

Abstract
A hallmark of infection with herpes simplex virus type 1 (HSV-1) is the establishment of latency in ganglia of the infected individual. During the life of the latently infected individual, the virus can occasionally reactivate, travel back to the eye, and cause recurrent disease. Indeed, a major cause of corneal scarring (CS) is the scarring induced by HSV-1 following reactivation from latency. In this study, we evaluated the relationship between the amount of CS and the level of the HSV-1 latency-associated transcript (LAT) in trigeminal ganglia (TG) of latently infected mice. Our results suggested that the amount of CS was not related to the amount of virus replication following primary ocular HSV-1 infection, since replication in the eyes was similar in mice that did not develop CS, mice that developed CS in just one eye, and mice that developed CS in both eyes. In contrast, mice with no CS had significantly less LAT, and thus presumably less latency, in their TG than mice that had CS in both eyes. Higher CS also correlated with higher levels of mRNAs for PD-1, CD4, CD8, F4/80, interleukin-4, gamma interferon, granzyme A, and granzyme B in both cornea and TG. These results suggest that (i) the immunopathology induced by HSV-1 infection does not correlate with primary virus replication in the eye; (ii) increased CS appears to correlate with increased latency in the TG, although the possible cause-and-effect relationship is not known; and (iii) increased latency in mouse TG correlates with higher levels of PD-1 mRNA, suggesting exhaustion of CD8+ T cells.
Herpes simplex virus type 1 (HSV-1) infections of the eye are among the most prevalent serious viral eye infections in developed countries. Estimates of the incidence of herpes ocular-disease episodes in these countries indicate up to 21 cases per 100,000 individuals (41, 68). In the United States, approximately 500,000 people have a history of recurrent ocular HSV necessitating doctor visits, medication, and, in severe cases, corneal transplants. It is estimated that 70 to 90% of American adults have antibodies to HSV-1 and/or HSV-2 and thus harbor latent herpesvirus infection and that 25% of these individuals have clinical symptoms upon routine clinical inquiry, with HSV-1 being responsible for greater than 90% of ocular HSV infections (13, 40, 41, 54, 55).
HSV infection generally begins at a mucosal surface. The virus can then move to the ganglionic sensory neurons and establish a latent infection that persists during the lifetime of the individual (20). Throughout the life of a latently infected individual, the virus has the potential to reactivate and travel back to the eye, causing recurrent disease. Indeed, reactivation of latent HSV-1 is a major cause of corneal scarring (CS) (4, 16), and this may correlate with the load of latent virus in the trigeminal ganglia (TG) (12, 58).
It is well established that HSV-1-induced eye disease, and thus HSV-1-induced corneal blindness, is the result of immune responses triggered by the virus (6, 19, 34, 47). Consistent with this, patients with enhanced immune responses develop the worst clinical manifestations of herpetic stromal disease (14, 59). However, the exact identity of the immune responses, including the fine specificity of the potentially harmful effector T cells expressing classic T-cell receptor αβ antigen receptors, which leads to CS, remains an area of intense controversy (2, 35, 69). It is thought that the preexisting inflammatory immune responses may contribute to the damaging effects that are manifested during recurrent infections. This would explain why CS in humans is much more likely to occur following recurrent ocular HSV infections rather than upon primary infections (8, 19, 20).
In this study, we sought to determine if the severity of CS in mice ocularly infected with HSV-1 is associated with (i) increased virus replication in the eye during primary infection; (ii) the load of latent virus, as determined by the amount of latency-associated transcript (LAT); and (iii) increased levels of various immune-related mRNAs in TG and/or corneas (as determined by TaqMan reverse transcription [RT]-PCR). To address these questions, we examined the corneas and TG of mice that survived for 30 days postinfection and compared the results obtained from mice that had not developed CS in either eye to those that had developed severe CS in both eyes. Our results suggest a strong correlation among the severity of eye disease, the load of latent virus in the TG, and the amount of PD-1 (programmed death 1) mRNA in TG. PD-1 is associated with exhaustion (inactivation) of CD8+ T cells, suggesting the possibility that increased PD-1 levels might result in decreased functional CD8+ T cells at the site of latency, thus resulting in more latent virus.
MATERIALS AND METHODS
Virus and cells.
Plaque-purified HSV-1 McKrae, a neurovirulent HSV-1 strain, was grown in rabbit skin (RS) cell monolayers in minimal essential medium containing 10% fetal calf serum, as described previously (29).
Mice.
BALB/cJ (female; 6-week-old) mice were obtained from the Jackson Laboratory (Bar Harbor, ME). The animals were handled in accordance with the Association for Research in Vision and Ophthalmology statement for the Use of Animals in Ophthalmic and Vision Research. The mice were infected with 1 × 105 PFU of HSV-1 McKrae per eye, administered as a 2-μl eye drop without corneal scarification.
Evaluation of CS.
Clinical eye disease patterns were determined by examining the eyes of the mice on day 28 postinfection. HSV-induced CS (epithelial keratitis) was evaluated by slit lamp biomicroscopy using 1% fluorescein stain. The magnitude of stromal disease was scored as 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, or 4, with 0, 1, 2, 3, and 4 representing no disease and disease involving 25, 50, 75, and 100% of the corneal surface, respectively.
Analysis of replication and clearance of HSV-1 from the eye.
Eyes were swabbed once daily on days 1, 3, and 5 post-ocular infection with a Dacron swab (Spectrum type 1). The swab was transferred to a 12- by 75-mm culture tube containing 1 ml of medium, frozen, and thawed, and the virus titers were determined using standard plaque assays on RS cells.
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
Corneas and TG from individual mice that survived ocular infection were collected on day 30 postinfection, immersed in RNAlater RNA stabilization reagent, and stored at −80°C until they were processed. Tissue processing of corneas and TG, total-RNA extraction, and RNA yield were carried out as we have described previously (48, 49). Following RNA extraction, 1,000 ng of total RNA was reverse transcribed using random hexamer primers and murine leukemia virus reverse transcriptase from the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA), in accordance with the manufacturer's recommendations.
The differences in the expression levels of LAT (stable 2-kb LAT intron), CD4, CD8, F4/80, CD11b, CD11c, CD45RO, interleukin-2 (IL-2), IL-4, gamma interferon (IFN-γ), lymphotoxin (LT), granzyme A (GzmA), GzmB, and PD-1 RNAs were evaluated using custom-made TaqMan Gene Expression primers described below. F4/80 was used as an estimate of the presence of macrophages in the corneas and TG, while CD11b was used to estimate the presence of granulocytes, macrophages, myeloid-derived dendritic cells, natural killer cells, microglia, and B-1 cells. Relative copy numbers for the LAT were calculated using standard curves generated from the plasmid pGEM5317. In all experiments, GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used for normalization of transcripts.
