface fluorescence was quantitated by flow cytometric analysis of live cells (>95% of the cells were viable and included in the acquisition gate, and >200,000 cells were collected) with a FACScalibur flow cytometer together with CellQuest software (BD Biosciences). For analysis of TLR4 cell surface expression levels, live cells were stained with Alexa 488-conjugated anti-CD284(TLR4) (MCA2061A488; AbD Serotec) and fluorescence intensity was determined by flow cytometry. Immunoblotting and subsequent detection with the ECL detection system (Amersham Pharmacia Biotech) were performed with anti-α-tubulin (B-5-1-2; Sigma), IκB-α (C-15, sc203; Santa Cruz Biotechnology), and affinity-purified rabbit anti-Sm16 and anti-IRAK1 (H-273; Santa Cruz Biotechnology, Inc.) as described previously (10). For quantitative analysis of immunoblots, the Bio-Rad ChemDoc (Bio-Rad) system was used with the Quantity One 4.4 program.
Circular-dichroism spectra and determinations of hydrodynamic parameters and molecular mass.
Circular-dichroism spectra were acquired by using a Jobin Yvon CD6 spectrodichrograph. The spectra between 190 and 260 nm were recorded by collecting data at 1-nm intervals with an integration time of 2 s (scan speed of 0.5 nm s−1). The Stokes radius was estimated by gel filtration chromatography on a Hi Load 16/60 Sephadex 200 or a Hi Load 16/60 Superdex 75 column with the ÄKTA purifier fast protein liquid chromatography system (Amersham Pharmacia Biotech). The elution buffer was phosphate-buffered saline, and columns were calibrated with standard proteins with known Stokes radii (cytochrome c, 1.70 nm; chymotrypsinogen A, 2.90 nm; ovalbumin, 3.05 nm; albumin, 3.55 mm; aldolase, 4.81 nm; catalase, 5.20 nm; ferritin, 6.10 nm). The sedimentation coefficient was estimated by velocity sedimentation on 5 to 20% sucrose gradients (4 ml) in phosphate-buffered saline at 42,000 rpm for 18 h in a Beckman SW60 rotor at 4°C. The sucrose gradients were individually calibrated by mixing Sm16 with standard proteins with known S values (cytochrome c, 1.86S; chymotrypsinogen A, 2.6S; ovalbumin, 3.5S; albumin, 4.6S; aldolase, 7.3S; catalase, 11.3S). Twenty fractions (∼200 μl each) were collected, and aliquots of each fraction were separated by 10 to 20% gradient SDS-PAGE. Peak fractions of the individual proteins were detected by densitometry of bands visualized by Coomassie blue staining. Sm16 in crude cercarial extract (kindly provided by Cecilia Thors, Stockholm, Sweden) was detected by immunoblotting with rabbit anti-Sm16 antibodies. Molecular masses were estimated by combined gel filtration and gradient sedimentation by using the equation M = S N0(6πηRS)/(1 − ν2ρ) = 4,205 S RS (21), in which M is molecular mass, N0 is Avogadro's number, η is the viscosity of the solvent (0.01 P), RS is the Stokes radius of the protein, ν2 is the partial specific volume of the protein (assumed to be 0.73 cm3/g), and ρ is density of the solvent (1.0). For the numerical formulation, S is in Svedberg units and the Stokes radius is in nanometers.
Cell cultures and analysis of inflammatory cytokines and cell proliferation.
TLR ligands were all purchased from InvivoGen (San Diego, CA). For analysis of cytokine production in human whole blood, freshly collected blood was diluted with serum-free RPMI 1640 medium (20% final blood concentration) and stimulated as indicated at 37°C. The human monocytic cell line Mono-Mac 6 was cultured in RPMI 1640 medium-5% FCS and for analysis of cytokine production; cells were incubated with various stimuli at 2.0 × 106/ml at 37°C. The cytokine contents of culture supernatants were assayed by using specific sandwich enzyme-linked immunosorbent assays as recommended by the manufacturer (R&D Systems, Minneapolis, MN). Human peripheral blood mononuclear cells were isolated from whole blood by Ficoll-Hypaque density gradient centrifugation and cultured at 1 × 106/ml in RPMI 1640 medium-5% FCS at 37°C. Cellular proliferation was analyzed by culturing cells (0.2 ml/culture, triplicate cultures) in flat-bottom microtiter plates in the presence of [3H]thymidine (1 μCi/well) for the indicated times. Cultures were terminated by precipitation on glass filters, and incorporated [3H]thymidine was determined by liquid scintillation.
Quantitative RT-PCR.
Total RNA was extracted with the TRIzol reagent (Invitrogen) by following the manufacturer's protocol. cDNA was synthesized from 1 μg total RNA with the RevertAid H Minus First Strand cDNA synthesis kit containing Moloney murine leukemia virus reverse transcriptase and random hexamer primers (Fermentas Life Sciences). Quantitative reverse transcription (RT)-PCR of triplicate samples with an interleukin-2 (IL-2)-specific primer pair (forward, 5′-AAC TCA CCA GGA TGC TCA CAT TT; reverse, 5′-TTA GCA CTT CCT CCA GAG GTT TG) or an IL-1β-specific primer pair (forward, 5′-AAA CAG ATG AAG TGC TCC TTC CAG G; reverse, 5′-TGG AGA ACA CCA CTT GTT GCT CCA) was performed with the Biotools QuantiMix EASY SYG kit and the iCycler iQ multicolor real-time PCR detection system (Bio-Rad). The cycling parameters were 95°C for 3 min, 40 amplification cycles (95°C for 10 s, 60°C for 45 s), and a 55-to-95°C melting gradient (10-s increments of 0.5°C).
RESULTS
Design of a recombinant version of the secreted form of Sm16 with a decreased aggregation propensity.
We have previously studied Sm16 secretion by transfection of cell lines and confirmed cleavage of the signal peptide between amino acids 22 and 23 during passage through the secretory pathway (9). Using E. coli to express the 95-residue secreted form of Sm16, termed Sm16(23-117) (Fig. 1A), we noted problems with poor expression and aggregation of the purified protein under physiological buffer conditions, which obstructed studies requiring a pure and soluble Sm16 protein. To overcome these problems, the Sm16 polypeptide was scanned for aggregation-promoting regions that may interfere with expression and solubility. Analysis of a series of C-terminally truncated derivatives indicated that poor Sm16 protein expression depends entirely on the short sequence Lys91-Ile-Leu-Gly94, as defined by the Sm16(23-94) and Sm16(23-90) proteins (Fig. 1B). We also found that a purified preparation of the highly expressed Sm16(23-90) protein remained soluble in physiological buffers (data not shown).
As a strategy to decrease the aggregation propensity of Sm16, the hydrophobicity within the identified region was decreased by replacing Ile-92 and Leu-93 with Ala residues; these substitutions are indicated by the AA superscript in the modified Sm16(23-117)AA protein name. As shown in Fig. 1C, immunoblot analysis of bacterial lysates shows that both of these epitope-tagged recombinant proteins migrate at ∼20 kDa but that Sm16(23-117)AA was expressed at 24-fold higher levels than the unmodified Sm16(23-117) protein.
While the pure unmodified Sm16(23-117) protein was soluble in 0.5 M imidazole-0.5 M NaCl, pH 8.0, it formed visible aggregates within 1 h in phosphate-buffered saline. However, the modified Sm16(23-117)AA protein was readily soluble in phosphate-buffered saline and purified preparations could be stored for prolonged periods without aggregating (data not shown). Thus, the present modification solved all of the problems we experienced with aggregation and consequent precipitation, which was a prerequisite for the present biochemical and functional characterization of a purified soluble Sm16 protein.
