

Abstract
Human immunodeficiency virus type 1 (HIV-1) Nef is a multifunctional protein that confers an ability to evade killing by cytotoxic T lymphocytes (CTLs) as well as other advantages to the virus in vivo. Here we exploited mathematical modeling and related statistical methods to estimate the impact of Nef activity on viral replication in vivo in relation to CTLs. Our results indicate that downregulation of major histocompatibility complex class I (MHC-I) A and B by wild-type Nef confers an advantage to the virus of about 82% in decreased CTL killing efficiency on average, meaning that abolishing the MHC-I downregulation function of Nef would increase killing by more than fivefold. We incorporated this estimate, as well as prior estimates of replicative enhancement by Nef, into a previously published model of HIV-1 and CTLs in vivo (W. D. Wick, O. O. Yang, L. Corey, and S. G. Self, J. Virol. 79:13579-13586, 2005), generalized to permit CTL recognition of multiple epitopes. A sequence database analysis revealed that 92.9% of HIV-1 epitopes are A or B restricted, and a previous study found an average of about 19 epitopes recognized (M. M. Addo et al., J. Virol. 77:2081-2092, 2003). We combined these estimates in the model in order to predict the impact of inhibiting Nef function in the general (chronically infected) population by a drug. The predicted impact on viral load ranged from negligible to 2.4 orders of magnitude, depending on the effects of the drug and the CTL dynamical scenario assumed. We conclude that inhibiting Nef could make a substantial reduction in disease burden, lengthening the time before the necessity of undertaking combination therapy with other antiretroviral drugs.
Nef is a protein that is unique to simian immunodeficiency virus (SIV) and human immunodeficiency virus (HIV) and is not found in other viruses (40). The importance of this protein in pathogenesis is clear from the striking attenuation of SIV- and HIV-1-induced disease in macaques and humans when Nef is defective (9, 11, 12, 13, 18, 26). However, the mechanism behind this phenomenon remains a topic of debate.
A bewildering plethora of cellular effects have been described, and it is unclear which of these play true physiologic roles in vivo, given the potential experimental artifacts of studying Nef expression in cells in vitro. Among the best-described effects of Nef is downregulation of the major histocompatibility complex class I (MHC-I) molecule from the surfaces of HIV-1-infected cells (14, 16). Given the pivotal protective role of HIV-1-specific CD8+ cytotoxic T lymphocytes (CTLs) in HIV-1 infection and the requirement for MHC-I presentation of viral epitopes for CTL function, it has been proposed that this is an important mechanism for viral immune evasion (8, 20).
Several studies of the interaction of HIV-1-specific CTLs with infected cells in vitro have confirmed that Nef confers relative resistance of HIV-1-infected cells to cytolysis by CTLs and interferes with the ability of CTLs to suppress viral replication (8, 42, 53). The in vivo impact of this mechanism, however, has been difficult to assess. Gross deletion of Nef affects several important functions (such as CD4 downregulation and activation of infected cells), and isolated point mutations that interfere with Nef function or expression are rapidly reverted (17, 23).
To date, one study has attempted to address the issue of MHC-I downregulation by Nef in vivo, using the SIV macaque model. Swigut et al. (41) devised mutations in SIV Nef that were difficult to revert (small deletions) and knocked out the MHC-I downregulatory function of Nef while preserving other functions. In that pilot study, four macaques were infected with SIV containing this defective Nef. After infection, a striking pattern of compensatory Nef evolution was observed, resulting in reconstitution of MHC-I downregulatory function through an alternative motif resembling that of HIV-1 Nef. Due to the small number of animals, however, it was impossible to assess the impact of this phenomenon on immune containment in the SIV-infected macaques.
Another hypothesized important effect of Nef is its ability to drive T-lymphocyte activation and to increase HIV-1 replication, given the dependence of the virus on host cell activation-dependent transcription factors. While this activity is not required for HIV-1 replication, particularly in immortalized T-cell lines, there is a marked upregulatory effect in primary cells. Nef boosts HIV-1 replication in primary peripheral blood mononuclear cells (PBMC) in vitro (5, 6, 34, 39) and has been observed to increase the target cell range in lymph nodes for SIV infection in vivo (40).
Thus, antagonizing Nef is a potential strategy to attenuate HIV-1 infection in vivo, by blocking the ability of Nef to downregulate MHC-I to circumvent CTL responses and/or blocking the ability of Nef to enhance HIV-1 replication, thereby reducing viremia and attenuating disease. Here we applied experimentally derived data on the reduction of CTL antiviral activity through Nef-mediated MHC-I downregulation in a mathematical model to explore the consequences of antagonizing Nef function in vivo.
MATERIALS AND METHODS
Assessment of Nef impact on HIV-1 inhibition by HIV-1-specific CTL clones.
Virus suppression assays comparing NL4-3.1 viruses containing wild-type Nef versus Nef with the M20A mutation (previously shown to selectively ablate MHC-I downregulation by Nef) were performed as previously described in detail (2). In brief, primary CTL clones (derived from the PBMC of HIV-1-infected persons) were cocultured with HIV-1-permissive T-cell lines that had been infected with NL4-3.1 containing wild-type Nef or NL4-3.1 containing M20A Nef at a ratio of 1:4. Enzyme-linked immunosorbent assay for p24 antigen was performed on about day 7, for comparison of viral replication in the absence and presence of CTLs.
Models and statistical techniques.