The expression levels of the various transcripts were evaluated using commercially available TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) with optimized primer and probe concentrations. Primer-probe sets consisted of two unlabeled PCR primers and the 6-carboxyfluorescein (FAM) dye-labeled TaqMan MGB probe formulated into a single mixture. Additionally, all cellular amplicons included an intron-exon junction to eliminate signal from genomic-DNA contamination. The assays used in this study were as follows: (i) CD4, ABI assay identifier (ID) Mm00442754_m1, amplicon length = 72 bp; (ii) CD8 (α chain), ABI assay ID Mn01182108_m1, amplicon length = 67 bp; (iii) IFN-γ, ABI assay ID Mm00801778_m1, amplicon length = 101 bp; (iv) GzmA, ABI assay ID Mm00439190_m1, amplicon length = 77 bp; (v) GzmB, ABI assay ID Mm00442834_m1, amplicon length = 95 bp; (vi) IL-2, ABI assay ID Mm00434256_m1, amplicon length = 82 bp; (vii) IL-4, ABI assay ID Mm00445259_m1, amplicon = 79 bp; (viii) CD11b (IntegrinαM), ABI assay ID Mm00434455_m1, amplicon length = 69 bp; (ix) CD11c (IntegrinαX), ABI assay ID Mm00498698_m1, amplicon length = 61 bp; (x) F4/80 (Emr1, EGF-like module containing mucin-like hormone receptor-like sequence 11), PD-1 (also known as CD279), ABI assay ID Mm00435532_m1, amplicon size = 65 bp; and (xi) GAPDH, ABI assay ID Mm999999.15_G1, amplicon length = 107 bp.
Additionally, custom-made primer and probe sets were used as follows: (i) HSV-1 LAT, forward primer (5′-GGGTGGGCTCGTGTTACAG-3′), reverse primer (5′-GGACGGGTAAGTAACAGAGTCTCTA-3′), and probe (5′-FAM-ACACCAGCCCGTTCTTT-3′), amplicon length = 81 bp, and (ii) CD45RO (three of seven exons), forward primer (5′-TTTGTCACAGGGCAAACACCTA-3′), reverse primer (5′-GCAGTCATGTAGCGAAAACTTGT-3′), and probe (5′-FAM-CACCCAGTGATGGGACTG-3′), amplicon length = 63 bp.
Quantitative real-time PCR was performed using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) in 384-well plates as we described previously (48, 49). Real-time PCR was performed in triplicate for each tissue sample. The threshold cycle values, which represent the PCR cycle at which there is a noticeable increase in the reporter fluorescence above baseline, were determined using SDS 2.2 software.
Statistical analysis.
Data are presented as the mean and standard error, unless otherwise indicated. Prior to analysis, all continuous data were assessed for normality of distribution using the Kolmogorov-Smirnov test. A two-sided Student's t test or Wilcoxon rank sum test was used, as appropriate, to test for significant differences between animals with and without CS. To assess the relationship of CS (an ordinal variable on a scale of 0 to 5) with the LAT copy number and the various outcome measures (Table 1), the nonparametric Spearman's coefficient of rank correlation (rs) was calculated. A P value of <0.05 was the criterion for significance. All analyses were performed using the SAS statistical software package, version 9.1 (SAS Institutes Inc., Cary, NC).
TABLE 1.
Correlations of RNA levels in the TG and corneas of mice with increased CSa
| RNA transcript | Correlation with CS of increased levels in:
|
|
|---|---|---|
| TG | Cornea | |
| LAT | Yesb | Not detected |
| IL-2 | No | Yesb |
| IL-4 | Yesb | Yesb |
| IFN-γ | No | Yesc |
| CD4 | Yesb | Yesb |
| CD8 | Yesc | Yesb |
| F4/80 | Yesb | Yesb |
| CD11b | Yesb | No |
| CD11c | No | Yesc |
| CD45RO | No | Yesc |
| GzmA | Yesc | Yesc |
| GzmB | Yesc | Yesc |
| PD-1 | Yesc | Not done |
Statistical analyses were performed using Spearman's coefficient of rank correlation as described in Materials and Methods. Yes means that there was a statistically significant correlation between increased levels of the RNA transcripts and mice having CS in both eyes (rather than no CS in both eyes). No means no correlation.
Correlations were significant at a P value of <0.05.
Correlations were significant at a P value of <0.01.
RESULTS
Severity of CS after ocular infection of mice with HSV-1.
Two hundred mice from two separate experiments (100 mice/experiment) were infected ocularly with 1 × 105 PFU/eye of HSV-1 strain McKrae. Tear films were collected from all infected mice on days 1, 3, and 5 post-ocular infection for determination of virus replication in the eye. On day 30 postinfection, 63 of the 200 (31%) infected mice had survived ocular infection. The eyes of the surviving mice were evaluated for CS and its severity. We found that (i) 10 mice had no CS in either eye, (ii) 33 mice had CS in one eye, and (iii) 20 mice had CS in both eyes. As would be expected, the average CS score for mice with CS in both eyes was significantly higher than for mice with CS in one eye (Fig. 1A) (P < 0.0001), and the CS score in mice with no CS in either eye was lower than that of either of the other groups (Fig. 1A) (P < 0.0001). Thus, we were able to establish at least three classes of HSV-1-associated eye disease in BALB/c mice.
FIG. 1.

CS and virus replication in infected mice. (A) CS in ocularly infected mice. Mice were ocularly infected with HSV-1, and on day 30 postinfection, the surviving mice were evaluated for the presence or absence of CS and the severity of the CS. Based on the presence of CS, the mice were separated into three groups: those with no CS (n = 20 eyes), those with CS in one eye (n = 66 eyes), and those with CS in both eyes (n = 40 eyes). The CS score represents the average severity score for CS ± standard error of the mean (SEM) for each group. (B) Virus replication in eye swabs from surviving mice. Tear films were collected from the infected mice on days 1, 3, and 5 postinfection. The virus titers in the eye swabs that had been obtained from the mice that survived were determined by standard plaque assays. Each point represents the mean titer of 20, 66, and 40 eyes from two separate experiments ± SEM for the groups with no CS, CS in one eye, and CS in both eyes, respectively.
Lack of correlation of virus replication in mouse tears with severity of CS.
The virus titers in the tear films that had been collected on days 1, 3, and 5 post-ocular infection from the 63 surviving mice were determined using plaque assays on RS cells. There were no significant differences among the virus titers in the tear films of mice with CS in both eyes, one eye, or neither eye (Fig. 1B) (P > 0.05). Thus, it appeared that there was no direct correlation between acute virus replication in the eye on day 1, 3, or 5 postinfection and the severity of CS 30 days postinfection.
Correlation between severity of CS and LAT RNA levels in the TG.