The amino acid composition and sequence of Sm16 suggest a high propensity to form an α-helical secondary structure. This propensity was experimentally confirmed by measurement of the far-UV circular-dichroism spectra, which revealed that ∼70% of the polypeptide exists in an α-helical configuration at 20°C (Fig. 1D).
Sm16 is an approximately nine-subunit oligomer.
When analyzed by SDS-PAGE, Sm16(23-117)AA migrates at ∼19 kDa under reducing conditions (Fig. 2A), which is slower than the deduced 13.6-kDa molecular mass of this epitope-tagged recombinant protein. However, by immunoblotting of cercarial extract, we could confirm that the native Sm16 protein also migrated slowly on SDS-PAGE compared with its deduced molecular mass of 11.3 kDa (Fig. 2D).
FIG. 2.

Hydrodynamic parameters and molecular masses of recombinant Sm16(23-117)AA and native Sm16 in crude cercarial extract. (A) The Sm16(23-117)AA protein was expressed in E. coli with an N-terminal His tag and an eight-residue C-terminal Flag tag. The metal ion affinity-purified protein was analyzed by SDS-PAGE in the presence (+) or absence (−) of the reducing agent 2-mercaptoethanol. Proteins were visualized by Coomassie blue staining, and arrows indicate the positions of standards with the indicated molecular weights (103). (B) Sephadex 200 gel filtration chromatography of purified Sm16(23-117)AA. Fractions were analyzed by SDS-PAGE, and proteins were visualized and quantitated by Coomassie blue staining, followed by scanning of gels. Open and filled symbols indicate the elution peaks of protein standards with known Stokes radii and Sm16, respectively. (C) Sucrose gradient sedimentation of purified Sm16(23-117)AA. Fractions were analyzed by SDS-PAGE as described for panel B. Open and filled symbols indicate the positions of protein standards with known Stokes radii and Sm16, respectively. (D) Crude cercarial extract was separated by SDS-PAGE as described for panel A, and Sm16 was detected by immunoblotting. (E) Crude cercarial extract was gel filtered together with protein standards as described for panel B. The Sm16 content of each fraction was quantified by immunoblotting and a comparison of signal intensity with a serial dilution of cercarial extract. (F) Crude cercarial extract was sedimented on a sucrose gradient together with protein standards as described for panel C. Fractions were analyzed as described for panel E. The data plotted are representative of at least three independent analyses, and the interexperimental variations in the estimations of Stokes radii and sedimentation coefficients were <7%. The values to the left of panels A and D are molecular sizes in kilodaltons. a.u., arbitrary units; Seph, Sephadex.
To characterize the engineered version of secreted Sm16 and to compare it with native Sm16 from cercarial extract, we employed a combination of gel filtration chromatography and sucrose gradient sedimentation. This provides estimates of the Stokes radius and the sedimentation coefficient (S), which allows calculation of the molecular masses of native complexes with the formula given in Materials and Methods. As shown in Fig. 2B and C, analysis of purified Sm16(23-117)AA indicates a 5.5-nm Stokes radius and a 5.4S sedimentation coefficient, which reveals that the protein forms an oligomer with an estimated molecular mass of ∼122 kDa. Importantly, parallel analysis of cercarial extracts (Fig. 2E and F), combined with immunodetection of Sm16 in fractions, revealed hydrodynamic parameters similar to those of the pure Sm16(23-117)AA protein (Stokes radii and sedimentation coefficients are indicated in the panels of Fig. 2).
The combined evidence from analysis of purified Sm16(23-117)AA and native Sm16 in crude cercarial extracts indicates that Sm16 is an oligomeric protein with a defined number of subunits. Based on the deduced 13.6-kDa molecular mass of Sm16(23-117)AA, an estimated molecular mass of ∼122 kDa of the oligomer, and the ∼10% uncertainty of the method (6), it can be estimated that Sm16 is a 9 ± 1-subunit oligomer.
Characterization of a biochemically distinct Sm16 trimer in a preparation of C-terminally truncated Sm16 protein.
To evaluate the importance of the C terminus for the oligomeric state of Sm16, a truncated derivative was analyzed by gel filtration. Interestingly, the purified Sm16(23-98)AA protein, which lacks the C-terminal 19 residues, was resolved into two distinct peaks on a Sephadex 200 column (Fig. 3A; SDS-PAGE analysis of pooled peak fractions is shown as an insert). Pooled peak fractions of the 5.5-nm species and the 3.1-nm species were further analyzed by sucrose gradient sedimentation (Fig. 3B and C). By this strategy, the Stokes radius and sedimentation coefficient were estimated for each of the two oligomeric forms of the C-terminally truncated Sm16(23-98)AA derivative. The data indicate that the Sm16(23-98)AA protein exists in two stable oligomeric forms which, based on their hydrodynamic properties, have estimated molecular masses of ∼92 kDa (5.5 nm and 4.0S) and ∼29 kDa (3.1 nm and 2.2S). Given the deduced 11.2-kDa molecular mass of Sm16(23-98)AA and the general ∼10% uncertainty of the method, the molecular mass of the large oligomer is consistent with a 9-mer, which implies the same oligomeric state as the full-length Sm16(23-117)AA protein. Moreover, the estimated 29-kDa molecular mass of the small oligomer indicates that a large proportion of the expressed Sm16(23-98)AA proteins also form stable approximately three-subunit oligomers. This result reveals a propensity of Sm16 to also form stable trimers, which may be relevant for the structure of the approximately nine-subunit native oligomer.
FIG. 3.

Identification of two oligomeric forms in a preparation of the C-terminally truncated Sm16(23-98)AA protein. (A) The Sm16(23-98)AA protein contains Ala substitutions for Ile-92 and Leu-93 to prevent aggregation. The metal ion affinity-purified protein was separated by Sephadex 200 gel filtration, and fractions were analyzed and quantified as described in the legend to Fig. 2B. Open and filled symbols indicate the elution peaks of protein standards with known Stokes radii and Sm16(23-98)AA, respectively. The estimated Stokes radii of the two peaks are indicated. Peak fractions corresponding to Stokes radii of 5.5 and 3.1 nm, respectively, were pooled and analyzed by SDS-PAGE as described in the legend to Fig. 2A (insert). (B and C) Sedimentation properties of pooled peak fractions (insert in panel A) of Sm16(23-98)AA species with Stokes radii of 5.5 nm (panel B) and 3.1 nm (panel C) were analyzed as described in the legend to Fig. 2C. (D and E) Heparin binding of pooled peak fractions (insert in panel A) of Sm16(23-98)AA species with Stokes radii of 5.5 nm (panel D) and 3.1 nm (panel E). Proteins diluted in phosphate-buffered saline (0.15 M NaCl) were passed over a column containing heparin coupled to Sepharose. Columns were eluted with a linear gradient of 0.15 to 1.0 M NaCl. Proteins in the fractions were analyzed and quantitated as described in the legend to Fig. 2B. The data plotted are representative of at least two independent analyses, and the experimental errors in the estimations of Stokes radii and sedimentation coefficients were <7%. a.u., arbitrary units; Seph, Sephadex. The values to the left of the inserts in panel A are molecular sizes in kilodaltons.