We derived estimates of the factor expressing increase in CTL activity (ICA) due to loss of MHC-I downregulation in Nef− mutants from in vitro data as follows. We employed a two-compartment linear ordinary differential equation (ODE) model of viral growth in vitro. (In 7 days, no evidence of saturation of the growth curve was observed, so the linear model was satisfactory.) Using X and Y for numbers of infected target cells in eclipse and productive phases, respectively, the ODE model is dX/dt = ιY − (η + δ)X; dY/dt = ηX − (δ + ξ)Y. Here, ι, η, and δ are, respectively, the infection rate, progression rate (inverse of eclipse period), and death rate other than from CTL killing (inverse of infected-cell lifetime). Variable ξ is the product of a coefficient, κ, representing killing efficiency of the CTL clone, times either the ICA, which we designate ƒ in equations, for the ΔNef virus or 1 for the wild-type virus. Absent CTL killing, the basic reproductive number, R0, is given by ιη/[δ(δ + η)], from which we can derive the infection rate constant, ι, given the other parameters. Let Nwt (no CTLs), Nwt (CTLs), Nmut (no CTLs), and Nmut (CTLs) denote the amount (picograms) of HIV p24 in a glass well infected by wild-type or mutant virus in the presence or absence of CTLs. These numbers were derived from separate experiments. Let rwt [calculated as Nwt (CTLs)/Nwt (no CTLs)] and rmut [calculated as Nmut (CTLs)/Nmut (no CTLs)] be the suppression ratios for the two viruses. In order to estimate the ICA ƒ, we utilized a regression setup, as follows. We assumed that the principal source of variation between experiments with the same clone resided in coefficient κ rather than measurement error. (Some CTL clones did not exhibit significant suppression of wild-type virus in some experiments.) Let function F(ξ) stand for V(ξ)/V(0), where V (assumed to be proportional to Y) is given by the analytical solution of the model equations at 7 days, with arbitrary initial conditions (does not affect the ratios), and other parameter values as follows: η = 1.0, δ = 0.3, and R0 = 7.0 (generates 1-log growth every 2.5 days; taken as standard in vitro). Defining G(·) as the functional inverse of F(·), we have the relationships G(rwt;i) = κmean + zi and G(rmut;i) = f(κmean + wi), where i (index) = 1, …, N indicates experiment number, and zi and wi represent independent, and identically distributed noise terms. The (maximum-likelihood) regression estimates derived from this setup are
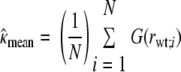 |
and
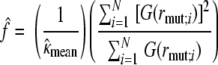 |
To construct confidence intervals, we combined a bootstrap technique with a sensitivity analysis. We stored residuals from the individual experiments as G(rwt;i) − κ̂mean = reswt;i and G(rmut;i)/f̂ − κ̂mean = resmut;i and resampled from all the residues of each type; then we derived new mock sets of ratios as r̃wt;i = F(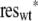 + κ̂mean and r̃mut;i = F(f̂
+ κ̂mean and r̃mut;i = F(f̂ 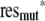 + f̂ κ̂mean), where a randomly resampled residual of the corresponding type was selected for each experiment. Finally, we redid the regression estimates using the bootstrapped data sets, simultaneously selecting nuisance parameters η, δ, and R0 independently from uniform distributions on [0.9; 1.2], [0.25; 0.3], and [5; 10], respectively. We repeated this procedure 200 times and determined the 0.025 and 0.975 percentiles; these are reported as 95% confidence intervals.
+ f̂ κ̂mean), where a randomly resampled residual of the corresponding type was selected for each experiment. Finally, we redid the regression estimates using the bootstrapped data sets, simultaneously selecting nuisance parameters η, δ, and R0 independently from uniform distributions on [0.9; 1.2], [0.25; 0.3], and [5; 10], respectively. We repeated this procedure 200 times and determined the 0.025 and 0.975 percentiles; these are reported as 95% confidence intervals.
The in vivo model of CTLs and HIV and the parameters are described in the appendix.
RESULTS
Estimation of Nef interference with the antiviral activity of HIV-1-specific CTLs.
To determine a modeling parameter for the impact of Nef on CTL antiviral activity, we utilized experimental data from an in vitro system. A previously reported data set (2) was analyzed to estimate the reduction of CTL activity by Nef when at least three independent measurements were available. MHC-I C-restricted CTLs were excluded from this analysis because Nef does not downregulate MHC-I C and therefore has no impact on the antiviral activity of those CTLs. Six MHC-I A- and B-restricted epitopes were examined. The estimation was carried out as explained in Materials and Methods.
Analysis of MHC-I restriction of known HIV-1 CTL epitopes.
Given that Nef does not downregulate MHC-I C molecules (7) and therefore has no effect on C-restricted CTLs (2), we estimated the distribution of A- and B- versus C-restricted CTL epitopes by analyzing the Los Alamos National Laboratory HIV Immunology Database. Scanning all epitopes for which MHC-I restriction was reported (as of 27 July 2007) (27), we found that 41.3% were A restricted, 51.7% were B restricted, and 7.1% were C restricted (Fig. 1). These percentages were similar to those reported in other studies (54, 24), which reported the relative dominance of MHC-I B and A over C.
FIG. 1.

Frequencies of MHC-I A, B, and C restriction of HIV-1 epitopes in the Los Alamos database.
Adjustment of an ODE model of CTL and HIV-1 interaction to include the influence of Nef in vivo.
We previously described a deterministic rate equation (ODE) model (47, 48, 49) of the in vivo relationship of CTLs with HIV-1, utilizing parameters derived from in vitro and in vivo experimental data (50). Whereas CTLs were previously considered a homogeneous population, we now expanded the model to include multiple distinct epitope-specific responses and MHC-I restrictions. Based on the known properties of MHC-I molecules and the described effects of Nef on MHC-I downregulation, these responses were assigned distinct activation and killing parameters based upon MHC-I A or B versus C restriction.
