

Abstract
Cellular homeostasis depends on an intricate balance of protein expression and degradation. The ubiquitin-proteasome pathway plays a crucial role in specifically targeting proteins tagged with ubiquitin for destruction. This degradation can be effectively blocked by both chemically synthesized and natural proteasome inhibitors. Poxviruses encode a number of proteins that exploit the ubiquitin-proteasome system, including virally encoded ubiquitin molecules and ubiquitin ligases, as well as BTB/kelch proteins and F-box proteins, which interact with cellular ubiquitin ligases. Here we show that poxvirus infection was dramatically affected by a range of proteasome inhibitors, including MG132, MG115, lactacystin, and bortezomib (Velcade). Confocal microscopy demonstrated that infected cells treated with MG132 or bortezomib lacked viral replication factories within the cytoplasm. This was accompanied by the absence of late gene expression and DNA replication; however, early gene expression occurred unabated. Proteasomal inhibition with MG132 or bortezomib also had dramatic effects on viral titers, severely blocking viral replication and propagation. The effects of MG132 on poxvirus infection were reversible upon washout, resulting in the production of late genes and viral replication factories. Significantly, the addition of an ubiquitin-activating enzyme (E1) inhibitor had a similar affect on late and early protein expression. Together, our data suggests that a functional ubiquitin-proteasome system is required during poxvirus infection.
The Poxviridae family is a large family of DNA viruses that replicate entirely within the cytoplasm of the cell. The best-characterized member of the poxvirus family is vaccinia virus, a member of the Orthopoxvirus genus, which also includes ectromelia virus (the causative agent of mousepox), cowpox virus, monkeypox virus, and variola virus, which caused the devastating illness smallpox (35, 57). Vaccinia virus was successfully employed in a vaccination program resulting in the eventual eradiation of smallpox (57). Despite the successful eradication of variola virus, poxvirus infections continue to elicit clinically relevant diseases in humans and other animals (20, 27, 30, 33, 41, 42). Aspects of the poxvirus life cycle and virus-host interaction are active areas of research, since efforts to improve and expand poxvirus-based therapies are often hampered by our incomplete understanding of poxvirus biology.
The poxvirus replication cycle is complicated due to the existence of two infectious forms of the virus, intracellular mature virus (IMV) and extracellular enveloped virus (EEV), which differ in the numbers of phospholipid bilayers surrounding their cores (56, 58). Upon infection, both IMV and EEV release virion cores into the cytosol. Early viral mRNA is synthesized within viral cores, and these typically encode products required for immune evasion, core uncoating, release of genomic DNA, and DNA replication (35). Late gene synthesis follows DNA replication, producing both structural and nonstructural proteins that initiate virion assembly, a process that also takes place in the cytoplasm (35). Viral DNA replication, as well as intermediate and late gene transcription, occurs in perinuclear sites within the cytoplasm referred to as “viral factories” (26). Unsuccessful viral DNA replication, as in the presence of the DNA synthesis inhibitor cytosine arabinose (AraC), results in failure to initiate late gene transcription (3, 12). Following a series of morphological changes and the acquisition of genomic viral DNA, immature virions mature to form fully infectious IMV, and a proportion of IMV is further wrapped by additional lipid bilayers derived from the trans-Golgi network and subsequently released from the cell as EEV (56, 58).
The ability of poxviruses to undergo DNA replication and assembly within the cytoplasm is a unique feature among DNA viruses and requires extensive regulation and modulation of many cellular systems. Aside from the desire to address the potential health threats that some poxviruses pose, much interest in poxvirus research stems from the ability of these viruses to employ a vast array of strategies to regulate the immune response and host cell signaling pathways (24, 53). One such cellular system known to have a role in poxvirus infection is the ubiquitin-proteasome pathway. The ubiquitin-proteasome system tightly regulates important cellular functions, such as the cell cycle, transcription, antigen presentation, signal transduction, and DNA repair (44, 45, 65). The activation of ubiquitin by the E1 ubiquitin-activating enzyme, followed by the covalent addition of ubiquitin onto substrates by a variety of ubiquitin ligases, results in either protein degradation or modification of protein function in the absence of degradation (44, 45, 65). Proteins tagged for degradation are targeted to the 26S proteasome, a multicatalytic protease consisting of a 20S catalytic core capped at both ends by 19S regulatory subunits (14, 18). In this way, cells can regulate protein levels and efficiently degrade aberrantly folded proteins to ensure cellular homeostasis.
Since the ubiquitin-proteasome system plays a crucial role in many cellular pathways, many viruses, including poxviruses, have evolved strategies to regulate the process of ubiquitination and protein degradation by the 26S proteasome (4, 5, 54). For example, we recently demonstrated that the RING finger proteins p28 and M143, encoded by orthopoxviruses and myxoma virus, respectively, function as ubiquitin ligases (22, 37). Although not formally proven, the avipoxviruses encode an expanded family of RING finger proteins that are predicted to function as ubiquitin ligases (1, 63), while canarypox virus and entomopoxiruses also encode ubiquitin molecules (1, 63). Numerous proteins encoded by poxviruses have retained F-box-like domains, which may function with cullin-1-based ubiquitin ligases (34, 59, 64). Additionally, myxoma virus regulates the levels of CD4 and major histocompatibility complex class I on the surfaces of cells via expression of a virus-encoded, membrane-bound ubiquitin ligase (6, 19, 32). More recently, we have found that BTB/kelch proteins encoded by ectromelia virus interact with cullin-3 ubiquitin ligases (67).
While it is apparent that poxviruses encode a battery of proteins to regulate the ubiquitin-proteasome system, the role of the 26S proteasome during poxvirus infection has not been established. Here we show that proteasomal inhibition impedes viral-factory formation and inhibits late protein production and viral DNA replication during vaccinia virus infection. Early gene expression, however, remained unaltered in the presence of proteasome inhibitors. Additionally, the use of an inhibitor of the E1 ubiquitin-activating enzyme also dramatically affected late protein production. Virus growth was severely inhibited in the presence of proteasome inhibitors; however, reversal of the proteasomal block could restore viral titers, viral-factory formation, and late protein production. Our data indicate that the proteasome and the ubiquitin cascade are required for orthopoxvirus infection and that inhibitors of the ubiquitin-proteasome system have the ability to potently inhibit poxvirus replication.
MATERIALS AND METHODS
Cells and viruses.