Previous studies have shown that cell surface binding of the secreted form of full-length Sm16 is not inhibited by the polyanionic substance heparin (9). Consistent with this result, the approximately nine-subunit oligomer of Sm16(23-98)AA with a 5.5-nm Stokes radius was found not to bind immobilized heparin at a physiological salt concentration (i.e., 0.15 M NaCl, Fig. 3D). Interestingly, however, the approximately three-subunit oligomer with a 3.1-nm Stokes radius bound avidly to heparin and was eluted at ∼0.5 M NaCl (Fig. 3E). Hence, the approximately three-subunit oligomer exposes polyanion binding surfaces that appear to be hidden in the native approximately nine-subunit Sm16 oligomer, which indicates a differential polypeptide arrangement within these two oligomeric forms. The presence of these two oligomeric forms may be interpreted to suggest that Sm16 is a nine-subunit trimer of trimers.
The C-terminal part of Sm16 is required for both oligomerization and binding to the surface of human cells.
Analysis of a series of truncated engineered versions of Sm16 proteins by Sephadex 200 gel filtration shows that consecutive C-terminal truncations of Sm16 result in a stepwise decrease in the fraction of approximately nine-subunit oligomers (Fig. 4A). Consistent with the analysis of Sm16(23-98)AA (Fig. 3), we observed apparent trimers in these preparations of truncated Sm16 and the proportion of trimers depended on the extent of the truncation (data not shown). Thus, it appears that multiple physically separated C-terminal regions of Sm16 contribute to the assembly of the approximately nine-subunit oligomer.
FIG. 4.
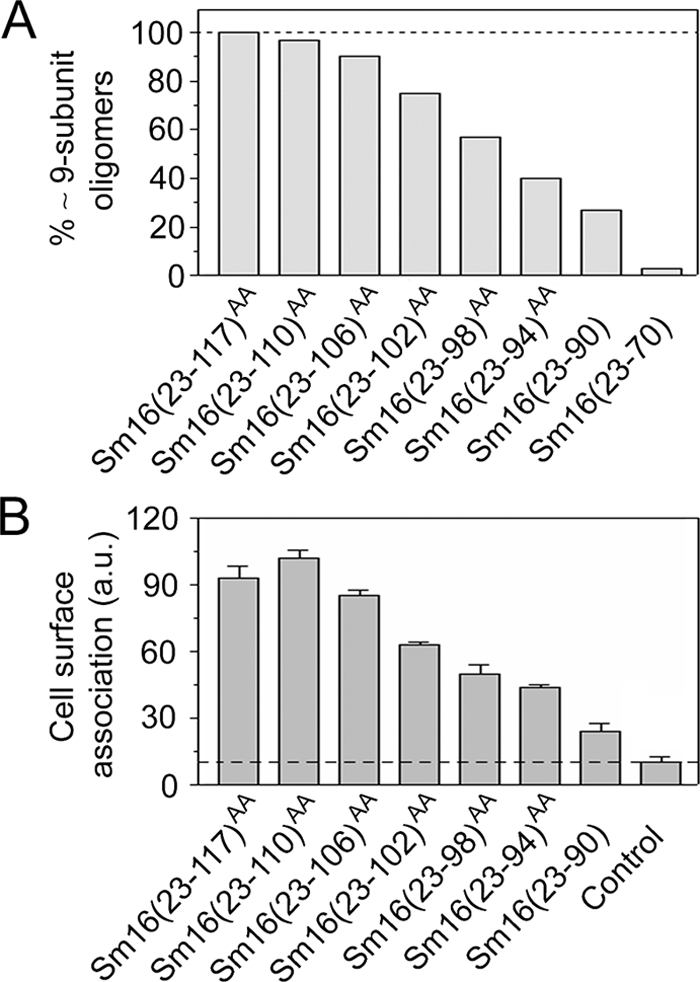
Importance of C-terminal regions for Sm16 oligomerization and cell surface binding. (A) The fraction of approximately nine-subunit oligomers, defined as a >4.5-nm Stokes radius, in metal ion affinity-purified preparations of the truncated Sm16 derivatives depicted in Fig. 1A was determined by Sephadex 200 gel filtration. Where indicated by the superscript AA, amino acid residues Ile-92 and Leu-93 were replaced with Ala to prevent protein aggregation. (B) Cell surface binding of approximately nine-subunit oligomers isolated by Sephadex 200 gel filtration of the indicated Sm16 derivative. The Sm16(23-70) protein does not show detectable affinity for cells (data not shown) but was not included in the present analysis since it does not form detectable amounts of approximately nine-subunit oligomers (panel A). Binding to live K562 erythroleukemia cells was analyzed as described in Materials and Methods. Background binding, i.e., anti-Sm16 and fluorescein-conjugated swine anti-rabbit immunoglobulin, was ∼12% (dotted line). The presented data are representative of at least three independent experiments. a.u., arbitrary units.
We have previously found that Sm16 binds both to protein-free liposomes and to the surfaces of various human cell lines. Binding does not involve polyanion interactions with cell surface proteoglycans, as evidenced by a lack of inhibition by heparin (9), which indicates that Sm16 binds to the cell surface through interaction with the lipid bilayer of the plasma membrane. To analyze the role of C-terminal regions in cell surface binding of the approximately nine-subunit oligomer, Sephadex 200 gel filtration was used to isolate the fraction of large oligomers of the relevant Sm16 proteins [i.e., all derivatives except Sm16(23-70)]. This ensured that this analysis of cell binding is unbiased by a differential oligomerization propensity, and it is notable that the isolated oligomers in all cases appeared stable, as confirmed by a second round of gel filtration (data not shown). Cell surface binding was determined by incubation with human K562 cells, followed by detection of cell surface-associated Sm16 with specific antibodies and flow cytometry. The data revealed that consecutive C-terminal truncations of Sm16 result in a stepwise decrease in cell surface binding and that truncation of 27 residues severely reduced binding (Fig. 4B). Thus, multiple small regions of the C-terminal part of Sm16 contribute to cell surface binding independently of their importance for oligomerization.
Biological activities of Sm16 on blood cells.
Interpretations of immunomodulatory activities exerted by a bacterially expressed lipid-binding protein is likely to be confounded by both formylation of the N-terminal methionine and trace amounts of endotoxin/pyrogen contamination. We therefore used the methylotrophic yeast P. pastoris for the expression of Sm16(23-117)AAP (the letter P denotes Pichia) and developed a stringent washing protocol during purification to eliminate pyrogenic contaminants (see Materials and Methods). The purified Sm16(23-117)AAP protein was found to be indistinguishable from the E. coli-derived protein with respect to the Stokes radius, migration on SDS-PAGE, and binding to the surfaces of human cells (data not shown).
Anti-CD3 antibodies activate T lymphocytes through the CD3/antigen receptor complex, which induces T-lymphocyte proliferation by a mechanism that depends on collaboration with monocytic cells (2). This collaboration induces cytokine production by monocytic cells, e.g., IL-1β, which is required for subsequent production of the lymphokine IL-2 by T lymphocytes. To evaluate potential immunosuppressive activities of Sm16(23-117)AAP, the effects on isolated blood mononuclear cells were analyzed in the presence or absence of CD3 stimulation. As shown in Fig. 5A and B, a high concentration of Sm16(23-117)AAP (50 μg/ml) had no effect by itself and did not inhibit anti-CD3 antibody-stimulated IL-1β or IL-2 expression. Moreover, we did not detect inhibition of DNA synthesis and the basal proliferation actually appeared to be somewhat increased in the presence of Sm16(23-117)AAP (Fig. 5C). Thus, we found no evidence that Sm16(23-117)AAP exerts general immunomodulatory activities.