These parameters considered two MHC-I-specific observations. First, levels of MHC-I C complexes are 10-fold lower than levels of A or B complexes, due to either low concentrations of high-affinity epitopes for the MHC-I-C molecule (28) or preselection of peptides transported to nascent MHC-I molecules (43). However, we assumed that presented C-restricted epitopes had immunogenicity equivalent to that of A- and B-restricted epitopes (for the justification, see Discussion). Second, Nef downregulates cell surface MHC-I A and B but not C molecules (7) and has thus been shown to reduce the antiviral efficiency of A- and B- but not C-restricted HIV-1-specific CTLs (2).
Another aspect of Nef that was now incorporated into the model was Nef-mediated enhancement of HIV-1 replication in primary CD4+ T lymphocytes. A detailed study of HIV-1 replication in a single-cycle system demonstrated that Nef doubled replication (5) and thus suggested that R0 (replication over the 2-day life cycle of HIV-1) is increased by a factor of 2. Other studies reported growth curves, which allow estimation of the difference between R0 for wild-type and Nef-defective strains by approximation with a simple model for growth rate: (R0 − 1)/g, where g is generation time. Analysis of published data indicates that this difference ranges from 0.74 to 2.2; assuming that R0 is not less than 2.0 for a Nef-defective strain, this implies a somewhat smaller impact on replication (Table 1). These studies suggest that the effect of Nef is most pronounced in resting PBMC that are subsequently activated—as further demonstrated by the Nef independence of HIV-1 growth in immortalized cell lines (53). Thus, in vivo, this enhancement likely depends on the activation status of the target cells; with this caveat, we incorporated into some simulations a potential Nef enhancement effect of R0(nonfunctional Nef) = 0.5R0(functional Nef).
TABLE 1.
Estimates of a replicative enhancement factor
| Study |
R0
|
|
|---|---|---|
| Difference | Ratio (range)a | |
| Chowers et al. (6) | 2.2 | 1:36, 2:10 |
| Miller et al. (34) | 0.79 | 1:12, 1:37 |
| Spina et al. (39) | 0.74 | 1:31, 1:40 |
Assumes R0 of the Nef-defective strain in the interval [2:0, 6:0].
Use of this modified ODE model to predict the in vivo impact of blocking Nef.
This model was now utilized to derive theoretical estimates of the impact on viremia level (VL) and CTL frequencies that would result from antagonizing the various functions of Nef. We modeled the impact of blocking the effect of Nef on A- and B-restricted CTL epitopes, with or without also blocking direct enhancement of replication, in several in vivo scenarios of CD8 T-cell activation and activity. The negative effect of MHC-I downregulation on CTL killing of target cells was assumed to be a factor of 5.6 (Table 2) (i.e., about 82% loss, relative to a Nef-defective strain) for A- and B-restricted epitopes and none for C-restricted epitopes. We considered two previously described “defect” scenarios for CTL functionality in vivo (48), which we call here defective memory (DM) and defective killing (DK); they were motivated by studies of immunological dysfunction and the evident inability of CTLs to clear an HIV infection. In the former, HIV is hypothesized to be controlled by a pool of short-lived effector-memory CTLs; in the latter, long-lived resting memory cells form but activation or killing rates are diminished relative to levels seen for infections by other viruses. Both models are capable of replicating the observed timing and response levels seen in primary HIV viremia. (For the mathematical details, see the appendix.) We also considered two hypotheses about the impact of Nef on resting CD8 T-cell activation. As downregulation of MHC-I by Nef is not observed in dendritic cells (8, 31, 45, 52), no effect of Nef was assumed for naïve-cell activation (which requires an antigen-presenting cell). For memory cell activation, we permitted two scenarios: no impact of Nef on activation of resting memory cells (NIM) and the same impact (SIM) as on killing, because memory CD8 T cells may not require an antigen-presenting cell to become activated but rather are activated directly by encounter with a target cell displaying antigen.
TABLE 2.
Estimates of Nef interference with CTL activity
| na | Epitope | Protein | Restriction | ICAb | CIc |
|---|---|---|---|---|---|
| 8 | ILKEPVHGU | RTd | A*02 | 8.29 | 2.76, 14.4 |
| 15 | SLYNTVATL | Gagp17 | A*02 | 8.08 | 3.93, 11.7 |
| 3 | HTQGYFPDW | Nef | B*57 | 2.47 | 0.77, 6.76 |
| 4 | YFPDWQNYT | Nef | B*57 | 2.12 | 0.91, 3.82 |
| 3 | KAFSPEVIPMI | Gag | B*57 | 2.34 | 0.74, 5.56 |
| 4 | RPAEPVPLQL | Rev | B*07 | 9.36 | 3.39, 40.7 |
| 37 | 5.59 | 3.92, 7.37 |
Number of experiments.
Estimated.
Confidence interval (95% bootstrap statistical and model sensitivity confidence).
RT, reverse transcriptase.