HeLa, baby green monkey kidney (BGMK), and CV-1 cells were obtained from the ATCC. Mouse embryonic fibroblasts (MEF) and baby mouse kidney (BMK) cells were provided by S. Korsmeyer (Dana-Farber Cancer Institute, MA) and E. White (Rutgers University, Piscataway, NJ), respectively. HeLa cells, MEF, CV-1 cells, and BMK cells were maintained in Dulbecco's modified Eagle medium (DMEM) (GIBCO Invitrogen Corp.) containing 10% fetal bovine serum (GIBCO Invitrogen Corp.), 50 U of penicillin (GIBCO Invitrogen Corp.)/ml, 50 μg of streptomycin (GIBCO Invitrogen Corp.)/ml, and 200 μM glutamine (GIBCO Invitrogen Corp.). BGMK cells were routinely maintained in DMEM containing 10% newborn calf serum (GIBCO Invitrogen Corp.), 50 U of penicillin/ml, 50 μg of streptomycin/ml, and 200 μM glutamine. Vaccinia virus strain Copenhagen expressing lacZ (VV65) was provided by G. McFadden (University of Florida, Gainesville). Ectromelia virus strain Moscow and cowpox virus strain Brighton Red were generously provided by R. M. Buller (St. Louis University, St. Louis, MO) and R. Moyer (University of Florida, Gainesville), respectively.
Treatment with proteasome and E1 inhibitors.
Prior to infection, cells were pretreated for 1 h with either 10 μM MG132 (Sigma-Aldrich), 10 μM MG115 (Sigma-Aldrich), 10 μM lactacystin (Sigma-Aldrich), or 1 μM bortezomib (Velcade; Millennium Pharmaceuticals). Alternatively, cells were treated with a 25 μM concentration of the E1 inhibitor Pyr-41 (Biogenova) for 8 h prior to infection, as previously described (69). Following pretreatment, inhibitors were removed by washing the cells with phosphate-buffered saline (PBS), and cells were infected with VV65 at a multiplicity of infection (MOI) of 5. After 1 h of infection, cells were again treated with proteasome inhibitors or Pyr-41 for the times indicated in Fig. 8. Alternatively, in some experiments, cells were treated with MG132 2, 4, 6, and 8 h after virus infection. Washout experiments were performed by treating cells with 10 μM MG132 1 h after virus infection and by removing MG132 at 4, 8, and 12 h after infection prior to their harvesting at 16 h. As a control, cells were treated with 40 μM AraC (Sigma-Aldrich) to inhibit DNA replication. For analysis of the plaque phenotype in the presence of MG132, BGMK cells were infected with VV65 and treated at 1 h postinfection with 10 μM MG132. Plaques were fixed and visualized by staining them with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal).
FIG. 8.

Inhibition of the E1-activating enzyme inhibits late gene expression. HeLa cells were infected with VV65 at an MOI of 5 and pretreated with 25 μM Pyr-41. Total cellular lysates were harvested at the indicated times and blotted for I5L, I3L, and Bak. α, anti.
Metabolic labeling.
HeLa cells (7 × 106) were pretreated with 10 μM MG132 or a similar volume of dimethyl sulfoxide for 1 h, and cells were either mock infected or infected with VV65 at an MOI of 5. After 1 h of infection, 10 μM MG132 or dimethyl sulfoxide was added back to the cells. As a control, DNA replication was inhibited by treating infected cells with 40 μg/ml AraC. At 4.5 h postinfection, cells were starved in DMEM media lacking l-methionine and l-cysteine (GIBCO Invitrogen Corp.) for 30 min before the addition of 33 μCi [35S]methionine and [35S]cysteine (PerkinElmer). Cells were harvested after 1 h of labeling, and proteins were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and visualized by autoradiography.
Slot blotting.
HeLa cells were infected with VV65 at an MOI of 5. To examine the effects of MG132 on virus replication, cells were mock treated or preincubated with 10 μM MG132 for 1 h prior to infection, MG132 was added back after 1 h, and DNA was harvested at 2, 4, 6, or 8 h postinfection. As a control, cells were also treated with 40 μg/ml of AraC. Cells were washed and resuspended in 10× saline sodium citrate containing 1 M ammonium acetate and stored at −80°C. The samples were frozen and thawed three times and clarified by centrifugation, and 25-μl aliquots were mixed with an equal volume of 0.8 M NaOH-20 mM EDTA, boiled for 10 min, cooled on ice, and diluted with 0.4 M NaOH and 10 mM EDTA. The samples were applied in triplicate to a Zeta-Probe membrane (Bio-Rad) by using a vacuum manifold, washed, and immobilized with UV light. A 3.1-kb probe spanning the DNA polymerase E9 gene was prepared by PCR, purified, and labeled with [α-32P]dATP (PerkinElmer) with a random-priming labeling kit (Roche Diagnostics) (2, 70). The membrane was processed using a Southern blot hybridization procedure and label detected by using a Typhoon phosphorimager (GE Healthcare) (50).
Single-step growth curves.
Single-step growth curves were determined in triplicate by infecting HeLa cells with VV65, cowpox virus strain Brighton Red, or ectromelia virus strain Moscow at an MOI of 1. Prior to infection, cells were pretreated with either 10 μM MG132 or 1 μM bortezomib. Following pretreatment, inhibitors were removed by washing the cells with PBS, and cells were infected with VV65. After 1 h of infection, cells were again treated with proteasome inhibitors. Cells were harvested at 0, 4, 8, 12, and 24 h postinfection, and the titers of samples were determined in duplicate on BGMK cells.
Confocal microscopy.
For fixed-cell confocal microscopy, coverslips were seeded with 2.5 × 105 HeLa cells. Cells were infected with VV65 at an MOI of 5 in the presence and absence of 10 μM MG132, 1 μΜ bortezomib, or 40 μM AraC. Sixteen hours postinfection, cells were fixed with 4% paraformaldehyde and permeabilized with 1% NP-40. Coverslips were incubated with anti-I3L antibody (diluted 1:100), provided by D. Evans, University of Alberta, for 3 h, followed by staining with Alexa Fluor 488 goat anti-mouse. Coverslips were washed with PBS containing 1% fetal bovine serum and mounted with 4 mg/ml of N-propyl gallate (Sigma-Aldrich) in 50% glycerol containing 250 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen Corporation) to visualize nuclei and cytoplasmic viral factories.
Western blotting.
Cell lysates were subjected to SDS-PAGE and transferred to nitrocellulose or polyvinylidene fluoride membranes. The following antibodies were used for detection: mouse anti-I3L (1:20), rabbit anti-I5L (1:5,000), rabbit anti-M2L (1:10,000; provided by J. Shisler, University of Illinois), mouse anti-B5R (1:5,000; provided by S. Isaacs, University of Pennsylvania), rabbit anti-BakNT (1:2,000; Upstate), and rabbit anti-Mcl-1 clone RC13 (1:250; Cedarlane Inc.). Proteins were visualized by enhanced chemiluminescence according to the manufacturer's directions (GE Healthcare).