FIG. 5.

Effect of P. pastoris-expressed Sm16 on cytokine expression and proliferation of peripheral blood mononuclear cells stimulated by an antibody directed to the T-lymphocyte antigen/CD3 complex. Density gradient-purified blood mononuclear cells were cultured in medium alone or stimulated with the anti-CD3 antibody OKT3 (50 ng/ml) as indicated. IL-1β (A) or IL-2 (B) mRNA expression levels determined by quantitative RT-PCR after 18 h of incubation in the absence (open bars) or presence (filled symbols) of Sm16(23-117)AAP (50 μg/ml) are shown. (C) Cell proliferation as determination by [3H]thymidine uptake during the last 6 h of a 78-h culture period in the presence of the indicated concentration of Sm16(23-117)AAP. The data plotted are representative of at least three independent experiments.
To search for Sm16-dependent effects on innate immunity, we analyzed the cytokine production of human blood in the absence or presence of the bacterial product lipopolysaccharide (LPS). As expected, Sm16(23-117)AAP alone did not stimulate cytokine production (Fig. 6, open squares). Interestingly, however, the full-length Sm16(23-117)AAP protein potently suppressed LPS-induced production of IL-6, tumor necrosis factor alpha (TNF-α), and IL-1β and 50% inhibition was observed at around 2 μg/ml in all cases (Fig. 6, filled squares). As a control, we also studied the effect of the C-terminally truncated Sm16(23-70)P protein, which does not bind to cell surfaces (Fig. 4B). As expected, this extensively truncated Sm16 protein did not inhibit the LPS response (Fig. 6, filled triangles). It should be noted that the dose response of Sm16(23-117)AAP-mediated inhibition was similar over a broad range of LPS concentrations (1 to 100 ng/ml; data not shown), which indicates that Sm16 does not inhibit by sequestering of LPS or by competing for TLR4 binding.
FIG. 6.
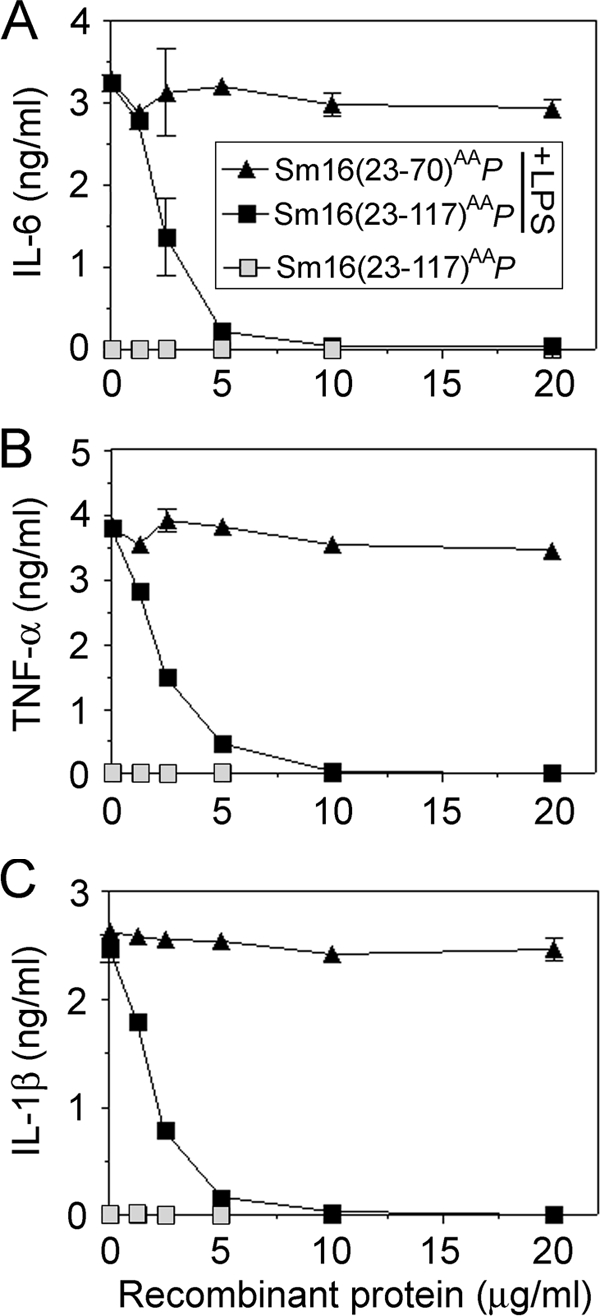
Effect of P. pastoris-expressed Sm16 proteins on LPS-induced production of proinflammatory cytokines in whole blood. Human blood was preincubated for 10 min with graded concentrations of P. pastoris-expressed Sm16(23-70)P (triangles) or Sm16(23-117)AAP (squares). Cells were then cultured for 6 h in the absence (open symbols) or presence (filled symbols) of LPS (50 ng/ml), followed by analysis of IL-6 (A), TNF-α (B), and IL-1β (C) in the supernatants. The data plotted are representative of at least 10 independent analyses performed in triplicate. Blood cells from four healthy donors were analyzed and found to be indistinguishable with regard to the effect of purified Sm16 derivatives.
Sm16 inhibits cytokine production in response to structurally distinct TLR ligands.
As outlined above, IL-1β production during an anti-CD3 antibody-induced mitogenic response depends on T-lymphocyte-dependent stimulation of monocytes (reviewed in reference 2). Thus, our finding that Sm16(23-117)AAP does not inhibit IL-1β expression in response to anti-CD3 antibodies (Fig. 5A) while LPS-induced production of IL-6, TNF-α, and IL-1β is potently suppressed (Fig. 6) indicates that Sm16 does not exert a general inhibition of monocyte cytokine production. To further characterize inhibition of cytokine production by Sm16(23-117)AAP, human blood was stimulated with saturating concentrations of the TLR4 ligand LPS, the synthetic TLR3 ligand poly(I:C), and the TLR2 ligands peptidoglycan (PG) and synthetic lipopeptide Pam3CSK4. The data reveal efficient Sm16(23-117)AAP-mediated inhibition of the response to either LPS or poly(I:C), while the responses to PG and Pam3CSK4 were only partially inhibited (Fig. 7, upper panels). Thus, Sm16(23-117)AAP inhibits the responses to several types of TLR ligands but with large differences in the apparent efficiency.
FIG. 7.
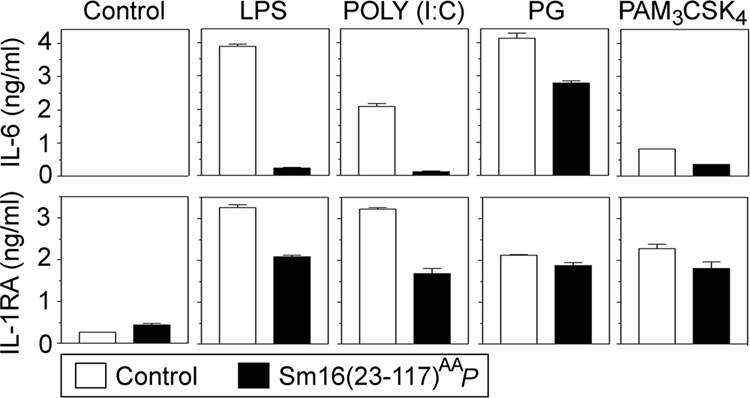
Effect of Sm16(23-117)AAP on cytokine production in response to various TLR ligands. Human blood was preincubated for 10 min with a buffer control (open bars) or P. pastoris-expressed Sm16(23-117)AAP (20 μg/ml) (filled bars). The buffer control, LPS (50 ng/ml), poly(I:C) (20 μg/ml), PG (5 μg/ml), or Pam3CSK4 (5 μg/ml) was then added as indicated for 6 h of incubation, followed by analysis of either IL-6 (upper panel) or IL-1RA (lower panels) in the supernatants. The data plotted are representative of at least four independent analyses performed in triplicate with blood from three donors.