First, we modeled the effect of blocking Nef activity in a hypothetical individual with a typical number of CTL epitope responses of 19 (1), including one C-restricted epitope. (The activation and killing parameters were assigned randomly [see below]; other parameters in the model are listed in Tables A1 and A2 in the appendix.) Figure 2 was generated assuming the DM scenario and shows the effect on VL of a 100%-effective drug, given at day 100, that interferes with MHC-I downregulation by Nef in infected target cells but without assuming an enhancement effect on R0. [VL is expressed as log(infected cells), rather than the more-usual virions per ml.] Figure 2a shows no impact on activation in memory cells (NIM; in model terms, the ICA factor applies to the killing but not the activation parameter); Fig. 2b shows the same impact on activation in memory cells as for killing target cells (SIM; ICA applies to both). Figure 3a and b show similar curves for the DK scenario. Figures 4 and 5 illustrate the predicted impact of the drug, assuming now a 50% enhancement of replication by Nef, blocked by the drug.
TABLE A1.
Parameters in the in vivo infection model
| Parameter | Symbol | Value (for rates, per day) |
|---|---|---|
| Basic reproductive no. | R0 | 6.0 |
| IT nonimmune death | δ | 0.33 |
| Immune killing | κ | (2-5) × 10−10 |
| Progression | η | 1.0 |
| Infection | ι | See reference 2 |
aIT, infected T cell.
TABLE A2.
Parameters in the in vivo CD8 modelb
| Parameter | Symbol | Value (for rates, per day) |
|---|---|---|
| Resting activation | α | (2-5) × 10−10 |
| NR “immigration” | β | See text |
| NR death rate | δNR | 0.00017 |
| MR death rate | δMR | 0.00017 or 0.333 |
| CTL death rate | δCTL | 0.333 |
| Reversion fraction | Revert | 0.05 |
| No. of doublings | ηd | 8 |
| Naïve cell cycles/day | 2.0 | |
| Memory cell cycles/day | 4.0 | |
| Naïve cell cycles before ES | 4 | |
| Memory cell cycles before ES | 1 | |
| Memory speed-up factor | 7.0 |
Defined quantities: NR, naïve resting cells; MR, memory resting cells; ηd, number of programmed-proliferation divisions; ES, effector status.
FIG. 2.

Impact in vivo of blocking MHC-I downregulation by Nef (DM scenario). (a) NIM scenario; (b) SIM scenario.
FIG. 3.

Impact in vivo of blocking MHC-I downregulation by Nef (DK scenario). (a) NIM scenario; (b) SIM scenario.
FIG. 4.

Impact in vivo of blocking MHC-I downregulation and enhanced replication by Nef (DM scenario). (a) NIM scenario; (b) SIM scenario.
FIG. 5.

Impact in vivo of blocking MHC-I downregulation and enhanced replication by Nef (DK scenario). (a) NIM scenario; (b) SIM scenario.
We then generalized the model to predict the likely benefit across a typical population of individuals that were assigned a random (binomial) distribution of epitopes modeled after that in untreated, chronically infected individuals, reported by Addo et al. (1) to have a median of 18.5, with a range of 8 to 42. The restriction of each epitope was also chosen randomly, with probabilities of 0.929 for A and B and 0.071 for C. In order to incorporate the observed half-log variation in steady-state VL observed in a population study (30), we randomized the immune parameters, multiplying activation and killing parameters by a log-normal random variable with a mean of 1 and standard deviation of 8 (we increased the latter relative to another study [47] because the larger number of epitopes produced an averaging effect on VL). The factor ICA produced by blocking Nef downregulation of MHC-I A and B was similarly randomized to have the mean reported in Table 2 (last line) and related standard deviation (0.76; not shown in the table). We proceeded to simulate 200 times (dropping runs in which VL was uncontrolled) under each of various combinations of assumptions about MHC-I downregulation of killing and/or activation of CTLs recognizing MHC-I A- or B-restricted epitopes, infectivity enhancement, and in vivo CTL scenario (DM or DK) (Table 3).
TABLE 3.
Theoretical impact of an anti-Nef drug on VL, from a simulated population study
| CSa | ASb | RFc | ICA | ΔlogVL
|
|
|---|---|---|---|---|---|
| Mean | SE | ||||
| 1 | 1 | 1.0 | 5.6 | 0.0021 | 0.0034 |
| 1 | 1 | 0.5 | 5.6 | 0.18 | 0.0083 |
| 1 | 1 | 0.5 | 1.0 | 0.18 | 0.0074 |
| 1 | 2 | 1.0 | 5.6 | 1.29 | 0.50 |
| 1 | 2 | 0.5 | 5.6 | 1.37 | 0.53 |
| 1 | 2 | 0.5 | 1.0 | 0.18 | 0.0078 |
| 2 | 1 | 1.0 | 5.6 | 0.62 | 0.17 |
| 2 | 1 | 0.5 | 5.6 | 1.20 | 0.18 |
| 2 | 1 | 0.5 | 1.0 | 0.63 | 0.021 |
| 2 | 2 | 1.0 | 5.6 | 1.81 | 0.84 |
| 2 | 2 | 0.5 | 5.6 | 2.4 | 0.81 |
| 2 | 2 | 0.5 | 1.0 | 0.63 | 0.022 |
CTL scenario: 1, DM; 2, DK.
Activation scenario: 1, downregulation of killing only; 2, downregulation also of activation in memory cells.
Replication factor (multiplies R0 in the Nef-defective strain).
DISCUSSION
Despite strong clinical evidence for a central enhancing role for Nef in the immunopathogenicity of HIV-1 infection, the precise mechanisms remain obscure. Among the multitude of effects that Nef has on cells, it is unclear to what extent individual effects might affect pathogenesis in vivo. In this study, we modeled the potential impact of blocking two well-defined Nef effects: downregulation of the MHC-I molecule, leading to immune evasion of CTLs, and enhancement of HIV-1 replication in primary CD4+ T lymphocytes.