RESULTS
MG132 treatment inhibits late protein production of orthopoxviruses.
The presence of multiple poxviral proteins that regulate the ubiquitin-proteasome system implies that this pathway plays an important role during poxvirus infection (19, 22, 32, 34, 37, 59, 60, 63, 64, 67); however, the role of the 26S proteasome during poxvirus infection has not been established. Therefore, to determine the effects of MG132 on vaccinia virus protein expression, we treated infected cells with MG132, a peptide aldehyde that reversibly inhibits the chymotrypsin-like activity of the proteasome, and examined protein expression over the course of infection (29, 36). Compared to untreated samples, HeLa cells infected with VV65 and treated with MG132 demonstrated a dramatic reduction of I5L, a small, membrane-bound, 8-kDa late protein (Fig. 1A) (61). Inhibition of I5L expression was also seen in cell lines other than HeLa, including MEF, BMK cells, and CV-1 cells (R. Shipclark and M. Barry, unpublished observation). In addition, the levels of B5R, another late protein important for the formation of enveloped virus (17, 68), were also greatly diminished over the course of vaccinia virus infection in the presence of MG132, indicating that proteasomal inhibition severely altered late gene expression (Fig. 1A). To verify that proteasomal activity was indeed inhibited over the course of infection, protein levels of the antiapoptotic Bcl-2 family member Mcl-1 were examined since Mcl-1 is tightly regulated and rapidly turned over by the ubiquitin-proteasome system (11, 40). Upon MG132 treatment, Mcl-1 protein expression increased over the course of infection, indicating that proteasomal function was indeed inhibited. Conversely, the levels of the proapoptotic protein Bak remained unaltered and served as a loading control (Fig. 1A). Treatment with AraC, an inhibitor of viral DNA replication (3, 12), confirmed that both I5L and B5R are late proteins requiring the replication of poxviral DNA in order to be expressed (Fig. 1B).
FIG. 1.

MG132 inhibits late protein expression. (A) HeLa cells were mock infected or infected with VV65 at an MOI of 5 in the presence or absence of 10 μM MG132. VV65-infected samples were harvested at 4, 8, 12, 16, and 24 h, and mock-infected samples were harvested at 4 h and Western blotted with anti-I5L, anti-B5R, anti-Mcl-1, and anti-Bak. α, anti. (B) HeLa cells were infected with VV65 in the presence and absence of 40 μg/ml AraC to determine late expression for I5L and B5R. (C) Metabolic labeling of VV65-infected cells. HeLa cells were mock infected or infected with VV65 at an MOI of 5 and treated with either 10 μM MG132 or 40 μg/ml AraC. Cells were pulse-labeled with [35S]methionine/cysteine at 4.5 h postinfection, and samples were analyzed by SDS-PAGE followed by autoradiography. Late proteins are marked with dots in the VV65-infected sample. (D) BGMK cells were infected with VV65 in the presence and absence of MG132, and plaques were visualized by staining with X-Gal.
To confirm that treatment of cells with MG132 had a global effect on the production of late proteins, we performed metabolic-labeling experiments. Infection with VV65 indicated the presence of a series of vaccinia virus gene products that were clearly inhibited in the presence of AraC, indicating that these proteins were expressed late during infection (Fig. 1C). The addition of MG132 resulted in a decrease in expression of these late gene products, suggesting that MG132 had a global effect on late protein synthesis. These observations were further supported by a clear reduction in plaque size in the presence of MG132 (Fig. 1D).
Early protein production of vaccinia virus is not affected by MG132.
Unlike late genes, which require DNA replication for synthesis, early gene expression is initiated in the virion core and precedes viral DNA replication (35). To determine whether early protein products were altered by MG132 treatment, we analyzed levels of the early poxviral proteins I3L and M2L. In contrast to late protein expression following MG132 treatment, which was severely reduced (Fig. 1), the production of I3L, a single-strand-DNA binding protein (16, 62, 66), and the production of M2L, an inhibitor of the NF-κB pathway (21), were not affected by MG132 treatment (Fig. 2A). To demonstrate that I3L and M2L were expressed early during infection, HeLa cells were subjected to AraC treatment. The expression of I3L and M2L was unaffected in the presence of AraC, indicating that both proteins were expressed early during infection (Fig. 2B). Together, these results imply that while proteasome inhibition hinders late protein production, the expression of early proteins was unaffected.
FIG. 2.

Early protein expression is unaffected by MG132. (A) HeLa cells were infected with VV65 at an MOI of 5 in the presence of 10 μM MG132. At the indicated times, samples were harvested and Western blotted with anti-I3L, anti-M2L, and anti-Bak. α, anti. (B) HeLa cells were infected with VV65 in the presence and absence of 40 μg/ml AraC to determine early expression for I3L and M2L.
Vaccinia virus is a member of the Orthopoxvirus genus, which also includes closely related species, such as cowpox virus, ectromelia virus, and variola virus (35). To determine if MG132 was able to inhibit late gene expression in other orthopoxviruses, we infected HeLa cells with ectromelia virus strain Moscow or cowpox virus strain Brighton Red in the presence and absence of MG132. Blotting for the late proteins I5L and B5R indicated that late protein production of both cowpox virus and ectromelia virus was greatly reduced in the presence of MG132. Expression of the early protein I3L during ectromelia virus and cowpox virus infection was unaffected by MG132 (Fig. 3), indicating that the effect of proteasome inhibitors on late gene expression was not specific to vaccinia virus.
FIG. 3.

MG132 inhibits late protein expression during cowpox virus and ectromelia virus infection. HeLa cells were infected with ectromelia virus Moscow or cowpox virus Brighton Red at an MOI of 5 in the presence of 10 μM MG132. At the indicated times, samples were harvested and Western blotted with anti-I5L, anti-B5R, anti-I3L, anti-Mcl-1, and anti-Bak. EVM, ectromelia virus Moscow; CPV, cowpox virus; α, anti.
Late protein production is inhibited by multiple proteasome inhibitors.
To ascertain whether the loss of late protein production was specific to MG132, we used a range of proteasome inhibitors, and I5L levels were assessed as an indication of late protein production during VV65 infection. MG115, an irreversible inhibitor that effectively inhibits the chymotrypsin-like activity of the proteasome (29, 36), was shown to significantly reduce late protein expression (Fig. 4A). I5L production was also inhibited by lactacystin, a natural metabolite from Streptomyces spp. that irreversibly inhibits the 26S proteasome by modification of the β-subunit (Fig. 4B) (29, 36). In addition, bortezomib, a boron-based proteasome inhibitor (25, 49), also inhibited the expression of I5L (Fig. 4C). In contrast, treatment of virally infected cells with MG115, lactacystin, and bortezomib had no effect on the production of the early protein I3L (Fig. 4A, B, and C).
