

Abstract
The BGLF4 protein kinase of Epstein-Barr virus (EBV) is a member of the conserved family of herpesvirus protein kinases which, to some extent, have a function similar to that of the cellular cyclin-dependent kinase in regulating multiple cellular and viral substrates. In a yeast two-hybrid screening assay, a splicing variant of interferon (IFN) regulatory factor 3 (IRF3) was found to interact with the BGLF4 protein. This interaction was defined further by coimmunoprecipitation in transfected cells and glutathione S-transferase (GST) pull-down in vitro. Using reporter assays, we show that BGLF4 effectively suppresses the activities of the poly(I:C)-stimulated IFN-β promoter and IRF3-responsive element. Moreover, BGLF4 represses the poly(I:C)-stimulated expression of endogenous IFN-β mRNA and the phosphorylation of STAT1 at Tyr701. In searching for a possible mechanism, BGLF4 was shown not to affect the dimerization, nuclear translocation, or CBP recruitment of IRF3 upon poly(I:C) treatment. Notably, BGLF4 reduces the amount of active IRF3 recruited to the IRF3-responsive element containing the IFN-β promoter region in a chromatin immunoprecipitation assay. BGLF4 phosphorylates GST-IRF3 in vitro, but Ser339-Pro340 phosphorylation-dependent, Pin1-mediated downregulation is not responsible for the repression. Most importantly, we found that three proline-dependent phosphorylation sites at Ser123, Ser173, and Thr180, which cluster in a region between the DNA binding and IRF association domains of IRF3, contribute additively to the BGLF4-mediated repression of IRF3(5D) transactivation activity. IRF3 signaling is activated in reactivated EBV-positive NA cells, and the knockdown of BGLF4 further stimulates IRF3-responsive reporter activity. The data presented here thus suggest a novel mechanism by which herpesviral protein kinases suppress host innate immune responses and facilitate virus replication.
The innate immune response is the first-line defense against viral infection. The production of interferons (IFNs) and other cytokines to prevent virus replication and spread is at the center of the antiviral response and requires the activation of multiple transcription activators. The family of IFN regulatory factors (IRFs) is defined by a highly conserved amino-terminal DNA binding domain (DBD) containing five tryptophan repeats and a unique C-terminal domain, the IRF association domain (IAD) (29). IRF3 and IRF7 are two major direct transducers of virus-mediated signaling that induce type I IFNs. IRF3 is a constitutively expressed phosphoprotein of 427 amino acids, which can shuttle into and out of the nucleus in its inactive form. Upon virus infection, cellular TBK-1- and IKKɛ-mediated phosphorylation of serines 385 and 386 and the serine/threonine cluster between amino acids 396 and 405 of IRF3 lead to its conformational change and activation (19, 29, 65).
The activated IRF3 then undergoes homodimerization or heterodimerization with IRF7, nuclear localization, and association with the coactivator CBP/P300 (29). The phosphorylated IRF3/CBP/P300 complex is retained in the nucleus and induces the transcription of the IFN-β gene and other IFN-stimulated genes (ISGs) by binding to the promoter regions of IFN-stimulated response elements (54). In the context of the IFN-β gene, IRF3 cooperates with another two transcription factors, NF-κB and ATF-2/c-Jun, and the chromatin architectural protein HMGI(Y) to form an enhanceosome on the nucleosome-free positive regulatory domain (PRD) regions of the promoter. To achieve fully active transcription, the enhanceosome recruits, in order, histone acetyltransferases, SWI/SNF, and basal transcription factors to modify and reposition a nucleosome that blocks the formation of a transcriptional preinitiation complex on the IFN-β promoter. The recruitment of the GCN5 complex acetylates the nucleosome, leading to the recruitment of the CBP-polymerase II holoenzyme complex. Subsequently, the nucleosome structure is modified further by SWI/SNF remodeling activity, which in turn permits the recruitment of transcription factor IID to the promoter and the activation of transcription (29, 30). The binding of secreted IFNs to their cellular receptor induces the phosphorylation and activation of the Janus kinases JAK1 and Tyk2 and the subsequent phosphorylation of signal transducer and activators of transcription 1 (STAT1) and STAT2, which can cooperate with IRF9 to form ISG factor 3 complexes to promote the expression of downstream genes (30).