Sm16 has previously been described as an IL-1 receptor antagonist (IL-1RA)-inducing protein (16). However, analysis of the anti-inflammatory cytokine IL-1RA revealed only a marginal increase in the basal level in the presence of Sm16(23-117)AAP (Fig. 7, lower panels). Importantly, while inhibition of IL-1RA secretion was not as potent as observed for IL-6 (Fig. 7, compare upper and lower panels), IL-1RA secretion was still clearly inhibited by Sm16(23-117)AAP in cells stimulated by either LPS or poly(I:C). Consistent with only a partial inhibition of PG- and Pam3CSK4-induced IL-6 production, the presence of Sm16(23-117)AAP caused only a minor decrease in IL-1RA secretion in response to either of these two TLR2 ligands (Fig. 7).
The finding that Sm16(23-117)AAP significantly inhibits both LPS- and poly(I:C)-induced IL-1RA secretion, as well as very potently inhibits the IL-6 response to these distinct TLR ligands, suggests that Sm16 has the potential to interfere with TLR signaling. While we cannot explain why the PG and Pam3CSK4 response is poorly inhibited by Sm16, these results still illustrate the specificity of Sm16-mediated inhibition of the LPS and poly(I:C) response.
Sm16 inhibits a TLR-proximal signaling event.
To address the mechanism behind the inhibitory action of Sm16, we used a clonal human monocytic leukemia cell line, Mono-Mac-6, as a model system. These monocytic cells respond to TLR stimulation by induction of cytokine expression and by maturation to macrophages, which is associated with decreased cell proliferation (25). As shown in Fig. 8A, Sm16(23-117)AAP alone has no detectable effect on the proliferation of Mono-Mac-6. Significantly, however, the presence of Sm16(23-117)AAP partially counteracts the decrease in cell proliferation observed after 4 days of stimulation with 2 ng/ml LPS. Moreover, flow cytometry did not indicate that Sm16 modulates the surface expression of the LPS-binding TLR4 complex (Fig. 8B). These results establish that Sm16(23-117)AAP does not cause any general inhibitory/cytotoxic effects on Mono-Mac-6 cells and reveal counteraction of LPS-induced cell proliferation inhibition without detectable effects on TLR4 surface expression.
FIG. 8.
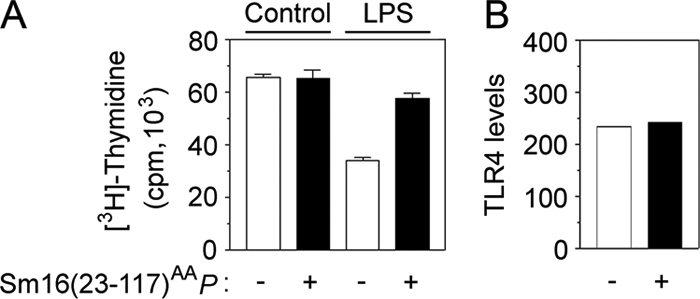
Effect of Sm16(23-117)AAP on the clonal monocytic cell line Mono-Mac-6. (A) Mono-Mac-6 cells seeded at 0.5 × 105/ml and cultured for 4 days with either a buffer control or LPS (2 ng/ml), as indicated, in the absence (open bars) or presence (filled bars) of Sm16(23-117)AAP (20 μg/ml). Cell proliferation was analyzed by determination of [3H]thymidine uptake during the last 24 h of culture. (B) Mono-Mac-6 cells were cultured for 5 h in the absence (open bars) or presence (filled bars) of Sm16(23-117)AAP (20 μg/ml). TLR4 surface expression was then analyzed by flow cytometry and expressed as the mean fluorescence signal.
To further characterize the action of Sm16(23-117)AAP on Mono-Mac-6 cells, we analyzed the production of the proinflammatory cytokine IL-6 in response to LPS, poly(I:C), and PG. As shown in Fig. 9, top panels, while the response to LPS and poly(I:C) was efficiently blocked, the response to PG was only partially inhibited. We also observed similar levels of inhibition by analysis of TNF-α and IL-1β (data not shown).
FIG. 9.
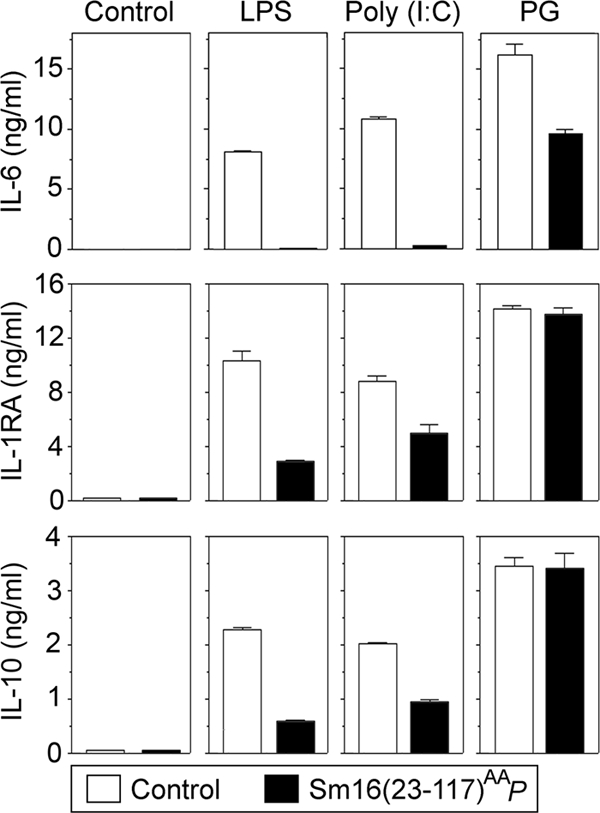
Effect of Sm16(23-117)AAP on cytokine production by Mono-Mac-6 cells in response to various TLR ligands. Mono-Mac-6 cells were preincubated for 10 min with a buffer control (open bars) or 20 μg/ml Sm16(23-117)AAP (filled bars). The buffer control, LPS (50 ng/ml), poly(I:C) (20 μg/ml), or PG (5 μg/ml) was then added as indicated for 8 h of incubation, followed by quantification of the proinflammatory cytokine IL-6 or the anti-inflammatory cytokines IL-1RA and IL-10 in the supernatants. The data plotted are representative of at least four independent analyses performed in triplicate.
We also analyzed the production of the two anti-inflammatory cytokines IL-1RA and IL-10 and found a partial inhibition by Sm16(23-117)AAP in the presence of either LPS or poly(I:C) (Fig. 9). It was also evident that the production of both IL-1RA and IL-10 is essentially unaltered in PG-stimulated Mono-Mac-6 cells. Thus, Sm16 appears to have similar actions on TLR ligand-induced responses of a clonal monocytic cell line and whole blood cells (compare Fig. 7 and 9).