FIG. 4.

Multiple proteasome inhibitors block late protein expression. HeLa cells were infected with VV65 at an MOI of 5 in the presence of 10 μM MG115 (A), 10 μM lactacystin (B), and 1 μM bortezomib (C). At the indicated times, samples were harvested and Western blotted with anti-I5L, anti-I3L, and anti-Bak. α, anti.
Viral factories fail to form in the presence of proteasome inhibitors.
Given that inhibition of the proteasome selectively inhibits late protein production while leaving early proteins unaffected, we sought to determine if viral-factory formation was affected. To this end, we infected HeLa cells with VV65 in the presence and absence of MG132 or bortezomib and visualized viral-factory formation by staining cells with DAPI and an antibody specific for I3L. HeLa cells infected with VV65 demonstrated the presence of DAPI-positive perinuclear viral factories (Fig. 5A) that colocalized with I3L, a single-strand-DNA binding protein that localizes to the virus factory (Fig. 5A, panels a to c) (16, 62, 66). Upon MG132 treatment, viral factories were conspicuously absent from VV65-infected cells and I3L expression was notably cytoplasmic in appearance (Fig. 5A, panels d to f). Similarly, bortezomib treatment also disrupted the formation of viral factories, resulting in cytoplasmic I3L localization (Fig. 5A, panels g to i). As a control, we treated cells with AraC to actively block viral DNA replication in infected cells, and I3L localization was examined. As expected, DNA replication was inhibited upon treatment with AraC, as demonstrated by the lack of DAPI-positive viral factories and the cytoplasmic localization of I3L (Fig. 5A, panels j to l).
FIG. 5.

MG132 and bortezomib block virus factory formation. (A) HeLa cells were infected with VV65 at an MOI of 5 in the presence or absence of 10 μM MG132, 1 μM bortezomib, or 40 μg/ml AraC. Sixteen hours postinfection, cells were fixed and stained with DAPI to visualize nuclei and virus factories and stained with anti-I3L (αI3L). (a to l) HeLa cells infected with VV65 (a to c), with VV65 in the presence of MG132 (d to f), with VV65 in the presence of bortezomib (g to i), or with VV65 in the presence of AraC (j to l). (B) A slot blot assay was used to assess the accumulation of VV65 DNA using a 32P-labeled probe specific for the E9 DNA polymerase gene in the presence and absence of MG132 or AraC.
The presence of cytoplasmic I3L in cells treated with proteasome inhibitors or AraC suggested that the early viral protein I3L was produced in the presence of proteasome inhibitors but that DNA replication was inhibited. Therefore, to determine if proteasome inhibitors had an effect on DNA replication, we performed a slot blot assay. HeLa cells were infected with VV65 in the presence and absence of MG132, and vaccinia virus DNA was isolated at various times and visualized using a probe specific for the vaccinia virus DNA polymerase. In the absence of MG132, vaccinia virus DNA replication was detected at 6 and 8 h postinfection (Fig. 5B). The presence of MG132, however, clearly inhibited vaccinia virus DNA replication, similarly to treatment with AraC (Fig. 5B).
To elucidate whether the proteasome-dependent arrest during poxviral infection could be rescued through restoration of proteasome activity, we performed washout experiments with the reversible proteasome inhibitor MG132 (29, 36). HeLa cells were infected with VV65 and treated with MG132 for 16 h, and cell lysates were subjected to Western blotting with anti-I3L, anti-I5L, or anti-Bak. Alternatively, HeLa cells were infected with VV65 in the presence of MG132 for 4, 8, or 12 h, after which MG132 was washed out and the infection allowed to resume for a total of 16 h. Washout of MG132 had no effect on the expression of I3L, as expected; however, washout of MG132 at 4, 8, and 12 h postinfection resulted in a rebound of I5L levels by 16 h postinfection, indicating that late protein production could be rescued by lifting the inhibition of the proteasome (Fig. 6A). Furthermore, examination by confocal microscopy revealed that removal of MG132 at 4, 8, and 12 h restored both viral-factory formation and the localization of I3L to viral factories at 16 h postinfection (Fig. 6B).
FIG. 6.

The effect of MG132 is reversible. (A) HeLa cells were infected with VV65 at an MOI of 5. Cells were either pretreated (Pre) or treated with MG132 for 12, 8, or 4 h, at which point cells were washed with PBS to remove MG132 and harvested at 16 h postinfection. Additionally, cells were not treated with MG132 [(−)]. I3L, I5L, and Bak expression was monitored by Western blotting. α, anti. (B) HeLa cells were infected with VV65 in the presence and absence of MG132, and at 4, 8, and 12 h, cells were washed with PBS to remove MG132. Cells were fixed at 16 h postinfection, stained with DAPI to visualize nuclei and virus factories, and stained with anti-I3L. (a to o) HeLa cells infected with VV65 (a to c), with VV65 in the presence of MG132 followed by a washout at 4 h (d to f), with VV65 in the presence of MG132 followed by washout at 8 h (g to i), with VV65 in the presence of MG132 followed by washout at 12 h (j to l), or with VV65 in the presence of MG132 (m to o).
Proteasome inhibitors act at an early step during infection.
In our previous experiments, proteasome inhibition was achieved through pretreatment of cells with proteasome inhibitors 1 h prior to viral infection. To further elucidate the mechanism by which proteasome inhibitors hinder viral processes, MG132 and bortezomib were added either 1 h prior to infection or at various times postinfection, and late protein production was examined by Western blotting for I5L 16 h postinfection. The addition of MG132 or bortezomib at 2 and 4 h postinfection significantly inhibited I5L expression comparably to what occurred in cells that had been pretreated with MG132 (Fig. 7A). In contrast, the addition of MG132 at 6 or 8 h postinfection had only a moderate effect on the inhibition of I5L expression (Fig. 7A). In addition, treatment of infected cells with bortezomib at 6 and 8 h postinfection showed an expression of I5L that was comparable to I5L expression in untreated cells (Fig. 7B). These observations indicated that the obstruction of the proteasome was most effective when the inhibitor was administered early during infection, further supporting the idea that a functional proteasome is likely required for an early viral process during infection.
FIG. 7.