Clinically, HSV-1-induced eye disease is usually a recurrent disease and hence is associated with viral reactivation in the TG and the return of virus to the cornea. It is well established that LAT plays an important role in HSV-1 reactivation in animal models of ocular HSV-1 infection (12, 23, 57, 58, 66). During HSV-1 neuronal latency, only the LAT is consistently expressed at high levels (21, 57). In this study, we sought to determine whether the severity of CS correlated with a higher load of latent virus in the TG, as judged by LAT expression levels. After euthanization on day 30, the corneas and TG from the surviving mice were harvested, and the levels of LAT were determined by TaqMan RT-PCR performed on the total RNA isolated from the two pooled TG from each individual mouse and on the two pooled corneas from each individual mouse. The amount of LAT RNA detected in the TG of mice with no CS was significantly smaller than the amount detected in the TG of mice that had CS in both eyes (Fig. 2) (P = 0.002). In addition, the severity of CS in mice having CS in both eyes was significantly associated with LAT copy numbers (rs = 0.65; P < 0.001). These results showed a correlation between the severity of CS and the level of LAT RNA in TG. In contrast, no LAT RNA or viral DNA was detected in the corneas of any mice at this time (not shown), suggesting that HSV-1 does not establish latency in mouse corneas.
FIG. 2.
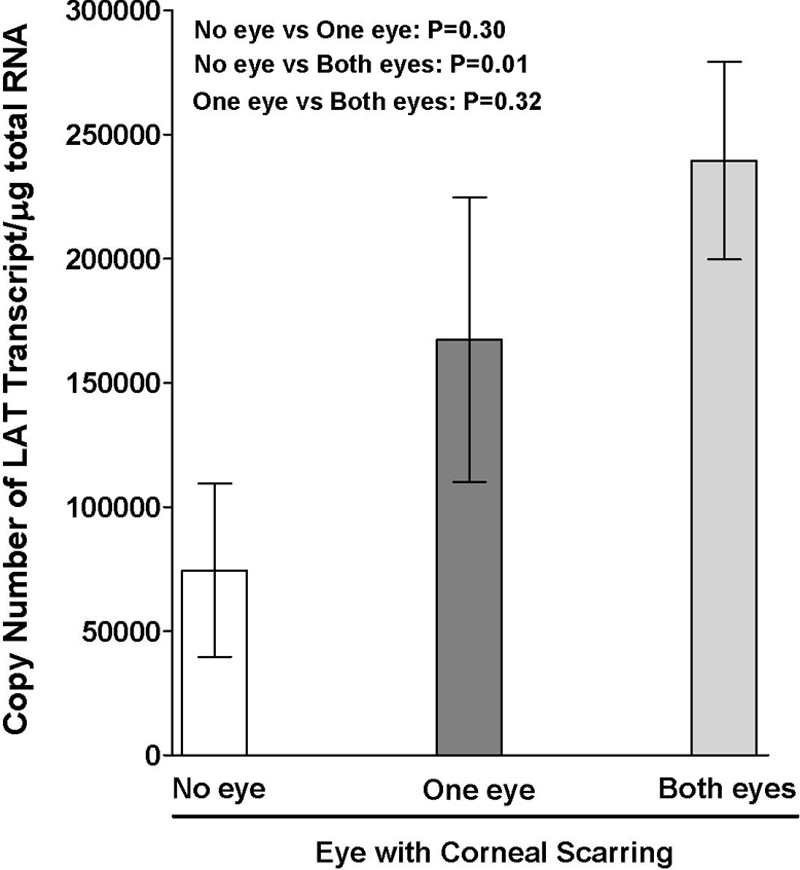
Detection of LAT in TG of latently infected mice. TG were harvested from the latently infected surviving mice, as described in the legend to Fig. 1, at day 30 postinfection. Quantitative RT-PCR was performed on the TG of the individual mice. In each experiment, an estimated relative copy number of LAT was calculated using standard curves generated from pGem-5317. Briefly, the DNA template was serially diluted 10-fold so that 5 μl contained from 103 to 1011 copies of LAT and then subjected to TaqMan PCR with the same set of primers. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standard, the copy number for each reaction was determined. GAPDH expression was used to normalize the relative expression of LAT in the TG. Each point represents the mean ± standard error of the mean for mice with no CS (n = 10), CS in one eye (n = 33), and CS in both eyes (n = 20).
Although the amounts of LAT RNA in the TG of mice with CS in one eye appeared to be greater than in mice with no CS and less than in mice with CS in both eyes, these results did not reach statistical significance. This may have resulted from lack of statistical power or from pooling and processing of RNAs from both TG. Regardless of the reason, in subsequent experiments we did not include the group of mice with CS in one eye and focused on comparisons of mice with no CS versus mice with CS in both eyes.
Mice with CS in both eyes had enhanced inflammatory responses (T cells, antigen-presenting cells, TH1/TH2 cytokines, and cytotoxic molecules) in corneas and TG.
Both T-cell-dependent immune responses (22, 28, 32, 33, 46) and T-cell-independent immune responses (7, 25, 53, 64, 65) have been implicated in HSV-1-induced CS. In HSV-1, either TH1 responses (1, 52), or TH2 responses (30, 31, 36), or both responses (26), have been reported to play a role in the development of herpes stromal keratitis (HSK). To investigate potential differences in TG and corneas from mice with no CS and those from mice with CS in both eyes, we performed real-time PCR in the same TG and corneal samples described above to determine expression levels of mRNAs characteristic of T cells (CD4 and CD8), antigen-presenting cells (CD11b, CD11c, and F4/80), and the TH1/TH2 cell ratio (IL-2, IL-4, and IFN-γ), as well as the expression levels of cytotoxic molecules (LT, GzmA, and GzmB).
The levels of IL-2 transcripts in the corneas of mice with CS were significantly higher than the levels in mice with no CS (Fig. 3A, cornea) (P < 0.0001). In contrast, the apparent difference in IL-2 mRNA levels in the TG did not reach significance (Fig. 3A, TG) (P = 0.32). The levels of IL-4 (Fig. 3B) and IFN-γ (Fig. 3C) transcripts were significantly higher in both the TG and corneas of mice with CS in both eyes than in mice with no CS. The levels of IL-2 transcripts and IFN-γ transcripts were higher in the TG than in the corneas of infected mice, although the reverse was true for IL-4 (Fig. 3A, B, and C). This set of experiments revealed a correlation between CS in both eyes and higher expression of IL-4 and IFN-γ transcripts in the corneas and TG. Since the group of mice with CS in both eyes had higher levels of LAT expression, there was also a correlation between higher levels of IL-4 and IFN-γ mRNAs in the cornea and TG, as well as the amount of latency (as judged by the amount of LAT RNA).
FIG. 3.