The family of NFκB transcription factors is central for TLR-stimulated expression of proinflammatory cytokines. NFκB-activating signals lead to phosphorylation, ubiquitination, and subsequent degradation of the associated inhibitor IκB, which results in nuclear translocation of released NFκB. To evaluate the effect of Sm16(23-117)AAP on NFκB-activating signals in Mono-Mac-6 cells, we analyzed the phosphorylation and degradation of IκB-α. As shown in Fig. 10A, immunoblot analysis of IκB-α revealed both phosphorylation (manifested as decreased electrophoretic mobility) (12) and decreased amounts of protein in cells stimulated with either LPS or PG. It is also shown that the presence of Sm16(23-117)AAP causes a partial inhibition of both phosphorylation and degradation of IκB-α in the presence of LPS. However, consistent with the inefficient inhibition of PG stimulation shown above, Sm16(23-117)AAP did not detectably interfere with PG-induced IκB-α phosphorylation and degradation (Fig. 10A). Thus, Sm16(23-117)AAP-mediated inhibition of an LPS-induced cytokine response is associated with a partial inhibition of NFκB-activating signals.
FIG. 10.
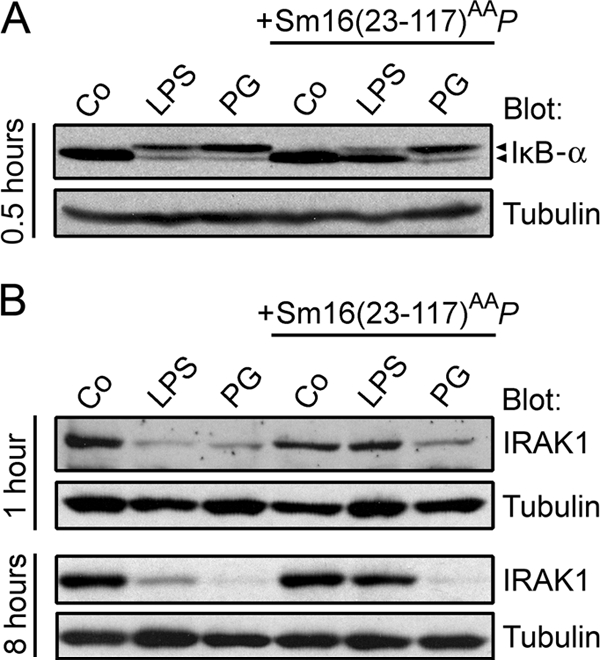
Effect of Sm16(23-117)AAP on TLR ligand-induced intracellular signaling events. Mono-Mac-6 cells were preincubated for 10 min in the absence or presence of Sm16(23-117)AAP (20 μg/ml) as indicated. A buffer control (Co), LPS (50 ng/ml), or PG (5 μg/ml) was then added as indicated. Cells were incubated at 37°C for 0.5 h (A), 1 h (B, top), or 8 h (B, bottom) and processed for immunoblot analysis. Filters were probed with either IκB-α (A) or anti-IRAK1 (B). Anti-α-tubulin was used as a control for equal loading. The presented data are representative of at least three independent analyses.
Activation of TLR2 or TLR4 triggers binding of the MyD88 adaptor to the intracellular portion of the receptors via the bridging adaptor MAL. This initiates a signaling cascade that involves recruitment of IRAK4 and IRAK1 proximal to the TLR complex and subsequent phosphorylation and degradation of IRAK1 (1). Accordingly, to evaluate whether Sm16 inhibits TLR signaling proximal to the receptor complex, we analyzed LPS- and PG-mediated IRAK1 degradation in the absence or presence of Sm16(23-117)AAP. As shown by immunoblotting in Fig. 10B, LPS induced a persistent reduction of IRAK1 protein content that was efficiently inhibited by Sm16(23-117)AAP. Significantly, however, the presence of Sm16(23-117)AAP did not significantly inhibit IRAK1 degradation in PG-stimulated Mono-Mac-6 cells, which seems consistent with the notion that PG-stimulated cytokine secretion is only partially inhibited under the same conditions (Fig. 9). These results confirm the specificity of the observed Sm16(23-117)AAP-mediated inhibition of IRAK1 degradation in LPS-stimulated cells.
The secreted Sm16 protein binds to the cell surface but lacks apparent cell membrane-penetrating properties (9), which makes it unlikely that the mechanism behind Sm16-mediated inhibition of TLR signaling involves direct interactions with cytosolic signaling proteins. Thus, it seems more likely that Sm16 exert its inhibitory action by interfering with ligand activation of the TLR complex. This implies that Sm16 acts upstream of the TLR-proximal IRAK1 protein, which is indeed shown in Fig. 10B.
DISCUSSION
Sm16 has been described as a stathmin-like protein (SmSLP), which implies similarity to a cytosolic microtubule regulator in animal cells (23). The evidence includes ∼26% identity of amino acid sequences and reported tubulin-binding and microtubule-destabilizing activities of a partially purified Sm16 preparation. However, by comparing recombinant Sm16 and stathmin proteins, we have reported a failure to reproduce stathmin-like activities of Sm16 and also found that stathmin does not share the demonstrated lipid bilayer-binding properties of Sm16. In addition, to introduce Sm16 into the cytosol of human cells, i.e., the same location as stathmin, we expressed a signal peptide-deficient Sm16 derivative, which was found to cause apoptotic cell death without evidence of stathmin-like microtubule regulatory activity (9). While these results illustrate a striking functional difference between Sm16 and stathmin, the proapoptotic effect of cytosolic Sm16 probably lacks a physiological significance since the secreted Sm16 protein evidently lacks cell membrane-penetrating properties (9). Both the Sm16 and stathmin sequences have characteristics indicating a high degree of α-helical secondary structure, which has also been experimentally confirmed (Fig. 1D and reference 24), but in the present study we demonstrate that Sm16 is an oligomeric protein, in contrast to the monomeric structure of stathmin (7, 22). Thus, Sm16 and stathmin are clearly very different with respect to both function and structure.
The present analysis of truncated Sm16 protein derivatives allowed us to design an engineered version of the secreted form of Sm16 with a decreased aggregation propensity and increased expression in E. coli (Fig. 1). This modified Sm16(23-117)AA protein was indeed also used in our previous study and was essential for in vitro studies of purified Sm16 in physiological buffers, which revealed efficient binding to cell surfaces and protein-free lipid bilayers (9). Here we show that the modified Sm16 protein forms a 9 ± 1-subunit oligomer with hydrodynamic properties similar to those of Sm16 in crude cercarial extract, thus indicating that the oligomeric state is shared by native Sm16 (Fig. 2).
The Sm16(23-117)AA protein was also functionally compared with the unmodified Sm16 protein by artificially expressing the proteins in the cytosol of transfected human cells and scoring for proapoptotic activity. While this activity is probably of no physiological significance, it still represents a distinctive and potent action of the Sm16 protein. In this experimental setting, which implies modest expression levels, we found that the Sm16(23-117)AA protein was expressed as strongly as the unmodified protein and exerted the same level of apoptosis-promoting activity (data not shown; 9). Thus, given evidently the same oligomeric structure and an unaltered proapoptotic activity, it seems reasonable to assume that that replacement of Ile-92 and Leu-93 with Ala residues does not cause any qualitative difference in biological activity. However, the modified Sm16(23-117)AA protein may still be less active than the wild-type protein. Thus, the present estimate that 2 μg/ml Sm16(23-117)AAP is required for 50% inhibition of the LPS response may underestimate the potency of the native Sm16 protein.