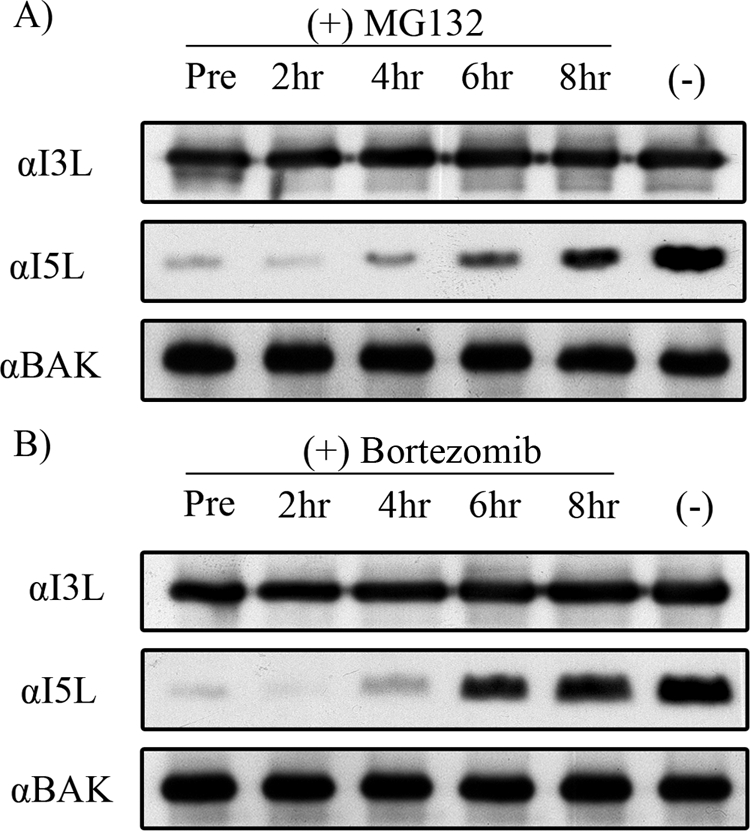
Proteasome inhibitors act at an early step during infection. (A) HeLa cells were infected with VV65 at an MOI of 5. Cells were either pretreated (Pre); treated with MG132 at either 2, 4, 6, or 8 h postinfection; or not treated with MG132 [(−)]. I3L, I5L, and Bak expression was monitored by Western blotting. α, anti. (B) HeLa cells were infected with VV65 at an MOI of 5. Cells were pretreated with bortezomib (Pre); treated with bortezomib at either 2, 4, 6, or 8 h postinfection; or not treated with bortezomib [(−)]. I3L, I5L, and Bak expression was monitored by Western blotting.
Inhibition of the E1 ubiquitin-activating enzyme inhibits late gene expression.
Prompted by the ability of proteasome inhibitors to block the orthopoxvirus cycle at an early stage, we sought to determine if inhibition of an upstream component of the ubiquitin-proteasome system displayed a similar effect. HeLa cells were pretreated with a newly developed inhibitor of the E1 ubiquitin-activating enzyme Pyr-41, and the expression of I5L, I3L, and Bak were evaluated following virus infection (69). As with cells treated with proteasome inhibitors, we found that inhibition of the E1-activating enzyme had an inhibitory effect on I5L production but that I3L levels were unaffected, clearly indicating that both ubiquitination and the proteasome played a role in the poxvirus life cycle (Fig. 8).
Proteasome inhibitors potently block viral replication.
To investigate the ability of proteasome inhibitors to effectively block viral replication and propagation, we performed single-step growth curve analyses with HeLa cells. Cells were infected with VV65 in the absence and presence of either MG132 or bortezomib. Both MG132 and bortezomib greatly reduced the production of viral progeny (Fig. 9A). In comparison, vaccinia virus grown in the absence of proteasomal inhibitors demonstrated a 2-log increase in viral titers. Similar drops in virus growth in the presence of bortezomib were also observed for cowpox virus and ectromelia virus (Fig. 9B and C). To determine whether washout of MG132 could restore viral titers, MG132 was removed 10 h postinfection, and viral levels were nearly restored by 18 h postinfection (Fig. 9D). This, along with restoration of late protein expression and viral-factory production (Fig. 6), reinforced the concept that lifting proteasome inhibition could rescue orthopoxvirus infection.
FIG. 9.

Inhibition of the ubiquitin proteasome system dramatically affects poxvirus production. (A) HeLa cells were infected with VV65 at an MOI of 1, and samples were harvested at 4, 8, 12, and 24 h postinfection. Samples were collected in triplicate, and titers were determined on BGMK cells. (B) HeLa cells were infected with cowpox virus Brighton Red (CPV) at an MOI of 1. (C) HeLa cells were infected with ectromelia virus strain Moscow (EVM) at an MOI 1. (D) HeLa cells were infected with VV65 at an MOI 5 for 10 or 24 h or the presence of 10 μM MG132 for 10 h. Additionally, MG132 was removed by washout at 10 h and infection was allowed to progress for a further 8 h.
DISCUSSION
The poxvirus life cycle is a complex multistep process that requires the modulation of multiple host signaling pathways (24, 53). As such, poxviruses are renowned for encoding numerous effector proteins capable of intercepting and hijacking normal cellular processes (24, 53). Recently, it has become increasingly clear that poxviruses also exploit the ubiquitin-proteasome system to their own advantage. This is achieved through the expression of virus-encoded ubiquitin ligases and ubiquitin molecules, as well as the presence of virus-encoded substrate adaptors for cullin-1- and cullin-3-based cellular ubiquitin ligases (19, 22, 32, 34, 37, 59, 60, 63, 64, 67). Prior to this study, however, the role of the proteasome during poxvirus infection had not been investigated.
To determine the role of the proteasome during infection, we treated orthopoxvirus-infected cells with a range of proteasome inhibitors. This approached verified the crucial importance of the 26S proteasome during poxvirus infection. Our studies indicated that multiple proteasome inhibitors dramatically inhibited late protein synthesis (Fig. 4) but that early protein expression was unaffected by proteasome inhibition (Fig. 2 and 4). Moreover, through confocal microscopy analysis, we found that poxviral factories failed to form in the presence of bortezomib or MG132 (Fig. 5A and 6B) and that the addition of proteasome inhibitors had a dramatic effect on poxvirus DNA replication (Fig. 5B). Removal of MG132, however, restored viral replication, viral-factory formation, and late protein production, suggesting that the effects were not solely due to cytotoxicity (Fig. 6 and 9). The addition of an ubiquitin-activating enzyme (E1) inhibitor had similar effects on late and early protein expression, indicating that both ubiquitination and proteasomal degradation were necessary for the productive infection of orthopoxviruses (Fig. 8).