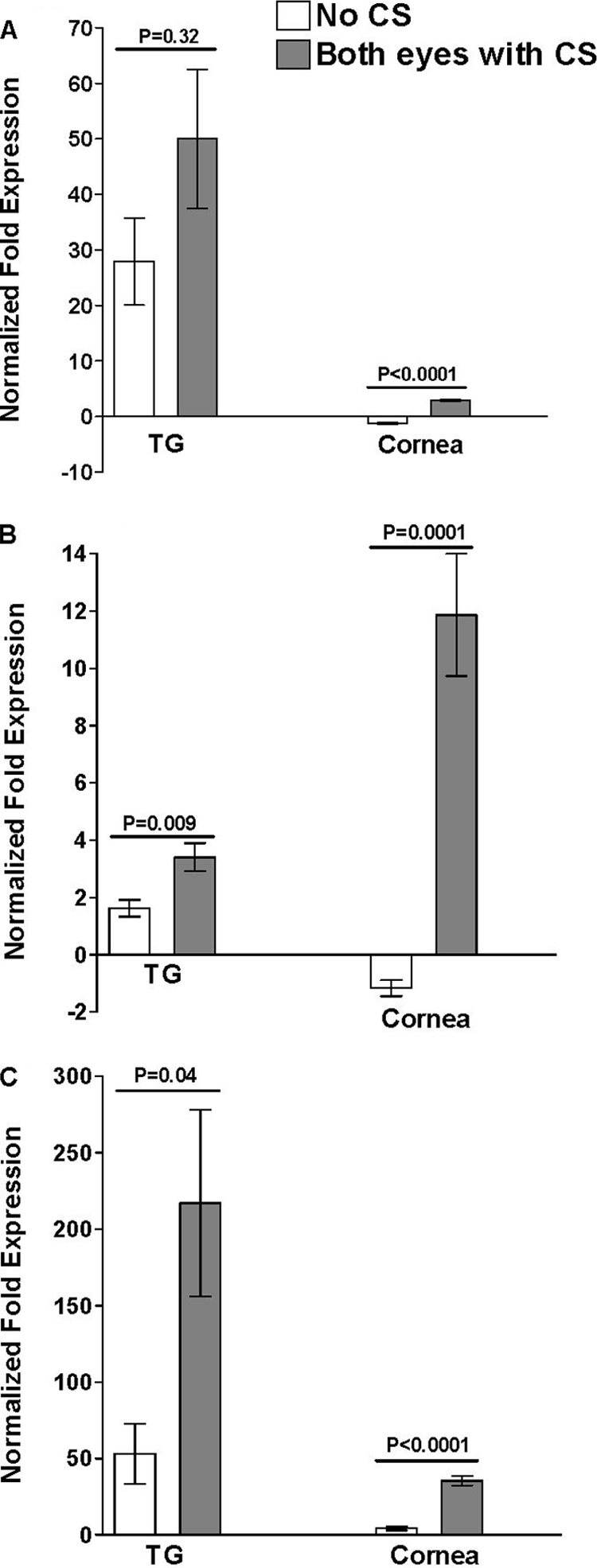
Effect of higher level of latency and CS on TH1/TH2 transcripts in TG and corneas of latently infected mice. The TG and corneas of surviving mice from the experiments described in the legends to Fig. 1 and 2 were isolated individually on day 30 postinfection, and quantitative RT-PCR was performed using total RNA. IL-2, IL-4, and IFN-γ expression in naive mice was used to estimate the relative expression of each transcript in the TG and corneas of mice with no CS and mice with CS in both eyes. GAPDH expression was used to normalize the relative expression of each transcript in the corneas and TG of latently infected mice. Each point represents the mean ± standard error of the mean from five and seven mice for no CS and mice with CS in both eyes, respectively. (A) IL-2 transcript. (B) IL-4 transcript. (C) IFN-γ transcript.
The levels of CD4 (Fig. 4A), CD8 (Fig. 4B), F4/80 (Fig. 4C), CD11b (Fig. 4D), CD11c (Fig. 4E), CD45RO (Fig. 4F), GzmA (Fig. 4G), and GzmB (Fig. 4H) transcripts were also significantly higher in the TG and corneas from mice with CS in both eyes than in those from mice with no CS. No LT mRNA was detected in the corneas or TG of mice with or without CS (not shown). In summary, we detected higher levels of IL-2, IL-4, IFN-γ, CD4, CD8, F4/80, CD11c, CD45RO, and GzmA and -B mRNAs in the corneas of mice with CS in both eyes than in those from mice with no CS (P < 0.05). In TG, mice with CS in both eyes appeared to have significantly higher levels of IL-4, IFN-γ, CD4, CD8, F4/80, CD11b, CD11c, PD1, GzmA, and GzmB mRNAs. No significant differences were detected in IL-2 or CD45RO mRNA levels in the TG. Comparison of the levels of the various transcripts in the corneas versus the TG of mice having CS in both eyes indicated three patterns: (i) the levels of IL-2, IFN-γ, CD8, CD11b, CD11c, and GzmB transcripts were higher in the TG than in the corneas; (ii) the levels of IL-4, CD4, CD11c, and CD45RO transcripts were higher in the corneas than in the TG; and (iii) the levels of F4/80 and GzmA transcripts were similar in the corneas and the TG.
FIG. 4.

Association of the level of latency and severity of CS with levels of CD4, CD8, CD11b, CD11c, F4/80, GzmA, GzmB, and CD45RO transcripts in TG and corneas of latently infected mice. Quantitative RT-PCR was performed using total RNA isolated from TG and corneas of surviving mice from the experiments described in the legends to Fig. 1, 2, and 3. CD4, CD8, CD11b, CD11c, F4/80, CD45RO, GzmA, and GzmB expression in naive mice was used to estimate the relative expression of each transcript in the TG and corneas of mice with no CS and mice with CS in both eyes. GAPDH expression was used to normalize the relative expression of each transcript in corneas and TG of latently infected mice. Each point represents the mean ± standard error of the mean for mice with no CS (n = 5) and mice with CS in both eyes (n = 7). (A) CD4 transcript. (B) CD8 transcript. (C) F4/80 transcript. (D) CD11b transcript. (E) CD11c transcript. (F) CD45RO transcript. (G) GzmA transcript. (H) GzmB transcript.
Elevated expression of PD-1 mRNA in mice with CS in both eyes.
CD8+ T cells have been reported to provide active surveillance of HSV-1 gene expression in latently infected sensory neurons (38, 43, 44), thus preventing reactivation of virus. Recently, it was shown that exhaustion of CD8+ T cells is a major factor allowing persistent infection by lymphocytic choriomeningitis virus (LCMV) in mice (3). Since our experiments described above suggested that CD8 mRNA levels were significantly elevated in TG of mice with more severe CS and that these mice had more latent virus, it was of interest to look for evidence of exhausted CD8+ T cells. This was done by looking for elevated mRNA levels of PD-1, indicative of exhausted (or impaired) CD8+ T cells (3), in TG of mice with CS in both eyes compared to mice with no CS, as described above for other transcripts. At present, PD-1 is the main marker to identify exhausted CD8+ T cells (67). We found significantly more PD-1 mRNA in TG of mice with CS in both eyes than in TG from mice with no CS (Fig. 5) (P < 0.0001). There was a significant positive correlation between the levels of PD-1 and CD8 transcripts in the TG (rs = 0.727; P = 0.007). These results show a correlation between more severe CS and higher levels of PD-1 mRNA. This suggests that many of the CD8+ T cells in the TG of these mice might be exhausted and therefore not capable of playing a significant role in control of the virus. This, in turn, may account for the increased amount of latency detected in the TG of mice with CS in both eyes.