With respect to CTL functionality in vivo, we considered the DM and DK scenarios, adapted from reference 48. In the former, HIV-1 replication is controlled in the steady-state period by a pool of short-lived effector-memory cells; long-lived, resting memory cells are assumed not to be formed. Other scenarios, which permit the formation of long-lived central memory cells but assume a defect in activation or killing efficiency of HIV-specific CTLs (relative to normal CTLs, which eradicate other viral infections), can also produce the 2- to 3-log drop in VL observed after primary viremia. In the study reported in reference 48, an experiment was proposed (adoptive transfer of carboxyfluorescein succinimidyl ester-labeled SIV-specific CTLs to vaccinated and unvaccinated macaques) that might distinguish the cases, but to our knowledge it has not been performed. However, the observation that HIV-1-specific CTL responses tend to rapidly decay after epitope escape mutation (38, 19) or antiretroviral drug suppression of HIV-1 replication (22, 37) lends more support to the DM scenario, and our model incorporating this scenario yields more realistic viremia peaks and steady-state levels.
We ran models varying these assumptions. Assuming blocking of Nef effects on CTL activation and killing and direct viral replication enhancement, the DM scenario predicted a drop in viremia of 1.4 orders of magnitude, and the DK scenario predicted a drop of 2.4 orders of magnitude. Interestingly, and perhaps surprisingly, blocking of the Nef effect on CTL killing efficiency alone in the DM scenario yielded only a small and transient drop; this phenomenon results from the nonlinear dynamics that establishes steady-state viremia. These results suggest that both of these functions may have key roles in Nef enhancement of HIV-1 pathogenesis.
Our model incorporated the assumption that fewer HLA-I C epitopes are presented on the surface of infected cells but that those presented are as immunogenic as A and B epitopes, based on several biological observations. First, the cell surface expression of HLA-I C has been noted to be about a tenth of that of HLA-I B/C molecules, due to differences in intracellular tracking (36, 55). Second, CTLs can be highly sensitive for recognizing their cognate epitope, requiring as few as 10 peptide-HLA complexes on the cell surface for triggering of the T-cell receptor (21). Third, the relative abundance of HLA-I C versus A and B molecules on the cell surface (36, 55) correlates well with the previously reported distribution of C- versus A- and B-restricted HIV-1-specific CTL epitopes in detailed studies of infected persons (24, 54) and across all reported HIV-1 CTL epitopes (see Results). Finally, although the frequencies of appearance of epitopes in the Los Alamos database may be questioned as being subject to reporting bias, the distribution of A, B, and C epitopes matches the detailed studies of individuals we have cited, and the laboratory methods for detecting epitope specificities are not intrinsically biased, using the same methodology for A versus B versus C.
We have assumed 100%-efficient blocking of Nef activity in the various modeled scenarios. A less efficient blockade would have a smaller impact; in our simulations, the order of magnitude of diminishment in viremia was roughly proportional to the efficiency of the drug. Thus, a drug that was 50% efficient at blocking MHC-I downregulation or HIV-1 enhancement functions of Nef would be predicted to mediate about half the log unit reduction in viremia of a fully efficient drug.
There are at least two reasons that our models may underestimate the impact of Nef on immune control of HIV-1 in vivo. First, Nef may not only enhance virion production by infected cells but also increase the cell range of the virus by activating otherwise insufficiently activated cells to promote replication (40). Thus, blocking the enhancing activity of Nef may have a greater impact on HIV-1 replication than described in the models. Second, reduction of HIV-1 replication may indirectly improve the antiviral activity of CTLs, through increased preservation of CD4+ helper cells and/or reduced immune activation, and thus, CTL function may not be constant, as assumed in the models.
Another point that could not be easily considered in our models is the observation that Nef activities probably vary depending on the stage of disease. For example, the MHC-I-downregulatory ability of Nef appears to be lost in immunosuppressed pediatric and late-stage AIDS patients (4, 15, 25, 46) and varies according to the breadth of the CTL response in vivo (29). Nef therefore appears to adapt its function to various selective pressures, and its activities are not fixed; in particular, the MHC-I downregulation function may be traded off to some degree for enhanced viral replication (3). This suggests that blockade of certain activities such as MHC-I downregulation may have degrees of impact that vary according to the clinical status of a patient. In late-stage disease (AIDS), the ICA may decrease toward 1 and the RF may increase; since most of the theoretical impact of blocking Nef (Table 3) arises from the first factor, the impact on viral load may be lowered (by as much as several logs).
Because disease progression is tightly linked with VL (33), reducing viremia by blocking the activities of Nef in vivo could offer a therapeutic avenue to retard disease in HIV-1-infected persons. Based on observations regarding the rate of peripheral blood CD4+-T-cell decline and VL (32), a 1.4- to 2.4-order-of-magnitude drop in viremia through Nef blockade would translate to a reduction in the rate of CD4+-T-cell decline by at least 40 cells/mm3 per year. This would delay the onset of need for antiretroviral therapy (currently recommended at a drop to a CD4+-T-cell count of 350 cells/mm3 blood or lower) and/or development of clinical AIDS by years for the average infected individual. Given the problems of increasing drug resistance and negative metabolic sequelae of antiretroviral drug treatment, such a delay could have a great impact on the clinical management of HIV-1.
Development of Nef inhibitors as therapeutic approaches would certainly face the major hurdle of HIV-1 resistance mutation, given the high sequence variability of Nef. Still, some of the most potent antiretroviral drugs currently available are highly susceptible to resistance through point mutations in their targets but remain effective when used in proper combinations. Thus, it is likely that a Nef inhibitor would require administration in combination with other drugs that reduce viral replication to limit the emergence of resistance mutations.