Viruses have coevolved over millions of years with their hosts, resulting in many examples of viral exploitation of the ubiquitin-proteasome system (4, 5, 54). Evidence indicates that proteasome inhibitors effectively inhibit viral processes across a variety of virus species, including human immunodeficiency virus (52), human cytomegalovirus (46), herpes simplex virus (13), murine hepatitis virus (72), vesicular stomatitis virus (39), coxsackievirus (55), infectious bursal disease virus (31), and avian reovirus (9). Proteasome inhibitors effectively inhibit viral propagation of these species through a variety of distinct mechanisms. For example, proteasome inhibitors have been demonstrated to block the release of murine hepatitis virus from an endocytic compartment (72), suppress RNA transcription and protein synthesis during coxsackievirus infection (55), and inhibit entry of herpes simplex virus into the nucleus (13). These observations indicate that multiple viruses have established an intricate relationship with the cellular ubiquitin-proteasome system, and orthopoxviruses are no exception. In the presence of proteasome inhibitors, we found that the block imposed during orthopoxvirus infection occurred at early stages (Fig. 7). This conclusion fits well with the observations that late protein expression and DNA replication were inhibited but that early protein expression was unaffected (Fig. 1 to 4 and 5B). Upon orthopoxvirus entry into cells, the viral core is released into the cytoplasm, resulting in the production of early mRNA and early protein synthesis, which is followed by virion uncoating and DNA replication (35). Since the obstruction occurred early during infection, it is tempting to speculate that core-associated and/or viral proteins involved in DNA replication may be targets for the ubiquitin-proteasome pathway to assist in either core uncoating or DNA replication. Mass spectrometry approaches have identified more than 70 distinct proteins in vaccinia virus virions (10, 48, 71). Specific components of the core are not well defined; however, the major components include A10L, A4L, A3L, L4R, and F17R (23, 43). Of these, A4L (p39), A10L (p4a), and A3L (p4b) may be accessible for ubiquitination based upon localization at the core surface (43). Intriguingly, one study indicated that ubiquitin accounted for approximately 3% of total virion protein, suggesting that virion components may be substrates for ubiquitination (10). Given the recent advancements in the use of mass spectrometry to identify ubiquitinated proteins, it is likely that ubiquitinated virion proteins could be identified, furthering our understanding of the intricate relationship between poxvirus replication and the ubiquitin-proteasome system (28).
Our data suggest that the ubiquitin-proteasome system may be an attractive target for the development of effective antivirals since many viruses, including poxviruses, hijack this cellular process to target specific cellular and viral proteins for ubiquitination and degradation (4, 5, 54). Currently used poxviral therapeutics include cidofovir, a nucleotide analogue that successfully inhibits orthopoxvirus infections both in vivo and in vitro (7, 38). Additionally, screening approaches have yielded other compounds that inhibit orthopoxvirus infection, including ST-246, which prevents successful virus formation and is effective in animal models (8, 47), and mitoxantrone, an anticancer agent that inhibits a late-stage assembly step of vaccinia virus (15). Our data indicate that the treatment of orthopoxvirus-infected cells with proteasome inhibitors or an E1 inhibitor dramatically affects late gene expression, suggesting that proteasome inhibitors or E1 inhibitors could potentially be employed as antivirals. The proteasome inhibitor bortezomib has recently been approved for treatment of multiple myeloma (25, 49); thus, in addition to having this previously described function, it may have potential in the treatment of viral infections. While an obvious drawback of targeting cellular processes includes cytotoxicity, inhibitors of cellular pathways have some advantages over traditional antiviral drugs (51). For example, antiviral approaches that target cellular proteins could have broad actions across many virus species compared to conventional antivirals with narrow ranges of action, and resistance to a cellular target may be more difficult to develop than resistance to viral targets (51).
In conclusion, we have shown that orthopoxviruses require a functional ubiquitin-proteasome system for replication. Further studies will investigate the precise mechanism of how proteasome inhibitors interfere with the orthopoxvirus replication cycle. Such studies will undoubtedly further our understanding of poxvirus biology and viral manipulation of the ubiquitin-proteasome system.
Acknowledgments
We thank D. Evans, S. Isaacs, and J. Shisler for providing reagents and useful discussions.
Work in our laboratory is supported by grants from the Canadian Institutes of Health Research (CIHR) and the Howard Hughes Medical Institute (HHMI). S.C. is the recipient of a Canada Graduate Scholarship. M.B. is a CIHR New Investigator, an Alberta Heritage Foundation for Medical Research Senior Scholar, and an HHMI International Research Scholar.
Footnotes
Published ahead of print on 24 December 2008.
REFERENCES
- 1.Afonso, C. L., E. R. Tulman, Z. Lu, L. Zsak, G. F. Kutish, and D. L. Rock. 2000. The genome of fowlpox virus. J. Virol. 743815-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrei, G., D. B. Gammon, P. Fiten, E. De Clercq, G. Opdenakker, R. Snoeck, and D. H. Evans. 2006. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice. J. Virol. 809391-9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babiuk, L. A., B. Meldrum, V. S. Gupta, and B. T. Rouse. 1975. Comparison of the antiviral effects of 5-methoxymethyl-deoxyuridine with 5-iododeoxyuridine, cytosine arabinoside, and adenine arabinoside. Antimicrob. Agents Chemother. 8643-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banks, L., D. Pim, and M. Thomas. 2003. Viruses and the 26S proteasome: hacking into destruction. Trends Biochem. Sci. 28452-459. [DOI] [PubMed] [Google Scholar]
- 5.Barry, M., and K. Fruh. 2006. Viral modulators of cullin RING ubiquitin ligases: culling the host defense. Sci. STKE 2006pe21. [DOI] [PubMed] [Google Scholar]