The potential activation of IFN signaling or the subsequent upregulation of ISGs was detected during the attachment or entry process of herpesviruses (5, 55, 57). Nevertheless, herpesviruses have evolved with multiple immune evasion strategies to replicate successfully in host cells. Because the synthesis and release of alpha IFN (IFN-α) or IFN-β are initiated mainly by the activation of IRF3 and/or IRF7, it is not surprising that many viruses counteract IFN production by preventing their activation or signaling pathway. For example, herpes simplex virus (HSV), human cytomegalovirus, human herpesvirus 6 (HHV-6), and HHV-8 impair IFN-β production by interfering with IRF3 (1, 20, 35, 51). Moreover, IFN signaling is also impaired in HSV-1-, human cytomegalovirus-, and HHV-8-infected cells to ensure successful virus replication (10, 21, 52, 76).
Epstein-Barr virus (EBV) is a widespread human gammaherpesvirus. EBV is the causative agent of infectious mononucleosis and is closely associated with several human malignant diseases including lymphoma, nasopharyngeal carcinoma, gastric carcinoma, and lymphoproliferative diseases in immunocompromised patients (58). Upon transmission to a naïve host, EBV replicates in a permissive cell type, leading to lifelong persistence with occasional reactivation, which may release infectious virions that are transmissible to a new host. It is believed that EBV also develops multiple mechanisms to counteract the host innate immune response. Indeed, IRF7 is negatively regulated by the immediate-early gene BZLF1 through their physical interaction (24). Other than that, we noticed that the UL protein kinase of HSV-1, UL13, has been implicated as a possible contributor to anti-IFN signaling, while the mechanism involved remains unclear (66). We were then curious to know whether the EBV UL kinase BGLF4 is also involved in regulating the IFN signaling pathway.
BGLF4 kinase is a virion-associated kinase and is expressed at the early stage of the lytic cycle (36, 69). Several viral and cellular substrates including BMRF1, BZLF1, EBNA2, EBNA-LP, and translation factor EF-1δ were found to be phosphorylated by BGLF4 at Cdk1 target sites (2, 38, 39, 72, 74). BGLF4 colocalizes with the viral DNA polymerase processivity factor BMRF1 at the viral DNA replication compartment in virus-replicating cells and phosphorylates BMRF1 at multiple sites in vitro and in vivo (69, 72). BGLF4 kinase also phosphorylates lamin A/C to promote the reorganization of the nuclear lamina to facilitate virion maturation, and the knockdown of BGLF4 resulted in an accumulation of viral nucleocapsids in the nucleus, suggesting that BGLF4 may regulate the process of nuclear egress in virus-replicating cells (23, 44). Our study also indicates that BGLF4 recruits the nucleotide excision repair protein XPC to the viral replication compartment, enhancing viral DNA replication (48). The expression of BGLF4 alone induces premature chromosome condensation and phosphorylates the cellular replication origin binding complex MCM4-MCM6-MCM7, leading to an inhibition of its helicase activities (41, 43). Both events suggest the ability of BGLF4 to inhibit cellular DNA replication and thus save resources for efficient viral DNA replication.
While searching for candidate cellular substrates of BGLF4, an IRF3 splicing variant was identified in a yeast two-hybrid screening. Because phosphorylation contributes to multiple regulatory effects on IRF3, we explored the possible function of BGLF4 in regulating IRF3 signaling.
MATERIALS AND METHODS
Plasmids.