In summary, we have modeled the effects of blocking two major Nef activities in vivo, finding that such a blockade could reduce viremia by more than an order of magnitude through direct reduction of viral replication and in creased efficiency of CTL responses. The results suggest that designing a drug targeted against Nef could have significant clinical effects in delaying disease progression. In addition, having such a molecule could also be helpful to experimentally distinguish between the DM and DK models. Seeking Nef antagonists could therefore be a worthwhile pursuit for clinical and research purposes.
Acknowledgments
This work was supported by NIAID grant AI054280 (W.D.W.) and grants AI051970 and AI043203 (O.O.Y.) from the National Institutes of Health.
APPENDIX
For the HIV infection process we assumed the two-compartment model (1). The parameters, before randomization, were as shown in Table A1. The model of HIV-specific CD8s in vivo was introduced in reference 48. In brief, the model has compartments representing naïve and memory, resting and activated, and effector status of cells and compartments for cells in the cell cycle that are derived from an activated cell that has undergone a number of divisions. We assumed the programmed-proliferation scenario (activated cells undergo at least eight divisions without the necessity of re-encountering antigen). The thymopoiesis rate (β in Table A2) was chosen to establish before infection a steady-state precursor CD8 compartment of 105 naïve, resting, HIV-specific cells (per epitope recognized).
For this work, the DM model was defined as follows: α and κ = 5 × 10−10 (omitting downregulation or randomization factors) and δMR = 0.33. The DK model was defined as follows: α and κ = 2 × 10−10 and δMR = 0.00017. We generalized the model to include arbitrary numbers of epitopes. The killing parameter, κe (and, in the second activation scenario, the parameter αe for memory cells), associated with an epitope was diminished by 1 over the ICA factor for MHC-I A- and B-restricted epitopes and Nef functional strains.
Footnotes
Published ahead of print on 17 December 2008.
REFERENCES
- 1.Addo, M. M., X. G. Yu, A. Rathod, D. Cohen, R. L. Eldridge, D. Strick, M. N. Johnston, C. Corcoran, A. G. Wurcel, C. A. Fitzpatrick, M. E. Feeney, W. R. Rodriguez, N. Basgoz, R. Draenert, D. R. Stone, C. Brander, P. J. Goulder, E. S. Rosenberg, M. Altfeld, and B. D. Walker. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 772081-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adnan, S., A. Balamurugan, A. Trocha, M. S. Bennett, H. L. Ng, A. Ali, C. Brander, and O. O. Yang. 2006. Nef interference with HIV-1-specific CTL antiviral activity is epitope specific. Blood 1083414-3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carl, S., T. C. Greenough, M. Krumbiegel, M. Greenberg, J. Skowronski, J. L. Sullivan, and F. Kirchhoff. 2001. Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J. Virol. 753657-3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmichael, A., X. Jin, P. Sissons, and L. Borysiewicz. 1993. Quantitative analysis of the human immunodeficiency virus type 1 (HIV-1)-specific cytotoxic T lymphocyte (CTL) response at different stages of HIV-1 infection: differential CTL responses to HIV-1 and Epstein-Barr virus in late disease. J. Exp. Med. 177249-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowers, M. Y., C. A. Spina, T. J. Kwoh, N. J. Fitch, D. D. Richman, and J. C. Guatelli. 1994. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 682906-2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chowers, M. Y., M. W. Pandori, C. A. Spina, D. D. Richman, and J. C. Guatelli. 1995. The growth advantage conferred by HIV-1 nef is determined at the level of viral DNA formation and is independent of CD4 down regulation. Virology 212451-457. [DOI] [PubMed] [Google Scholar]
- 7.Cohen, G. B., R. T. Gandhi, D. M. Davis, O. Mandelboim, B. K. Chen, J. L. Strominger, and D. Baltimore. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10661-671. [DOI] [PubMed] [Google Scholar]
- 8.Collins, K. L., and D. Baltimore. 1999. HIV's evasion of the cellular immune response. Immunol. Rev. 16865-74. [DOI] [PubMed] [Google Scholar]
- 9.Couillin, I., B. Culmann-Penciolelli, E. Gomard, J. Choppin, J. P. Levy, J. G. Guillet, and S. Saragosti. 1994. Impaired cytotoxic T lymphocyte recognition due to genetic variations in the main immunogenic region of the human immunodeficiency virus 1 NEF protein. J. Exp. Med. 1801129-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cramer, L. A., and J. A. Frelinger. 2001. Dendritic cells transduced with HIV Nef express normal levels of HLA-A and HLA-B class I molecules. J. Acquir. Immune. Defic. Syndr. 27417-425. [DOI] [PubMed] [Google Scholar]
- 11.Daniel, M. D., F. Kirchhoff, S. C. Czajak, P. K. Sehgal, and R. C. Desrosiers. 1992. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 2581938-1941. [DOI] [PubMed] [Google Scholar]
- 12.Deacon, N. J., A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting, D. J. Hooker, D. A. McPhee, A. L. Greenway, A. Ellett, C. Chatfield, V. A. Lawson, S. Crowe, A. Maerz, S. Sonza, J. Learmont, J. S. Sullivan, A. Cunningham, D. Dwyer, D. Dowton, and J. Mills. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270988-991. [DOI] [PubMed] [Google Scholar]