Our analysis of truncated Sm16 proteins revealed regions at the C-terminal end that are required for the formation of the nine-subunit oligomer (Fig. 3 and 4A). Analysis of the truncated Sm16(23-98)AA protein shows that the C-terminal 19 residues contribute to, but are not essential for, the assembly of the native approximately nine-subunit oligomer while more centrally located regions are sufficient for trimer formation. The differences in heparin binding between these oligomers (Fig. 3D and E) indicate that the polyanion binding surfaces of Sm16 trimers are masked in the approximately nine-subunit oligomer, which would be consistent with a model in which a nine-subunit Sm16 oligomer is built from three trimers. However, both of these oligomeric forms appeared stable after separation by gel filtration, as indicated by both sucrose gradient sedimentation (Fig. 3C) and a second round of gel filtration (data not shown). We therefore have no direct evidence that the observed trimers have the potential to form the approximately nine-subunit oligomer. Thus, while the present analyses of truncated Sm16 establish a potential to form stable trimers, the significance of trimer formation for the oligomeric configuration of native Sm16 remains to be established.
Short consecutive C-terminal truncations result in a gradual decrease in the approximately nine-subunit oligomer, but as revealed by the Sm16(23-70) protein, removal of the C-terminal half of Sm16 results in a complete failure to assemble the approximately nine-subunit oligomer Fig. 4A). Provided replacement of Ile-92 and Leu-93 with Ala in applicable derivatives, all C-terminal truncation derivatives were efficiently expressed in E. coli, which also includes the Sm16(23-70) protein, and the purified proteins appeared equally stable (data not shown). Thus, assembly into the approximately nine-subunit oligomer is not of importance for the structural integrity of Sm16. By analyzing the cell surface binding of approximately nine-subunit oligomers isolated by gel filtration, we found a gradual decrease in binding that appeared to correlate with a decreased propensity of the truncated Sm16 protein derivatives to form approximately nine-subunit oligomers. However, since isolated approximately nine-subunit oligomers were used for analysis, our results show that the Sm16 C-terminal end is important not only for oligomerization but also for cell surface binding of the assembled approximately nine-subunit oligomer.
To aid the development of vaccines, both the innate and adaptive immune responses during Schistosoma infections have been extensively studied (for a review, see reference 11). However, although the parasite has evidently developed strategies to avoid the host immune response, the mechanisms and molecules involved remain poorly characterized. The original functional study of the Sm16 protein involved separations of secreted cercarial proteins by denaturing SDS-PAGE, excision of an ∼16-kDa region, and subsequent elution of proteins (16). This one-step protocol implies a partial purification of denatured Sm16 monomers, and it is not clear whether the reported activities reflect the activity of the pure native protein. For example, inhibition of antigen-specific T-lymphocyte proliferation was observed, in contrast to the present finding that not even high concentrations of Sm16 have any effect on the anti-CD3 response (Fig. 5). Moreover, it was also reported that Sm16 increased the production of the anti-inflammatory cytokine IL-1RA, which provoked the proposal that Sm16 acts through an IL-1RA-dependent mechanism. This is not supported by our study since recombinant Sm16 produced in P. pastoris actually inhibited the production of IL-1RA in LPS- or poly(I:C)-stimulated human blood cells (Fig. 7). In a subsequent study (17), it was reported that intradermal injection of an Sm16-encoding DNA construct into mice resulted in a detectable suppression of LPS-provoked cutaneous inflammation, as well as a general immunosuppressive activity manifested on the level of T-lymphocyte proliferation. While our analysis of the action of a purified Sm16 protein is certainly consistent with inhibition of a cutaneous inflammation, we did not find evidence of a lymphocyte-directed activity of Sm16.
The present study reveals a potent Sm16-mediated inhibition of the cytokine response induced by two structurally distinct TLR ligands, namely, LPS and the double-stranded RNA mimic poly(I:C). The molecular mechanism was approached by analysis of the IRAK1 protein, which is known to be degraded in response to the activation of a signaling complex directly associated with some of the plasma membrane-located TLRs, such as TLR2 and TLR4 (reviewed in reference 1). Based on specific inhibition of IRAK1 protein depletion in cells stimulated with the TLR4 ligand LPS (Fig. 10B), we present evidence that inhibition is exerted proximal to the TLR complex. Consistent with poor inhibition of the cytokine response to the TLR2 ligand PG (Fig. 9), we did not detect Sm16-mediated inhibition of PG-induced IκB-α or IRAK1 degradation (Fig. 10). While we cannot explain the large difference in the potency with which Sm16 inhibits the response to LPS and PG, these differences still serve to ensure the specificity of the potent inhibition of the LPS response.
Sm16 appears to bind equally well to the surfaces of diverse cell types, which would also be expected if binding were primarily mediated by the demonstrated affinity of Sm16 for lipid bilayers (9). This affinity for lipid bilayers seems likely to be functionally relevant, and our previous fluorescence microscopy analysis of cells cultured with Alexa 488-labeled Sm16(23-117)AA has indeed revealed efficient plasma membrane binding, followed by Sm16 uptake by endocytosis. The truncated Sm16(23-70) protein was also included in the analysis, and in this case we did not detect plasma membrane binding or endocytosis (9), which is consistent with the present finding that Sm16(23-70) does not detectably inhibit the LPS response (Fig. 6).
Analysis of cells cultured with Alexa 488-labeled Sm16(23-117)AA indicated that all intracellular fluorescence was confined to intracellular vesicles, and we obtained no evidence that Sm16 penetrates lipid bilayers to escape from endocytotic vesicles to the cytosol (9). This apparent absence of membrane-penetrating properties in Sm16 is indeed consistent with our persistent failure to reproduce the apoptosis-promoting effect of cytosolic expression of Sm16 by adding Sm16 to the culture media of various types of human cells (no effect at 100 μg/ml Sm16; data not shown). Thus, we found no evidence that exogenous Sm16 gains access to the cytosol, which argues against a physiological significance of the previously noted apoptosis-promoting activity of Sm16 artificially introduced into the cytosol (9). Membrane binding in the absence of membrane penetration implies that Sm16 will remain on the extracellular side of cellular lipid bilayers.
We observed large differences in the potency of inhibition of the four distinct TLR ligands used in this study (Fig. 7 and 9), which suggests that Sm16 is not a general inhibitor of TLR signaling. However, it is notable that Sm16 is a potent inhibitor of the poly(I:C) response (Fig. 7 and 9), which is mediated by a distinct receptor only found in intracellular vesicles, namely, TLR3 (reviewed in reference 1). Given the efficiency with which membrane-bound Sm16 is taken up by endocytosis and the evidence of accumulation in intracellular vesicles (9), it seems reasonable to assume that Sm16 exerts its inhibition of TLR3 signaling from the endoplasmic luminal side of the vesicle by the same general mechanism by which Sm16 inhibits LPS signaling by plasma membrane-located TLR4. This implies that Sm16 may inhibit TLR function through some type of interference with ligand-induced TLR signaling. Such a mechanism of action appears consistent with our observation that various TLR ligands are inhibited with different degrees of potency.