FIG. 5.
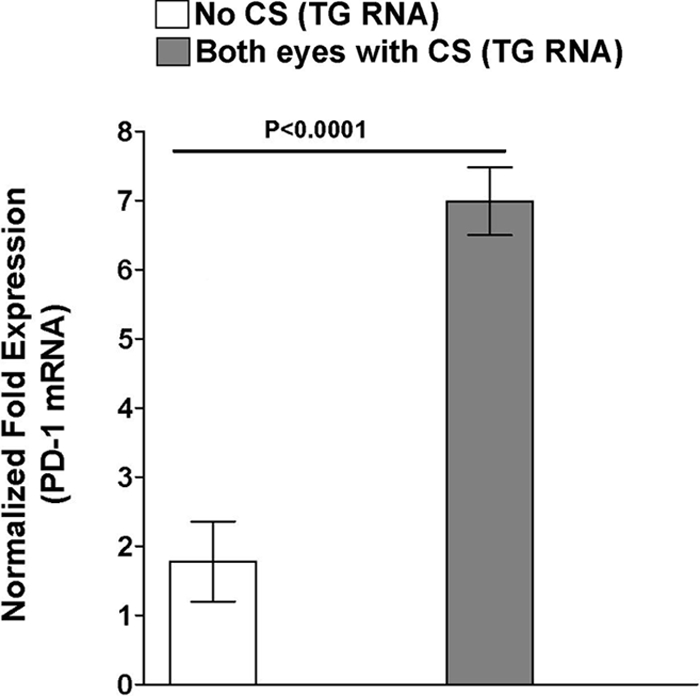
Association of latency and CS with level of PD-1 transcript in the TG of latently infected mice. Quantitative RT-PCR was performed on total RNA isolated from the TG of surviving mice from the experiments described in the legends to Fig. 1 to 4. PD-1 expression in TG of naive mice was used to estimate the relative expression of PD-1 transcript in TG of mice with no CS and mice with CS in both eyes. GAPDH expression was used to normalize the relative expression of each transcript in corneas and TG of latently infected mice. Each point represents the mean ± standard error of the mean from mice with no CS (n = 5) and mice with CS in both eyes (n = 7).
Association of LAT copy number with the severity of CS.
Finally, the correlation between the severity of CS and the levels of expression of the inflammatory responses was assessed in the TG and corneas of latently infected mice. The correlative results of the above-mentioned studies are summarized in Table 1. As shown in Table 1, in the TG, the LAT copy number and PD-1 mRNA levels, as well as IL-4, CD4, CD8, F4/80, CD11b, and GzmA and -B mRNA levels, were all significantly positively associated with the severity of CS. There was no difference in the IL-2, IFN-γ, or CD45RO mRNA levels in the TG. In the cornea, the IL-2, IL-4, IFN-γ, CD4, CD8, F4/80, CD11c, CD45RO, and GzmA and -B mRNA levels were all significantly positively associated with the severity of CS; however, there was no difference in CD11b mRNA levels in the cornea. These correlations suggest that there are strong relationships between the severity of CS and higher latency in the corneas and TG of latently infected mice.
DISCUSSION
In this study, we found a correlation between the severity of CS and the levels of many of the transcripts we examined in the TG of latently infected mice. These included the amount of LAT (which is indicative of the relative amount of HSV-1 latency) and the PD-1, IL-4, CD4, CD8, F4/80, CD11b, and GzmA and -B mRNA levels. In the cornea, the IL-2, IL-4, IFN-γ, CD4, CD8, F4/80, CD11c, CD45RO, and GzmA and -B mRNA levels also correlated with the severity of CS. Although these correlations do not demonstrate cause and effect, it is tempting to speculate that increased viral latency and increased HSK are functionally related in some manner.
Following ocular infection with HSV-1, the virus travels on microtubules toward neuronal cell bodies in a retrograde direction and establishes latency in the TG. The latent virus can be reactivated spontaneously and return to the eye by traveling on the microtubules in an anterograde direction (18). The vast majority of primary HSV infections are either asymptomatic or so mildly symptomatic as to go almost completely unrecognized (11, 14, 20, 61). In contrast, reactivation of virus from latency and return to the original site of infection can cause significant recurrent disease. Recurrent eye disease after HSV-1 reactivation is a major cause of corneal disease and blindness (5, 41, 42). It is well established that HSV-1-induced CS, which also is broadly referred to as HSK, is due to immune responses triggered by the virus (19, 34, 47).
Following experimental ocular infection of mice, the virus is cleared from the eye by day 7 to 10 postinfection. HSK first appears after day 10 (i.e., after the acute infection has been cleared) and may increase up to day 20 postinfection, after which the resultant CS remains unchanged. The most commonly used animal model of HSK is mice. However, in contrast to HSV-1-infected humans or rabbits, spontaneous virus reactivation in mice is rare (45). The evidence generated using the mouse model suggests that eye disease may be associated with immune responses triggered by the virus, rather than the virus directly. One approach to studying the anterograde transport of infiltrates and/or virus from the TG to the cornea is ablation of the TG. Several compounds, including kainic acid, caspazine, and resiniferatoxin, have been used for ablation of the TG without affecting vision (10, 51); however, such studies are difficult to perform.
To our knowledge, there have been no previous reports in which the amount of latency in the TG and the levels of various immune factors in the TG and the cornea have been systematically compared to the amounts of HSK in different groups of identically infected, but otherwise untreated, mice. In this study, we ocularly infected a large number (200) of inbred mice (BALB/c) under identical conditions and analyzed the data generated in terms of the incidence of CS in mice surviving 30 days postinfection. We grouped the mice into three groups: (i) mice with no CS in either eye, (ii) mice with CS in one eye, and (iii) mice with CS in both eyes. These groupings reflected the severity of CS in the eye. This approach formed a useful model to (i) determine whether mice with more severe CS had higher virus replication in the eye during the primary acute infection and/or a greater load of latent virus in their TG and (ii) analyze various factors that correlate with more severe CS and, as it turned out, higher latency in the TG.
We found a direct correlation between the severity of CS and higher levels of latency in the TG, as judged by the level of LAT RNA. In addition, we found a correlation in the levels of certain mRNAs between the corneas and TG of mice. This may suggest a bidirectional movement of infiltrates between the cornea and the TG. Previously, it has been reported that an HSV-1 US9 mutant virus caused keratitis, even though it was unable to return from the ganglia to the cornea (56). The authors concluded that herpes keratitis can occur without anterograde transport from the ganglia to the cornea through a process that was probably mediated by virus that persisted in the cornea. HSV-1 DNA has been found in the corneas of latently infected rabbits and individuals with a history of ocular herpes infection (9, 17, 37). However, it should be noted that in humans and rabbits, reactivation of virus in the TG followed by shedding of infectious virus in tears is common. Thus, detection of HSV-1 DNA in the corneas of rabbits and humans does not demonstrate latent or persistent infection by virus in the cornea. In this report, we did not detect any LAT RNA in the corneas of any of the infected mice, including those with CS. This is consistent with the extremely low level of HSV-1 spontaneous reactivation in the mouse and argues against latent or persistent infection in mouse corneas.