To generate pJTW2 [GAL4DBD-BGLF4(1-293)] and pJTW3 [GAL4DBD-BGLF4(201-429)] for yeast two-hybrid screening, a DNA fragment encoding amino acids (aa) 1 to 293 or 201 to 429 of BGLF4 was PCR amplified using pGEM3Z-EBVBamHI G as a template and subcloned into the BamHI site of pGBDUC1 (34). pJTW64 [GAL4AD-IRF3′(282-452)], which contains the cDNA sequence of an IRF3 splicing variant (GenBank accession no. AAH00660), was isolated from a human HeLa cDNA library constructed in pGAD GH/X (25). pSG5 (Promega Corp.)-based plasmids expressing wild-type BGLF4 (pYPW17), a kinase-dead mutant (pYPW20), and E1-tagged BGLF4 at positions 1 to 293 [BGLF4(1-293)] (pYPW18) were described previously (69). pCR3.1 (Invitrogen)-based plasmids expressing wild-type IRF3 (pCR3.1-huIRF3) and IRF3(5D) [pCR3.1-huIRF3(5D)] were kindly provided by Y. L. Lin (8). Hemagglutinin (HA)-IRF3′(282-452) (pJTW32) was generated by cloning a fragment at PCR-amplified codons 282 to 452 of IRF3′ from pJTW64 into the XhoI/HindIII sites of HA-containing pSG5 (pHY25). HA-IRF3(282-427) (pJTW28) was generated by cloning a fragment at PCR-amplified codons 282 to 427 of IRF3 from pCR3.1-huIRF3 into the XhoI/HindIII sites of pHY25. pIRES-hrGFP-1a (Stratagene)-based plasmids expressing IRF3-Flag and IRF3(5D)-Flag were gifts from Y. L. Lin (8). pGEX-3X (Amersham biosciences)-based plasmids expressing glutathione S-transferase (GST)-BGLF4 (pJTW52) and GST-K102I (pJTW53) were described previously (48). pJTW46 [GST-IRF3(282-427)] was generated by cloning a PCR-amplified DNA fragment encoding aa 282 to 427 from pCR3.1-huIRF3 into the EcoRI/SmaI sites of pGEX-4T-1 (Amersham biosciences). pJTW42 (GST-IRF3) was generated by subcloning the full-length IRF3 from pCR3.1-huIRF3 into the EcoRI site of pGEX-4T-1. pLuc-IFN-β and pLuc-PRD(III-I)3 reporter plasmids were kindly provided by K. Fitzgerald (19). pGal4-IRF3, which encodes a fusion protein containing the Gal4 DBD and codons 57 to 427 of IRF3, and pGal4-IRF3(S339A), in which Ser339 of Gal4-IRF3 is changed to Ala, were provided by S. Yamaoka (60). pTK-MH100x4-Luc (4xGal4-luc) was kindly provided by H.-M.Shih (Institute of Biomedical Sciences, Academia Sinica, ROC) (26). All site-directed IRF3(5D) mutants were generated by using a single primer-based in vitro mutagenesis strategy (49) with the primers and templates specified in Table S1 in the supplemental material. All of the constructs were confirmed by DNA sequencing.
Yeast two-hybrid screening.
pJTW2, which encodes GAL4DBD-BGLF4(1-293), was used as the bait to screen a human HeLa cDNA library as described previously (34). Briefly, the prey HeLa cDNA library and bait were cotransformed into strain PJ69-4A. Positive clones were selected on plates of synthetic complete medium lacking Ura, Leu, and Ade and confirmed for interactions on plates of synthetic complete medium lacking Ura, Leu, and His.
Cell culture, transfection, and double-stranded RNA stimulation.
The 293-TLR3 stable cell line was a gift of K. A. Fitzgerald (19). 293T, HeLa, 293-TLR3, and NA cells, which were established by in vitro infection of NPC-TW01 cells with the recombinant EBV Akata (9), were all maintained in Dulbecco's modified Eagle's medium supplemented with 8% fetal calf serum. EREV8 cells, which were established by in vitro infection of tetracycline-inducible Rta 293TRex cells with the recombinant EBV Akata, were maintained in Dulbecco's modified Eagle's medium supplemented with 6% of tetracycline-free fetal calf serum (44). All cells were cultured in the presence of penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C with 5% CO2. Plasmid DNA was transfected into 293T cells using the calcium phosphate-N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid method (11) or into HeLa, 293-TLR3, and NA cells with Lipofectamine 2000 (Invitrogen). For double-stranded RNA stimulation, HeLa cells were transfected with 1 μg/ml of poly(I:C) (Amersham) with Lipofectamine 2000, and 293-TLR3 cells were directly incubated with poly(I:C) at a final concentration of 50 μg/ml.