- 13.Dyer, W. B., A. F. Geczy, S. J. Kent, L. B. McIntyre, S. A. Blasdall, J. C. Learmont, and J. S. Sullivan. 1997. Lymphoproliferative immune function in the Sydney Blood Bank Cohort, infected with natural nef/long terminal repeat mutants, and in other long-term survivors of transfusion acquired HIV-1 infection. AIDS 111565-1574. [DOI] [PubMed] [Google Scholar]
- 14.Foster, J. L., and J. V. Garcia. 2006. HIV pathogenesis: Nef loses control. Cell 1251034-1035. [DOI] [PubMed] [Google Scholar]
- 15.Geffin, R., D. Wolf, R. Müller, M. D. Hill, E. Stellwag, M. Freitag, G. Sass, G. B. Scott, and A. S. Baur. 2000. Functional and structural defects in HIV type 1 nef genes derived from pediatric long-term survivors. AIDS Res. Hum. Retrovir. 161855-1868. [DOI] [PubMed] [Google Scholar]
- 16.Geyer, M., O. T. Fackler, and B. M. Peterlin. 2001. Structure-function relationships in HIV-1 Nef. EMBO Rep. 2580-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glushakova, S., J. Munch, S. Carl, T. C. Greenough, J. L. Sullivan, L. Margolis, and F. Kirchhoff. 2001. CD4 down-modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4+ T-cell depletion in human lymphoid tissue ex vivo. J. Virol. 7510113-10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, Y., L. Zhang, and D. D. Ho. 1995. Characterization of nef sequences in long-term survivors of human immunodeficiency virus type 1 infection. J. Virol. 6993-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamieson, B. D., O. O. Yang, L. Hultin, M. A. Hausner, P. Hultin, J. Matud, K. Kunstman, S. Killian, J. Altman, K. Kommander, B. Korber, J. Giorgi, and S. Wolinsky. 2003. Epitope escape mutation and decay of human immunodeficiency virus type 1-specific CTL responses. J. Immunol. 1715372-5379. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, W. E., and R. C. Desrosiers. 2002. Viral persistence: HIV's strategies of immune system evasion. Annu. Rev. Med. 53499-518. [DOI] [PubMed] [Google Scholar]
- 21.Kageyama, S., T. J. Tsomides, Y. Sykulev, and H. N. Eisen. 1995. Variations in the number of peptide-MHC class I complexes required to activate cytotoxic T cell responses. J. Immunol. 154567-576. [PubMed] [Google Scholar]
- 22.Kalams, S. A., P. J. Goulder, A. K. Shea, N. G. Jones, A. K. Trocha, G. S. Ogg, and B. D. Walker. 1999. Levels of human immunodeficiency virus type 1-specific cytotoxic T-lymphocyte effector and memory responses decline after suppression of viremia with highly active antiretroviral therapy. J. Virol. 736721-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kestler, H. W., III, D. J. Ringler, K. Mori, D. L. Panicali, P. K. Sehgal, M. D. Daniel, and R. C. Desrosiers. 1991. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65651-662. [DOI] [PubMed] [Google Scholar]
- 24.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder. 2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432769-775. [DOI] [PubMed] [Google Scholar]
- 25.Kirchhoff, F., P. J. Easterbrook, N. Douglas, M. Troop, T. C. Greenough, J. Weber, S. Carl, J. L. Sullivan, and R. S. Daniels. 1999. Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J. Virol. 735497-5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirchhoff, F., T. C. Greenough, D. B. Brettler, J. L. Sullivan, and R. C. Desrosiers. 1995. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332228-232. [DOI] [PubMed] [Google Scholar]
- 27.Korber, B. T. M., C. Brander, B. F. Haynes, R. Koup, J. P. Moore, B. D. Walker, and D. I. Watkins. 2007 2006. HIV molecular immunology. LA-UR 07-4752. Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, NM.
- 28.Kość, A., J. Dubis, I. Wojciechowska, Z. Makiewicz, W. Gorczyca, A. Myc, G. Kupryszewski, M. Maczak, P. Myc, and P. Kunierczyk. 1998. Studies on binding of HIV-1 p24gag peptide to HLA-Cw3+ cells. Immunol. Lett. 6457-62. [DOI] [PubMed] [Google Scholar]
- 29.Lewis, M. J., A. Balamurugan, A. Ohno, S. Kilpatrick, H. L. Ng, and O. O. Yang. 2008. Functional adaptation of Nef to the immune milieu of HIV-1 infection in vivo. J. Immunol. 1804075-4081. [DOI] [PubMed] [Google Scholar]
- 30.Lyles, R. H., A. Muoz, T. E. Yamashita, H. Bazmi, R. Detels, C. R. Rinaldo, J. B. Margolick, J. P. Phair, and J. W. Mellors. 2000. Natural history of human immunodeficiency virus type 1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. Multicenter AIDS cohort study. J. Infect. Dis. 181872-880. [DOI] [PubMed] [Google Scholar]
- 31.Maccormac, L. P., J. M. Jacque, and B. Chain. 2004. The functional consequences of delivery of HIV-1 Nef to dendritic cells using an adenoviral vector. Vaccine 22528-535. [DOI] [PubMed] [Google Scholar]
- 32.Mellors, J. W., A. Muoz, J. V. Giorgi, J. B. Margolick, C. J. Tassoni, P. Gupta, L. A. Kingsley, J. A. Todd, A. J. Saah, R. Detels, J. P. Phair, and C. R. Rinaldo, Jr. 1997. Plasma viral load and CD4+ lymphocytes as prognostic markers of HIV-1 infection. Ann. Intern. Med. 126946-954. [DOI] [PubMed] [Google Scholar]
- 33.Mellors, J. W., C. R. Rinaldo, Jr., P. Gupta, R. M. White, J. A. Todd, and L. A. Kingsley. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 2721167-1170. [DOI] [PubMed] [Google Scholar]
- 34.Miller, M. D., M. T. Warmerdam, I. Gaston, W. C. Greene, and M. B. Feinberg. 1994. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med. 179101-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reference deleted.