In infected mice, the development of HSK starts shortly after the virus is cleared from the eyes. This suggests that in mice, HSK is due to the immune response to remnants of the acute infection and/or movement of infiltrates and/or viral proteins from the TG into the cornea. Consistent with the movement of viral proteins from the TG to the cornea, the US9 protein, which is necessary for long-distance anterograde axonal transport of viral nucleocapsids, is not necessary for transport of virus envelope (39). Thus, it is possible that virus proteins returning to the mouse eye in the absence of reactivation of infectious virus could lead to inflammatory responses and corneal disease.
It is well accepted that HSV-1-induced CS is the result of immunopathology (41, 68). Patients with enhanced immune responses develop the worst clinical manifestations of herpetic stromal disease (14, 59). The cornea is a highly organized group of cells and proteins, but in contrast to most of the tissues in the body, it does not normally contain blood or lymphatic vessels (15). However, vascularization of the cornea and infiltration can occur following surgical manipulation, in certain corneal diseases, or on viral infection (63). Infiltrates can also be delivered into the eye following breakdown of the blood-eye barrier (62). In the course of this study, we examined the immune responses in the spleens of the infected mice, but we did not detect any differences in the levels of the transcripts between mice with no eye disease and mice having severe CS in both eyes (data not shown), which suggests that disruption of the blood-eye barrier did not contribute to the CS.
Our results indicate that there were higher levels of inflammatory infiltrates in the TG and corneas of the mice with CS in both eyes than in those of mice with no CS. We also found a correlation between increased levels of latent virus in the TG (as judged by the level of LAT) of mice with CS in both eyes compared to mice with no CS. Although this does not indicate any direct cause-and-effect relationship between latency in mouse TG and CS, it is possible that the latent virus in the mouse TG is involved in the initiation of HSK, as might be expected in humans and rabbits, in which HSK is known to be related to reactivation of virus in the TG.
One of the most interesting findings relates to the possible exhaustion of CD8+ T cells in the TG of mice with CS in both eyes and hence in mice with higher latency. We found higher CD8 mRNA levels, which may be indicative of more CD8+ T cells, in the TG of mice with greater CS and latency. CD8+ T cells play a major role in virus clearance during primary virus infection of mice (27). In addition, CD8+ T cells appear to provide active surveillance of HSV-1 gene expression in latently infected sensory neurons (38, 43, 44), thus preventing reactivation of virus. Interestingly, the exhaustion of CD8+ T cells appears to be a major factor leading to persistent LCMV infections in mice (3), suggesting the possibility that similar exhaustion of CD8+ T cells might lead to increased HSV-1 latency. The CD8+ T-cell exhaustion (impairment) was associated with upregulation of the PD-1 gene. This was shown to be functional, since in vivo administration of antibodies that blocked the interaction of this inhibitory receptor with its ligand, PD-L1 (also known as B7-H1 or CD274), enhanced T-cell responses and clearance of LCMV (3). Consistent with the LCMV studies, we found significantly higher levels of PD-1 transcript in TG of mice with higher HSV-1 latency (i.e., CS in both eyes). This suggested that these mice had elevated PD-1, and hence, elevated exhaustion of CD8+ T cells. Thus, even though the mice with higher levels of HSV-1 latency appeared to have more CD8+ T cells, many, if not most, of these CD8+ T cells may have been exhausted CD8+ T cells that were not able to clear the virus from the TG either prior to or after establishment of latency. However, since the studies presented here were done using total RNA from TG, it remains to be shown that the elevated CD8 mRNA levels are due to increased levels of CD8+ T cells and that the elevated PD-1 mRNA levels reflect increased expression of PD-1 in the CD8+ T cells.
Recently, we reported that the number of lymphoid-related (CD11c+ CD8α+) dendritic cells correlated with the amount of HSV-1 latency in mouse TG (50). PD-1 has two ligands, PD-L1 (B7-H1) (24) and PD-L2 (B7-DC) (60). Thus, it is possible that lymphoid-related dendritic cells may increase the engagement of PD-L1 and/or PD-L2 with PD-1, leading to impairment of CD8+ T cells and establishment of latency. Regardless of the mechanism, if elevated PD-1 leads to increased latency, blockade of PD-L/PD-1 interactions might be a potentially effective strategy for the prevention and/or reduction of latent herpesvirus infections and hence recurrent herpes disease.
Acknowledgments
This work was supported by Public Health Service grants EY14966 and EY13615 from the National Eye Institute to H.G.
Footnotes
Published ahead of print on 17 December 2008.
REFERENCES
- 1.Babu, J. S., S. Kanangat, and B. T. Rouse. 1995. T cell cytokine mRNA expression during the course of the immunopathologic ocular disease herpetic stromal keratitis. J. Immunol. 1544822-4829. [PubMed] [Google Scholar]
- 2.Banerjee, K., S. Deshpande, M. Zheng, U. Kumaraguru, S. P. Schoenberger, and B. T. Rouse. 2002. Herpetic stromal keratitis in the absence of viral antigen recognition. Cell Immunol. 219108-118. [DOI] [PubMed] [Google Scholar]
- 3.Barber, D. L., E. J. Wherry, D. Masopust, B. Zhu, J. P. Allison, A. H. Sharpe, G. J. Freeman, and R. Ahmed. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439682-687. [DOI] [PubMed] [Google Scholar]
- 4.Barron, B. A., L. Gee, W. W. Hauck, N. Kurinij, C. R. Dawson, D. B. Jones, K. R. Wilhelmus, H. E. Kaufman, J. Sugar, R. A. Hyndiuk, et al. 1994. Herpetic Eye Disease Study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 1011871-1882. [DOI] [PubMed] [Google Scholar]
- 5.Binder, P. S. 1977. Herpes simplex keratitis. Surv. Ophthalmol. 21313-331. [DOI] [PubMed] [Google Scholar]
- 6.Brandt, C. R. 2005. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Exp. Eye Res. 80607-621. [DOI] [PubMed] [Google Scholar]
- 7.Brandt, C. R., and C. A. Salkowski. 1992. Activation of NK cells in mice following corneal infection with herpes simplex virus type-1. Invest. Ophthalmol Vis. Sci. 33113-120. [PubMed] [Google Scholar]