Coimmunoprecipitation.
After transfection or treatment, 200 to 500 μg of whole-cell extracts of 293T, HeLa, or EREV8 cells was prepared in lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 5 mM EDTA [pH 8.0], 30 mM NaF, 1 mM Na3VO4, 40 mM β-glycerophosphate, 1× protease inhibitor mixture, 10% glycerol, and 1% NP-40) and then incubated overnight with 1 μg of EBNA-1 5C11 monoclonal antibody (MAb) (13), anti-HA MAb, anti-Flag MAb, anti-BGLF4 (2224) (69), or anti-CBP MAb (Santa Cruz Biotechnology). The mixtures were then incubated with 100 μl (20%) of a 1:1 slurry of protein A/G-, protein A-, or protein G-Sepharose beads (Amersham Biosciences) for 2 h at 4°C. The beads were washed four times with NP-40 lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 1 mM Na3VO4) and resuspended in 2× sodium dodecyl sulfate (SDS) sample buffer. The immunoprecipitated complexes and cell extracts were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and analyzed by immunoblotting.
Western blotting.
Western blotting was performed as described previously (12). The primary antibodies used were Flag MAb (Sigma-Aldrich), BGLF4 MAbs 2224 and 2616 (69), HA MAb (Covance), phospho-STAT1(Tyr701) antibody (Cell Signaling), STAT1 antibody (Cell Signaling), IRF3 antibody (Santa Cruz Biotechnology), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Biodesign), CBP MAb (Santa Cruz Biotechnology), Rta MAb 467 (32), and BMRF1 antibody (Capricorn).
Expression and purification of GST fusion proteins.
The expression and purification of GST fusion proteins were carried out according to the Amersham handbook, with slight modifications. Competent BL21(DE3) bacteria were transformed with expression plasmids, and the bacteria were then induced at exponential phase with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and grown at 24°C for 4 h. The protein sample was harvested by sonication and then incubated with glutathione-Sepharose 4B (Sigma) at 4°C for 1 h. After extensive washings with PBST (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 [pH 7.3], 1% Triton X-100), the protein was eluted with glutathione elution buffer (50 mM Tris [pH 8.0], 5 mM reduced glutathione). The eluates were dialyzed against dialysis buffer (50 mM Tris [pH 7.4], 50 mM NaCl, 10% glycerol) and stored in aliquots at −80°C.
In vitro transcription/translation.
Plasmid DNA was expressed in vitro and labeled with [35S]methionine using the TNT Quick Coupled Transcription/Translation system (Promega) according to the manufacturer's instructions. Briefly, 1.0 μg of the indicated plasmid was incubated with 40 μl of TNT Quick Master Mix and [35S]methionine (20 μCi) in a 50-μl reaction mixture volume at 30°C for 90 min.
GST pull-down assays.
For GST pull-down, 15 μl of 35S-labeled proteins was precleared with 20 μl of 50% slurry glutathione-Sepharose 4B at 4°C for 2 h. Subsequently, 20 μl of 50% slurry glutathione-Sepharose 4B was added together with either 2 μg of GST fusion proteins or GST. The reaction mixture volume was adjusted to 500 μl with binding buffer (20 mM Tris [pH 8.0], 100 mM NaCl, 10% glycerol, 1% NP-40), rotated on a vertical wheel at 4°C for 2 h, washed four times with binding buffer, boiled in 2× SDS sample buffer, and displayed by SDS-PAGE. The gels were then stained with Coomassie blue and subjected to autoradiography.
Reporter assays.