- 6.Barry, M., S. F. Lee, L. Boshkov, and G. McFadden. 1995. Myxoma virus induces extensive CD4 downregulation and dissociation of p56lck in infected rabbit CD4+ T lymphocytes. J. Virol. 695243-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buller, R. M., G. Owens, J. Schriewer, L. Melman, J. R. Beadle, and K. Y. Hostetler. 2004. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 318474-481. [DOI] [PubMed] [Google Scholar]
- 8.Byrd, C. M., T. C. Bolken, A. M. Mjalli, M. N. Arimilli, R. C. Andrews, R. Rothlein, T. Andrea, M. Rao, K. L. Owens, and D. E. Hruby. 2004. New class of orthopoxvirus antiviral drugs that block viral maturation. J. Virol. 7812147-12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, Y. T., C. H. Lin, W. T. Ji, S. K. Li, and H. J. Liu. 2008. Proteasome inhibition reduces avian reovirus replication and apoptosis induction in cultured cells. J. Virol. Methods 15195-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung, C. S., C. H. Chen, M. Y. Ho, C. Y. Huang, C. L. Liao, and W. Chang. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 802127-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuconati, A., C. Mukherjee, D. Perez, and E. White. 2003. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 172922-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Clercq, E., E. Darzynkiewicz, and D. Shugar. 1975. Antiviral activity of O′-alkylated derivatives of cytosine arabinoside. Biochem. Pharmacol. 24523-527. [DOI] [PubMed] [Google Scholar]
- 13.Delboy, M. G., D. G. Roller, and A. V. Nicola. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 823381-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demartino, G. N., and T. G. Gillette. 2007. Proteasomes: machines for all reasons. Cell 129659-662. [DOI] [PubMed] [Google Scholar]
- 15.Deng, L., P. Dai, A. Ciro, D. F. Smee, H. Djaballah, and S. Shuman. 2007. Identification of novel antipoxviral agents: mitoxantrone inhibits vaccinia virus replication by blocking virion assembly. J. Virol. 8113392-13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domi, A., and G. Beaud. 2000. The punctate sites of accumulation of vaccinia virus early proteins are precursors of sites of viral DNA synthesis. J. Gen. Virol. 811231-1235. [DOI] [PubMed] [Google Scholar]
- 17.Engelstad, M., and G. L. Smith. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194627-637. [DOI] [PubMed] [Google Scholar]
- 18.Forster, A., and C. P. Hill. 2003. Proteasome degradation: enter the substrate. Trends Cell Biol. 13550-553. [DOI] [PubMed] [Google Scholar]
- 19.Guerin, J. L., J. Gelfi, S. Boullier, M. Delverdier, F. A. Bellanger, S. Bertagnoli, I. Drexler, G. Sutter, and F. Messud-Petit. 2002. Myxoma virus leukemia-associated protein is responsible for major histocompatibility complex class I and Fas-CD95 down-regulation and defines scrapins, a new group of surface cellular receptor abductor proteins. J. Virol. 762912-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanson, D., and D. G. Diven. 2003. Molluscum contagiosum. Dermatol. Online J. 92. [PubMed] [Google Scholar]
- 21.Hinthong, O., X. L. Jin, and J. L. Shisler. 2008. Characterization of wild-type and mutant vaccinia virus M2L proteins' abilities to localize to the endoplasmic reticulum and to inhibit NF-kappaB activation during infection. Virology 373248-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, J., Q. Huang, X. Zhou, M. M. Shen, A. Yen, S. X. Yu, G. Dong, K. Qu, P. Huang, E. M. Anderson, S. Daniel-Issakani, R. M. Buller, D. G. Payan, and H. H. Lu. 2004. The poxvirus p28 virulence factor is an E3 ubiquitin ligase. J. Biol. Chem. 27954110-54116. [DOI] [PubMed] [Google Scholar]
- 23.Jensen, O. N., T. Houthaeve, A. Shevchenko, S. Cudmore, T. Ashford, M. Mann, G. Griffiths, and J. Krijnse Locker. 1996. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J. Virol. 707485-7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston, J. B., and G. McFadden. 2003. Poxvirus immunomodulatory strategies: current perspectives. J. Virol. 776093-6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanagasabaphy, P., G. J. Morgan, and F. E. Davies. 2007. Proteasome inhibition and multiple myeloma. Curr. Opin. Investig. Drugs 8447-451. [PubMed] [Google Scholar]
- 26.Katsafanas, G. C., and B. Moss. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2221-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kile, J. C., A. T. Fleischauer, B. Beard, M. J. Kuehnert, R. S. Kanwal, P. Pontones, H. J. Messersmith, R. Teclaw, K. L. Karem, Z. H. Braden, I. Damon, A. S. Khan, and M. Fischer. 2005. Transmission of monkeypox among persons exposed to infected prairie dogs in Indiana in 2003. Arch. Pediatr. Adolesc. Med. 1591022-1025. [DOI] [PubMed] [Google Scholar]
- 28.Kirkpatrick, D. S., C. Denison, and S. P. Gygi. 2005. Weighing in on ubiquitin: the expanding role of mass-spectrometry-based proteomics. Nat. Cell Biol. 7750-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, D. H., and A. L. Goldberg. 1998. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8397-403. [DOI] [PubMed] [Google Scholar]
- 30.Leite, J. A., B. P. Drumond, G. S. Trindade, Z. I. Lobato, F. G. da Fonseca, S. J. dos, M. C. Madureira, M. I. Guedes, J. M. Ferreira, C. A. Bonjardim, P. C. Ferreira, and E. G. Kroon. 2005. Passatempo virus, a vaccinia virus strain, Brazil. Emerg. Infect. Dis. 111935-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, J., L. Wei, T. Jiang, L. Shi, and J. Wang. 2007. Reduction of infectious bursal disease virus replication in cultured cells by proteasome inhibitors. Virus Genes 35719-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mansouri, M., E. Bartee, K. Gouveia, B. T. Hovey Nerenberg, J. Barrett, L. Thomas, G. Thomas, G. McFadden, and K. Fruh. 2003. The PHD/LAP-domain protein M153R of myxomavirus is a ubiquitin ligase that induces the rapid internalization and lysosomal destruction of CD4. J. Virol. 771427-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marris, E. 2007. Dramatic rescue relieves rare case of smallpox infection. Nat. Med. 13517. [DOI] [PubMed] [Google Scholar]
- 34.Mercer, A. A., S. B. Fleming, and N. Ueda. 2005. F-box-like domains are present in most poxvirus ankyrin repeat proteins. Virus Genes 31127-133. [DOI] [PubMed] [Google Scholar]
- 35.Moss, B. 2001. Poxviridae: the viruses and their replication, 4th ed., vol. 2. Lippincott Williams and Wilkins, Philadelphia, PA.