- 8.Brown, D. C., A. B. Nesburn, J. S. Nauheim, D. Pavan-Langston, and H. E. Kaufman. 1968. Recurrent herpes simplex conjunctivitis. Arch. Ophthalmol. 79733-735. [DOI] [PubMed] [Google Scholar]
- 9.Cantin, E., J. Chen, D. E. Willey, J. L. Taylor, and W. J. O'Brien. 1992. Persistence of herpes simplex virus DNA in rabbit corneal cells. Invest. Ophthalmol Vis. Sci. 33:2470-2475. [PubMed] [Google Scholar]
- 10.Caterina, M. J., M. A. Schumacher, M. Tominaga, T. A. Rosen, J. D. Levine, and D. Julius. 1997. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389816-824. [DOI] [PubMed] [Google Scholar]
- 11.Cobo, M. 1988. Ocular herpes simplex infections. Mayo Clin. Proc. 631154-1156. [DOI] [PubMed] [Google Scholar]
- 12.Cook, S. D., M. J. Paveloff, J. J. Doucet, A. J. Cottingham, F. Sedarati, and J. M. Hill. 1991. Ocular herpes simplex virus reactivation in mice latently infected with latency-associated transcript mutants. Invest. Ophthalmol Vis. Sci. 321558-1561. [PubMed] [Google Scholar]
- 13.Corey, L. 1994. The current trend in genital herpes. Progress in prevention. Sex. Transm. Dis. 21S38-S44. [PubMed] [Google Scholar]
- 14.Corey, L., and P. G. Spear. 1986. Infections with herpes simplex viruses. N. Engl. J. Med. 314686-691. [DOI] [PubMed] [Google Scholar]
- 15.Cursiefen, C., L. Chen, M. R. Dana, and J. W. Streilein. 2003. Corneal lymphangiogenesis: evidence, mechanisms, and implications for corneal transplant immunology. Cornea 22273-281. [DOI] [PubMed] [Google Scholar]
- 16.Dawson, C. R. 1984. Ocular herpes simplex virus infections. Clin. Dermatol. 256-66. [DOI] [PubMed] [Google Scholar]
- 17.Devi-Rao, G. B., J. S. Aguilar, M. K. Rice, H. H. Garza, Jr., D. C. Bloom, J. M. Hill, and E. K. Wagner. 1997. Herpes simplex virus genome replication and transcription during induced reactivation in the rabbit eye. J. Virol. 717039-7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diefenbach, R. J., M. Miranda-Saksena, M. W. Douglas, and A. L. Cunningham. 2008. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 1835-51. [DOI] [PubMed] [Google Scholar]
- 19.Dix, R. D. 2002. Pathogenesis of herpes simplex ocular disease, vol. 2. Lippincott, Williams and Wilkins, Philadelphia, PA.
- 20.Dix, R. D. 1987. Prospects for a vaccine against herpes simplex virus types 1 and 2. Prog. Med. Virol. 3489-128. [PubMed] [Google Scholar]
- 21.Dobson, A. T., T. P. Margolis, F. Sedarati, J. G. Stevens, and L. T. Feldman. 1990. A latent, nonpathogenic HSV-1-derived vector stably expresses beta-galactosidase in mouse neurons. Neuron 5353-360. [DOI] [PubMed] [Google Scholar]
- 22.Doymaz, M. Z., and B. T. Rouse. 1992. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Invest. Ophthalmol Vis. Sci. 332165-2173. [PubMed] [Google Scholar]
- 23.Fraser, N. W., T. M. Block, and J. G. Spivack. 1992. The latency-associated transcripts of herpes simplex virus: RNA in search of function. Virology 1911-8. [DOI] [PubMed] [Google Scholar]
- 24.Freeman, G. J., A. J. Long, Y. Iwai, K. Bourque, T. Chernova, H. Nishimura, L. J. Fitz, N. Malenkovich, T. Okazaki, M. C. Byrne, H. F. Horton, L. Fouser, L. Carter, V. Ling, M. R. Bowman, B. M. Carreno, M. Collins, C. R. Wood, and T. Honjo. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 1921027-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghiasi, H., S. Cai, G. C. Perng, A. B. Nesburn, and S. L. Wechsler. 2000. The role of natural killer cells in protection of mice against death and corneal scarring following ocular HSV-1 infection. Antivir. Res. 4533-45. [DOI] [PubMed] [Google Scholar]
- 26.Ghiasi, H., F. M. Hofman, S. Cai, G. C. Perng, A. B. Nesburn, and S. L. Wechsler. 1999. Vaccination with different HSV-1 glycoproteins induces different patterns of ocular cytokine responses following HSV-1 challenge of vaccinated mice. Vaccine 172576-2582. [DOI] [PubMed] [Google Scholar]
- 27.Ghiasi, H., G. Perng, A. B. Nesburn, and S. L. Wechsler. 1999. Either a CD4+or CD8+ T cell function is sufficient for clearance of infectious virus from trigeminal ganglia and establishment of herpes simplex virus type 1 latency in mice. Microb. Pathog. 27387-394. [DOI] [PubMed] [Google Scholar]
- 28.Ghiasi, H., D. C. Roopenian, S. Slanina, S. Cai, A. B. Nesburn, and S. L. Wechsler. 1997. The importance of MHC-I and MHC-II responses in vaccine efficacy against lethal herpes simplex virus type 1 challenge. Immunology 91430-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghiasi, H., S. Slanina, A. B. Nesburn, and S. L. Wechsler. 1994. Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein K. J. Virol. 682347-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghiasi, H., S. L. Wechsler, R. Kaiwar, A. B. Nesburn, and F. M. Hofman. 1995. Local expression of tumor necrosis factor alpha and interleukin-2 correlates with protection against corneal scarring after ocular challenge of vaccinated mice with herpes simplex virus type 1. J. Virol. 69334-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heiligenhaus, A., S. Jayaraman, S. Soukiasian, M. Dorf, and C. S. Foster. 1995. Glycoprotein D (5-23) specific Th2-T-cell line induces HSV-1 keratitis. Ophthalmologe 92484-491. [PubMed] [Google Scholar]
- 32.Hendricks, R. L., R. J. Epstein, and T. Tumpey. 1989. The effect of cellular immune tolerance to HSV-1 antigens on the immunopathology of HSV-1 keratitis. Invest. Ophthalmol. Vis. Sci. 30105-115. [PubMed] [Google Scholar]
- 33.Hendricks, R. L., M. Janowicz, and T. M. Tumpey. 1992. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J. Immunol. 1482522-2529. [PubMed] [Google Scholar]
- 34.Hendricks, R. L., and T. M. Tumpey. 1990. Contribution of virus and immune factors to herpes simplex virus type I-induced corneal pathology. Invest. Ophthalmol. Vis. Sci. 311929-1939. [PubMed] [Google Scholar]
- 35.Huster, K. M., V. Panoutsakopoulou, K. Prince, M. E. Sanchirico, and H. Cantor. 2002. T cell-dependent and -independent pathways to tissue destruction following herpes simplex virus-1 infection. Eur. J. Immunol. 321414-1419. [DOI] [PubMed] [Google Scholar]
- 36.Jayaraman, S., A. Heiligenhaus, A. Rodriguez, S. Soukiasian, M. E. Dorf, and C. S. Foster. 1993. Exacerbation of murine herpes simplex virus-mediated stromal keratitis by Th2 type T cells. J. Immunol. 1515777-5789. [PubMed] [Google Scholar]