Cells (3 × 105 HeLa or 5 × 105 293-TLR3 cells/well) were seeded into 12-well plates and incubated overnight. Cells were then cotransfected with reporter gene plasmid pLuc-PRD(III-I)3 or pLuc-IFN-β (19) using Lipofectamine 2000, along with each expression vector as indicated. The total DNA concentration was kept constant by supplementing empty vector pSG5. pWP1 (pCMV-Renilla reniformis) or pEGFP-C1 was transfected at the same time for normalizing transfection efficiencies. At 24 h posttransfection, poly(I:C) stimulation was carried out for 8 h, and luciferase activity was determined with the dual-luciferase assay system or Bright Glo luciferase assay system (Promega, Madison, WI). To assay IRF3 transactivation activity, cells were cotransfected with pGal4-Luc and expression plasmids encoding Gal4-IRF3 or Gal4-IRF3(S339A) with BGLF4 or K102I. pWP1 (pCMV-Renilla) was transfected at the same time for normalization of transfection efficiency. At 24 h posttransfection, luciferase activity was determined with the dual-luciferase assay system.
RNA purification and quantitative real-time RT-PCR.
Total cellular RNA was isolated with a REzol C&T RNA extraction kit (PROtech Technologies) according to the manufacturer's instructions. Reverse transcription (RT) of purified RNA was carried out using random primers. The quantification of gene transcripts was performed by real-time PCR using SYBR green I dye (Invitrogen) and the iCycler iQ apparatus (Bio-Rad). Expression values were normalized with control GAPDH. The primers used are as follows: sense primer 5′-AAGGAGGACGCCGCATTG-3′ and antisense primer 5′-GATAGACATTAGCCAGGAGGTTC-3′ for IFNB1 and sense primer 5′-CATCATCCCTGCCTCTACTG-3′ and antisense primer 5′-GCCTGCTTCACCACCTTC-3′ for GAPDH.
Analysis of IRF3 dimerization by native PAGE.
Native PAGE was performed as described previously (33), with a slight modification. Briefly, whole-cell extracts were prepared in native gel lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 50 mM NaF, 10% glycerol, 5.0 mM Na3VO4, 1% NP-40, 1× protease inhibitor mixture) and were separated on 7.5% native acrylamide gels. The electrophoresis buffers were composed of a cathode chamber buffer (25 mM Tris [pH 8.4], 192 mM glycine, and 0.5% sodium deoxycholate [Sigma-Aldrich]) and an anode chamber buffer (25 mM Tris [pH 8.4], 192 mM glycine). Proteins were then transferred onto membranes and probed for IRF3 dimerization with IRF3 antibody.
Immunofluorescence and microscopy.
Cells grown on slides were fixed with ice-cold methanol at −20°C for 20 min. Double staining was performed with the indicated primary antibodies BGLF4 MAb 2224 (69) and IRF3 antibody (Santa Cruz Biotechnology) at 37°C for 2 h, followed by fluorescein isothiocyanate-conjugated anti-rabbit immunoglobulin G (IgG) (1:100; Jackson) and rhodamine red-conjugated anti-mouse IgG (1:100; Jackson) antibodies at 37°C for 1 h. DNA was stained with Hoechst 33258 stain (Sigma-Aldrich) at room temperature for 30 s. Slides were mounted with medium (H1000; Vector) for fluorescence microscopy (Axioskop 40 FL; Zeiss).