- 36.Myung, J., K. B. Kim, and C. M. Crews. 2001. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 21245-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nerenberg, B. T., J. Taylor, E. Bartee, K. Gouveia, M. Barry, and K. Fruh. 2005. The poxviral RING protein p28 is a ubiquitin ligase that targets ubiquitin to viral replication factories. J. Virol. 79597-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neyts, J., P. Leyssen, E. Verbeken, and E. De Clercq. 2004. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob. Agents Chemother. 482267-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neznanov, N., E. M. Dragunsky, K. M. Chumakov, L. Neznanova, R. C. Wek, A. V. Gudkov, and A. K. Banerjee. 2008. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS ONE 3e1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nijhawan, D., M. Fang, E. Traer, Q. Zhong, W. Gao, F. Du, and X. Wang. 2003. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 171475-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nitsche, A., A. Kurth, and G. Pauli. 2007. Viremia in human Cowpox virus infection. J. Clin. Virol. 40160-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parker, S., A. Nuara, R. M. Buller, and D. A. Schultz. 2007. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 217-34. [DOI] [PubMed] [Google Scholar]
- 43.Pedersen, K., E. J. Snijder, S. Schleich, N. Roos, G. Griffiths, and J. K. Locker. 2000. Characterization of vaccinia virus intracellular cores: implications for viral uncoating and core structure. J. Virol. 743525-3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickart, C. M. 2001. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70503-533. [DOI] [PubMed] [Google Scholar]
- 45.Pickart, C. M., and D. Fushman. 2004. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 8610-616. [DOI] [PubMed] [Google Scholar]
- 46.Prosch, S., C. Priemer, C. Hoflich, C. Liebenthaf, N. Babel, D. H. Kruger, and H. D. Volk. 2003. Proteasome inhibitors: a novel tool to suppress human cytomegalovirus replication and virus-induced immune modulation. Antivir. Ther. 8555-567. [PubMed] [Google Scholar]
- 47.Quenelle, D. C., R. M. Buller, S. Parker, K. A. Keith, D. E. Hruby, R. Jordan, and E. R. Kern. 2007. Efficacy of delayed treatment with ST-246 given orally against systemic orthopoxvirus infections in mice. Antimicrob. Agents Chemother. 51689-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Resch, W., K. K. Hixson, R. J. Moore, M. S. Lipton, and B. Moss. 2007. Protein composition of the vaccinia virus mature virion. Virology 358233-247. [DOI] [PubMed] [Google Scholar]
- 49.Roccaro, A. M., A. Vacca, and D. Ribatti. 2006. Bortezomib in the treatment of cancer. Recent Patents Anticancer Drug Discov. 1397-403. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 51.Schang, L. M. 2002. Cyclin-dependent kinases as cellular targets for antiviral drugs. J. Antimicrob. Chemother. 50779-792. [DOI] [PubMed] [Google Scholar]
- 52.Schubert, U., D. E. Ott, E. N. Chertova, R. Welker, U. Tessmer, M. F. Princiotta, J. R. Bennink, H. G. Krausslich, and J. W. Yewdell. 2000. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 9713057-13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21377-423. [DOI] [PubMed] [Google Scholar]
- 54.Shackelford, J., and J. S. Pagano. 2005. Targeting of host-cell ubiquitin pathways by viruses. Essays Biochem. 41139-156. [DOI] [PubMed] [Google Scholar]
- 55.Si, X., G. Gao, J. Wong, Y. Wang, J. Zhang, and H. Luo. 2008. Ubiquitination is required for effective replication of coxsackievirus B3. PLoS ONE 3e2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith, G. L., and M. Law. 2004. The exit of vaccinia virus from infected cells. Virus Res. 106189-197. [DOI] [PubMed] [Google Scholar]
- 57.Smith, G. L., and G. McFadden. 2002. Smallpox: anything to declare? Nat. Rev. Immunol. 2521-527. [DOI] [PubMed] [Google Scholar]
- 58.Smith, G. L., A. Vanderplasschen, and M. Law. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 832915-2931. [DOI] [PubMed] [Google Scholar]
- 59.Sonnberg, S., B. T. Seet, T. Pawson, S. B. Fleming, and A. A. Mercer. 2008. Poxvirus ankyrin repeat proteins are a unique class of F-box proteins that associate with cellular SCF1 ubiquitin ligase complexes. Proc. Natl. Acad. Sci. USA 10510955-10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sperling, K. M., A. Schwantes, B. S. Schnierle, and G. Sutter. 2008. The highly conserved orthopoxvirus 68k ankyrin-like protein is part of a cellular SCF ubiquitin ligase complex. Virology 374234-239. [DOI] [PubMed] [Google Scholar]
- 61.Takahashi, T., M. Oie, and Y. Ichihashi. 1994. N-terminal amino acid sequences of vaccinia virus structural proteins. Virology 202844-852. [DOI] [PubMed] [Google Scholar]
- 62.Tseng, M., N. Palaniyar, W. Zhang, and D. H. Evans. 1999. DNA binding and aggregation properties of the vaccinia virus I3L gene product. J. Biol. Chem. 27421637-21644. [DOI] [PubMed] [Google Scholar]
- 63.Tulman, E. R., C. L. Afonso, Z. Lu, L. Zsak, G. F. Kutish, and D. L. Rock. 2004. The genome of canarypox virus. J. Virol. 78353-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Buuren, N., B. Couturier, Y. Xiong, and M. Barry. 2008. Ectromelia virus encodes a novel family of F-box proteins that interact with the SCF complex. J. Virol. 829917-9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weissman, A. M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2169-178. [DOI] [PubMed] [Google Scholar]
- 66.Welsch, S., L. Doglio, S. Schleich, and J. Krijnse Locker. 2003. The vaccinia virus I3L gene product is localized to a complex endoplasmic reticulum-associated structure that contains the viral parental DNA. J. Virol. 776014-6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilton, B. A., S. Campbell, N. Van Buuren, R. Garneau, M. Furukawa, Y. Xiong, and M. Barry. 2008. Ectromelia virus BTB/kelch proteins, EVM150 and EVM167, interact with cullin-3-based ubiquitin ligases. Virology 37482-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolffe, E. J., A. S. Weisberg, and B. Moss. 1998. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology 24420-26. [DOI] [PubMed] [Google Scholar]
- 69.Yang, Y., J. Kitagaki, R. M. Dai, Y. C. Tsai, K. L. Lorick, R. L. Ludwig, S. A. Pierre, J. P. Jensen, I. V. Davydov, P. Oberoi, C. C. Li, J. H. Kenten, J. A. Beutler, K. H. Vousden, and A. M. Weissman. 2007. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 679472-9481. [DOI] [PubMed] [Google Scholar]
- 70.Yao, X. D., and D. H. Evans. 2004. Construction of recombinant vaccinia viruses using leporipoxvirus-catalyzed recombination and reactivation of orthopoxvirus DNA. Methods Mol. Biol. 26951-64. [DOI] [PubMed] [Google Scholar]
- 71.Yoder, J. D., T. S. Chen, C. R. Gagnier, S. Vemulapalli, C. S. Maier, and D. E. Hruby. 2006. Pox proteomics: mass spectrometry analysis and identification of Vaccinia virion proteins. Virol. J. 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu, G. Y., and M. M. Lai. 2005. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 79644-648. [DOI] [PMC free article] [PubMed] [Google Scholar]