Inhibition of TLR signaling during skin penetration may represent a strategy for avoidance of inflammatory reactions caused by tissue damage and/or cercaria-derived TLR ligands. There is indeed evidence of both TLR stimulatory ligands (reviewed in reference 14) and activities that suppress TLR function (20) among helminth parasites. However, to our knowledge, the present studies of the Sm16 protein provide the first example of TLR inhibition by a purified parasite-derived protein. The Sm16 protein is abundant (3 to 4% of acetabular gland secretions) (5), and the demonstrated binding to cell membranes (9) will prevent diffusion. Thus, there are reasons to assume local concentrations of Sm16 sufficiently high to suppress activation of the innate immune response in the dermis proximal to the site of cercarial entry.
Acknowledgments
We are indebted to Cecilia Thors, Swedish Institute for Infectious Disease Control, Stockholm, for providing cercariae.
This work was supported by the Swedish Research Council.
Editor: J. F. Urban, Jr.
Footnotes
Published ahead of print on 5 January 2009.
REFERENCES
- 1.Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124783-801. [DOI] [PubMed] [Google Scholar]
- 2.Cantrell, D. A. 2002. T-cell antigen receptor signal transduction. Immunology 105369-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, L., K. V. Rao, Y. X. He, and K. Ramaswamy. 2002. Skin-stage schistosomula of Schistosoma mansoni produce an apoptosis-inducing factor that can cause apoptosis of T cells. J. Biol. Chem. 27734329-34335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherfas, J. 1991. New hope for vaccine against schistosomiasis. Science 251630-631. [DOI] [PubMed] [Google Scholar]
- 5.Curwen, R. S., P. D. Ashton, S. Sundaralingam, and R. A. Wilson. 2006. Identification of novel proteases and immunomodulators in the secretions of schistosome cercariae that facilitate host entry. Mol. Cell. Proteomics 5835-844. [DOI] [PubMed] [Google Scholar]
- 6.Erickson, H. P. 2004. Protein-protein interactions. Chapter 1. Protein structure—size and shape at the nm level. http://www.cellbio.duke.edu/Faculty/Erickson/pdf's/Prot-p%20biophys%20Ch1.pdf.
- 7.Gigant, B., P. A. Curmi, C. Martin-Barbey, E. Charbaut, S. Lachkar, L. Lebeau, S. Siavoshian, A. Sobel, and M. Knossow. 2000. The 4 Å X-ray structure of a tubulin:stathmin-like domain complex. Cell 102809-816. [DOI] [PubMed] [Google Scholar]
- 8.Hervé, M., V. Angeli, E. Pinzar, R. Wintjens, C. Faveeuw, S. Narumiya, A. Capron, Y. Urade, M. Capron, G. Riveau, and F. Trottein. 2003. Pivotal roles of the parasite PGD2 synthase and of the host D prostanoid receptor 1 in schistosome immune evasion. Eur. J. Immunol. 332764-2772. [DOI] [PubMed] [Google Scholar]
- 9.Holmfeldt, P., K. Brannstrom, M. E. Sellin, B. Segerman, S. R. Carlsson, and M. Gullberg. 2007. The Schistosoma mansoni protein Sm16/SmSLP/SmSPO-1 is a membrane-binding protein that lacks the proposed microtubule-regulatory activity. Mol. Biochem. Parasitol. 156225-234. [DOI] [PubMed] [Google Scholar]
- 10.Holmfeldt, P., K. Brannstrom, S. Stenmark, and M. Gullberg. 2003. Deciphering the cellular functions of the Op18/Stathmin family of microtubule-regulators by plasma membrane-targeted localization. Mol. Biol. Cell 143716-3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins, S. J., J. P. Hewitson, G. R. Jenkins, and A. P. Mountford. 2005. Modulation of the host's immune response by schistosome larvae. Parasite Immunol. 27385-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin, Y.-C., K. Brown, and U. Siebenlist. 1995. Activation of NF-κB requires proteolysis of the inhibitor IκB-α: signal-induced phosphorylation of IκB-α alone does not release active NF-κB. Proc. Natl. Acad. Sci. USA 92552-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKerrow, J. H. 1997. Cytokine induction and exploitation in schistosome infections. Parasitology 115S107-S112. [DOI] [PubMed] [Google Scholar]
- 14.Perrigoue, J. G., F. Marshall, and D. Artis. 2008. On the hunt for helminths: innate immune cells in the recognition and response to helminth parasites. Cell Microbiol. 101757-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ram, D., F. Lantner, E. Ziv, V. Lardans, and I. Schechter. 1999. Cloning of the SmSPO-1 gene preferentially expressed in sporocyst during the life cycle of the parasitic helminth Schistosoma mansoni1. Biochim. Biophys. Acta 1453412-416. [DOI] [PubMed] [Google Scholar]
- 16.Ramaswamy, K., B. Salafsky, S. Potluri, Y. X. He, J. W. Li, and T. Shibuya. 1995. Secretion of an anti-inflammatory, immunomodulatory factor by schistosomulae of Schistosoma mansoni. J. Inflamm. 4613-22. [PubMed] [Google Scholar]
- 17.Rao, K. V., Y. X. He, and K. Ramaswamy. 2002. Suppression of cutaneous inflammation by intradermal gene delivery. Gene Ther. 938-45. [DOI] [PubMed] [Google Scholar]
- 18.Rao, K. V., and K. Ramaswamy. 2000. Cloning and expression of a gene encoding Sm16, an anti-inflammatory protein from Schistosoma mansoni. Mol. Biochem. Parasitol. 108101-108. [DOI] [PubMed] [Google Scholar]
- 19.Segerman, B., P. Holmfeldt, J. Morabito, L. Cassimeris, and M. Gullberg. 2003. Autonomous and phosphorylation-responsive microtubule-regulating activities of the N-terminus of Op18/stathmin. J. Cell Sci. 116197-205. [DOI] [PubMed] [Google Scholar]
- 20.Semnani, R. T., P. Goel Venugopal, C. A. Leifer, S. Mostböck, H. Sabzevari, and T. B. Nutman. 2008. Inhibition of TLR3 and TLR4 function and expression in human dendritic cells by helminth parasites. Blood 1121290-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel, L. M., and K. J. Monty. 1966. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta 112346-362. [DOI] [PubMed] [Google Scholar]
- 22.Steinmetz, M. O., R. A. Kammerer, W. Jahnke, K. N. Goldie, A. Lustig, and J. van Oostrum. 2000. Op18/stathmin caps a kinked protofilament-like tubulin tetramer. EMBO J. 19572-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valle, C., A. Festucci, A. Calogero, P. Macri, B. Mecozzi, P. Liberti, and D. Cioli. 1999. Stage-specific expression of a Schistosoma mansoni polypeptide similar to the vertebrate regulatory protein stathmin. J. Biol. Chem. 27433869-33874. [DOI] [PubMed] [Google Scholar]
- 24.Wallon, G., J. Rappsilber, M. Mann, and L. Serrano. 2000. Model for stathmin/OP18 binding to tubulin. EMBO J. 19213-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziegler-Heitbrock, H. W., W. Schraut, P. Wendelgass, M. Strobel, T. Sternsdorf, C. Weber, M. Aepfelbacher, M. Ehlers, C. Schutt, and J. G. Haas. 1994. Distinct patterns of differentiation induced in the monocytic cell line Mono Mac 6. J. Leukoc. Biol. 5573-80. [DOI] [PubMed] [Google Scholar]