- 37.Kaye, S. B., C. Lynas, A. Patterson, J. M. Risk, K. McCarthy, and C. A. Hart. 1991. Evidence for herpes simplex viral latency in the human cornea. Br. J. Ophthalmol 75195-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khanna, K. M., R. H. Bonneau, P. R. Kinchington, and R. L. Hendricks. 2003. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18593-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaVail, J. H., A. N. Tauscher, A. Sucher, O. Harrabi, and R. Brandimarti. 2007. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 146974-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liesegang, T. J. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18127-143. [DOI] [PubMed] [Google Scholar]
- 41.Liesegang, T. J. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 201-13. [DOI] [PubMed] [Google Scholar]
- 42.Liesegang, T. J. 1988. Ocular herpes simplex infection: pathogenesis and current therapy. Mayo Clin. Proc. 631092-1105. [DOI] [PubMed] [Google Scholar]
- 43.Liu, T., K. M. Khanna, B. N. Carriere, and R. L. Hendricks. 2001. Gamma interferon can prevent herpes simplex virus type 1 reactivation from latency in sensory neurons. J. Virol. 7511178-11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu, T., K. M. Khanna, X. Chen, D. J. Fink, and R. L. Hendricks. 2000. CD8+ T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med. 1911459-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Margolis, T. P., F. L. Elfman, D. Leib, N. Pakpour, K. Apakupakul, Y. Imai, and C. Voytek. 2007. Spontaneous reactivation of herpes simplex virus type 1 in latently infected murine sensory ganglia. J. Virol. 8111069-11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mercadal, C. M., D. M. Bouley, D. DeStephano, and B. T. Rouse. 1993. Herpetic stromal keratitis in the reconstituted scid mouse model. J. Virol. 67:3404-3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metcalf, J. F., and H. E. Kaufman. 1976. Herpetic stromal keratitis—evidence for cell-mediated immunopathogenesis. Am. J. Ophthalmol. 82827-834. [DOI] [PubMed] [Google Scholar]
- 48.Mott, K. R., Y. Osorio, D. J. Brown, N. Morishige, A. Wahlert, J. V. Jester, and H. Ghiasi. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol. Vis. 131802-1812. [PubMed] [Google Scholar]
- 49.Mott, K. R., G. C. Perng, Y. Osorio, K. G. Kousoulas, and H. Ghiasi. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J. Virol. 8112962-12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mott, K. R., D. UnderHill, S. L. Wechsler, and H. Ghiasi. 2008. Lymphoid-related CD11c+ CD8a+ dendritic cells are involved in enhancing HSV-1 latency. J. Virol. 829870-9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neubert, J. K., A. J. Mannes, J. Keller, M. Wexel, M. J. Iadarola, and R. M. Caudle. 2005. Peripheral targeting of the trigeminal ganglion via the infraorbital foramen as a therapeutic strategy. Brain Res. Brain Res. Protoc. 15119-126. [DOI] [PubMed] [Google Scholar]
- 52.Niemialtowski, M. G., and B. T. Rouse. 1992. Phenotypic and functional studies on ocular T cells during herpetic infections of the eye. J. Immunol. 1481864-1870. [PubMed] [Google Scholar]
- 53.Oakes, J. E., C. A. Monteiro, C. L. Cubitt, and R. N. Lausch. 1993. Induction of interleukin-8 gene expression is associated with herpes simplex virus infection of human corneal keratocytes but not human corneal epithelial cells. J. Virol. 674777-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oh, J. O., S. J. Kimura, H. B. Ostler, C. R. Dawson, and G. Smolin. 1976. Oculogenital transmission of type 2 herpes simplex virus in adults. Surv. Ophthalmol. 21106-109. [DOI] [PubMed] [Google Scholar]
- 55.Pavan-Langston, D. 1983. Ocular viral infections. Med. Clin. N. Am. 67973-990. [DOI] [PubMed] [Google Scholar]
- 56.Polcicova, K., P. S. Biswas, K. Banerjee, T. W. Wisner, B. T. Rouse, and D. C. Johnson. 2005. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA 102:11462-11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L. Lewis, J. R. Lokensgard, and S. L. Wechsler. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol. 613820-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rootman, D. S., Y. Haruta, and J. M. Hill. 1990. Reactivation of HSV-1 in primates by transcorneal iontophoresis of adrenergic agents. Invest. Ophthalmol. Vis. Sci. 31597-600. [PubMed] [Google Scholar]
- 59.Schmid, D. S. 1988. The human MHC-restricted cellular response to herpes simplex virus type 1 is mediated by CD4+, CD8− T cells and is restricted to the DR region of the MHC complex. J. Immunol. 1403610-3616. [PubMed] [Google Scholar]
- 60.Sharpe, A. H., and G. J. Freeman. 2002. The B7-CD28 superfamily. Nat. Rev. Immunol. 2116-126. [DOI] [PubMed] [Google Scholar]
- 61.Stanberry, L. R. 1986. Herpesvirus latency and recurrence. Prog. Med. Virol. 3361-77. [PubMed] [Google Scholar]
- 62.Streilein, J. W. 1999. Immunoregulatory mechanisms of the eye. Prog. Retin. Eye Res. 18357-370. [DOI] [PubMed] [Google Scholar]
- 63.Streilein, J. W., M. R. Dana, and B. R. Ksander. 1997. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol. Today 18443-449. [DOI] [PubMed] [Google Scholar]
- 64.Tumpey, T. M., H. Cheng, D. N. Cook, O. Smithies, J. E. Oakes, and R. N. Lausch. 1998. Absence of macrophage inflammatory protein-1α prevents the development of blinding herpes stromal keratitis. J. Virol. 723705-3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tumpey, T. M., H. Cheng, X. T. Yan, J. E. Oakes, and R. N. Lausch. 1998. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J. Leukoc. Biol. 63486-492. [DOI] [PubMed] [Google Scholar]
- 66.Wechsler, S. L., A. B. Nesburn, R. Watson, S. M. Slanina, and H. Ghiasi. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J. Virol. 624051-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wherry, E. J., S. J. Ha, S. M. Kaech, W. N. Haining, S. Sarkar, V. Kalia, S. Subramaniam, J. N. Blattman, D. L. Barber, and R. Ahmed. 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27670-684. [DOI] [PubMed] [Google Scholar]
- 68.Wilhelmus, K. R., C. R. Dawson, B. A. Barron, P. Bacchetti, L. Gee, D. B. Jones, H. E. Kaufman, J. Sugar, R. A. Hyndiuk, P. R. Laibson, R. D. Stulting, P. A. Asbell, et al. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Br J. Ophthalmol. 80969-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao, Z. S., F. Granucci, L. Yeh, P. A. Schaffer, and H. Cantor. 1998. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science 2791344-1347. [DOI] [PubMed] [Google Scholar]