- 36.Neisig, A., C. J. Melief, and J. Neefjes. 1998. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J. Immunol. 160171-179. [PubMed] [Google Scholar]
- 37.Ogg, G. S., X. Jin, S. Bonhoeffer, P. Moss, M. A. Nowak, S. Monard, J. P. Segal, Y. Cao, S. L. Rowland-Jones, A. Hurley, M. Markowitz, D. D. Ho, A. J. McMichael, and D. F. Nixon. 1999. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J. Virol. 73797-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price, D. A., P. J. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 941890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spina, C. A., T. J. Kwoh, M. Y. Chowers, J. C. Guatelli, and D. D. Richman. 1994. The importance of nef in the induction of human immunodeficiency virus type 1 replication from primary quiescent CD4 lymphocytes. J. Exp. Med. 179115-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugimoto, C., K. Tadakuma, I. Otani, T. Moritoyo, H. Akari, F. Ono, Y. Yoshikawa, T. Sata, S. Izumo, and K. Mori. 2003. nef gene is required for robust productive infection by simian immunodeficiency virus of T-cell-rich paracortex in lymph nodes. J. Virol. 774169-4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swigut, T., L. Alexander, J. Morgan, J. Lifson, K. G. Mansfield, S. Lang, R. P. Johnson, J. Skowronski, and R. Desrosiers. 2004. Impact of Nef-mediated downregulation of major histocompatibility complex class I on immune response to simian immunodeficiency virus. J. Virol. 7813335-13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomiyama, H., H. Akari, A. Adachi, and M. Takiguchi. 2002. Different effects of Nef-mediated HLA class I down-regulation on human immunodeficiency virus type 1-specific CD8+ T-cell cytolytic activity and cytokine production. J. Virol. 767535-7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong, J. C., Z. H. Zhang, J. T. August, V. Brusic, T. W. Tan, and S. Ranganathan. 2007. In silico characterization of immunogenic epitopes presented by HLA-Cw*0401. Immunome Res. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trono, D. 1995. HIV accessory proteins: leading roles for the supporting cast. Cell 82189-192. [DOI] [PubMed] [Google Scholar]
- 45.Verhasselt, B., E. Naessens, C. Verhofstede, M. De Smedt, S. Schollen, T. Kerre, D. Vanhecke, and J. Plum. 1999. Human immunodeficiency virus nef gene expression affects generation and function of human T cells, but not dendritic cells. Blood 942809-2818. [PubMed] [Google Scholar]
- 46.Walker, P. R., M. Ketunuti, I. A. Choge, T. Meyers, G. Gray, E. C. Holmes, and L. Morris. 2007. Polymorphisms in Nef associated with different clinical outcomes in HIV type 1 subtype C-infected children. AIDS Res. Hum. Retrovir. 23204-215. [DOI] [PubMed] [Google Scholar]
- 47.Wick, W. D., P. B. Gilbert, and S. G. Self. 2006. On modeling HIV and T cells in vivo: assessing causal estimators in vaccine trials. PLoS Comput. Biol. 2e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wick, D., and S. G. Self. 2002. What's the matter with HIV-directed killer T-cells? J. Theor. Biol. 21919-31. [PubMed] [Google Scholar]
- 49.Wick, D., and S. G. Self. 2005. How fast can HIV escape from immune control? In W.-Y. Tan and H. Wu (ed.), Deterministic and stochastic models of AIDS epidemics and HIV infections with intervention. World Scientific, Singapore.
- 50.Wick, W. D., O. O. Yang, L. Corey, and S. G. Self. 2005. How many human immunodeficiency virus type 1-infected target cells can a cytotoxic T lymphocyte kill? J. Virol. 7913579-13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reference deleted.
- 52.Yamamoto, T., M. Isogai, K. Otake, and Y. Tsunetsugu-Yokota. 2006. High and inducible expression of human immunodeficiency virus type 1 (HIV-1) Nef by adenovirus vector does not disturb potent antigen presentation by monocyte-derived dendritic cells. Microbes Infect. 82522-2530. [DOI] [PubMed] [Google Scholar]
- 53.Yang, O. O., P. T. Nguyen, S. A. Kalams, T. Dorfman, H. G. Göttlinger, S. Stewart, I. S. Chen, S. Threlkeld, and B. D. Walker. 2002. Nef-mediated resistance of human immunodeficiency virus type 1 to antiviral cytotoxic T lymphocytes. J. Virol. 761626-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu, X. G., M. M. Addo, E. S. Rosenberg, W. R. Rodriguez, P. K. Lee, C. A. Fitzpatrick, M. N. Johnston, D. Strick, P. J. Goulder, B. D. Walker, and M. Altfeld. 2002. Consistent patterns in the development and immunodominance of human immunodeficiency virus type 1 (HIV-1)- specific CD8+ T-cell responses following acute HIV-1 infection. J. Virol. 768690-8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zemmour, J., and P. Parham. 1992. Distinctive polymorphism at the HLA C locus: implications for the expression of HLA-C. J. Exp. Med. 176937-950. [DOI] [PMC free article] [PubMed] [Google Scholar]