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were performed according to the protocols of Upstate Biotechnology, with minor modifications. HeLa cells were cross-linked with 1% formaldehyde at room temperature for 12 min, and glycine was added to a final concentration of 0.125 M at room temperature for 15 min. Cells were washed three times with ice-cold phosphate-buffered saline, resuspended in 1 ml of cell lysis buffer (5 mM HEPES [pH 8.0], 85 mM KCl, 0.5% NP-40, 1 mM dithiothreitol [DTT], 1× protease inhibitor mixture), incubated on ice for 30 min, and centrifuged at 2,300 × g at 4°C for 20 min. The pellets were resuspended in nucleus lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris [pH 8.1], 1 mM DTT, 1× protease inhibitor mixture, 50 mM NaF) on ice for 30 min, and resuspended chromatin was sheared by sonication to a mean size of 200 to 1,000 bp, followed by centrifugation at 17,600 × g at 4°C for 10 min. Supernatants were collected for determinations of protein concentration. Equal amounts of sheared chromatin (500 μg) were diluted in immunoprecipitation dilution buffer (16.7 mM Tris [pH 8.1], 167 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM DTT, 1× protease inhibitor mixture, 50 mM NaF) and incubated with 2 μg of specific antibodies at 4°C overnight. Immunoprecipitated complexes were collected with 60 μl of 50% slurry protein A/G-Sepharose at 4°C for 2 h, followed by sequential washes twice in radioimmunoprecipitation assay wash buffer A (0.1% SDS, 1% NP-40, 5 mM EDTA, 50 mM Tris [pH 8.0], 150 mM NaCl, 1% sodium deoxycholic acid), radioimmunoprecipitation assay wash buffer B (0.1% SDS, 1% NP-40, 5 mM EDTA, 50 mM Tris [pH 8.0], 300 mM NaCl, 1% sodium deoxycholic acid), LiCl wash buffer (0.1% SDS, 1% NP-40, 5 mM EDTA, 50 mM Tris [pH 8.0], 150 mM NaCl, 300 mM LiCl, 1% sodium deoxycholic acid), and Tris-EDTA buffer (10 mM Tris [pH 8.0], 1 mM EDTA). Precipitates were eluted twice with immunoprecipitation elution buffer (1% SDS, 50 mM NaHCO3) at 4°C for 30 min, and NaCl was added to a final concentration of 0.3 M to reverse cross-links at 67°C overnight. The chromatin was then incubated with proteinase K buffer (50 mM Tris [pH 7.5], 25 mM EDTA, 1.25% SDS, 250 μg/ml proteinase K) at 45°C for 2 h. DNA fragments were extracted using the phenol-chloroform method, precipitated with ethyl alcohol, and dried. The purified DNA was resuspended in 50 μl double-distilled water. A total of 2 to 4 μl of purified sample was used for PCR. The primers used for IFNB1 were sense primer 5′-CACAGTTTGTAAATCTTTTTCCC-3′ and antisense primer 5′-ATGGGTATGGCCTATTTATATGA-3′.
Immunoprecipitation kinase assay.
HeLa cells expressing BGLF4 or K102I were lysed with lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 5 mM EDTA [pH 8.0], 30 mM NaF, 1 mM Na3VO4, 40 mM β-glycerophosphate, 1× protease inhibitor mixture, 10% glycerol, and 1% NP-40). One hundred micrograms of cell extract was immunoprecipitated with 1 μg of anti-BGLF4 MAb. The immunocomplexes were washed twice in the order of NP-40 lysis buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 1 mM Na3VO4), low-salt buffer (20 mM HEPES [pH 7.9], 10 mM MgCl2, 0.1 mM sodium orthovanadate, 20 mM β-glycerolphosphate, 1 mM DTT, 50 mM NaCl), high-salt buffer (20 mM HEPES [pH 7.9], 10 mM MgCl2, 0.1 mM sodium orthovanadate, 20 mM β-glycerolphosphate, 1 mM DTT, 250 mM NaCl), LiCl wash buffer (20 mM HEPES [pH 7.9], 10 mM MgCl2, 0.1 mM sodium orthovanadate, 20 mM β-glycerolphosphate, 1 mM DTT, 50 mM NaCl, 0.5 M LiCl), and kinase buffer (20 mM HEPES [pH 7.9], 10 mM MgCl2, 0.1 mM sodium orthovanadate, 20 mM β-glycerolphosphate, 10 mM p-nitrophenyl phosphate, 1 mM DTT, 50 mM NaCl, 2 mM NaF). Immunocomplexes were then incubated with 1 μg purified recombinant GST-IRF3 or GST in the presence of 10 μCi [γ-32P]ATP and kinase buffer for 30 min at 30°C. To stop the reaction, 2× SDS sample buffer was added, and the reaction mixtures were boiled for 5 min before analysis by immunoblotting, Coomassie blue staining, and autoradiography.
Reporter assay of IRF3-responsive activity in EBV-positive NA cells and design of siBGLF4.
NA cells (3 × 105 cells/well) were seeded into 12-well plates, incubated overnight, and transfected with Rta-expressing plasmid, green fluorescent protein (GFP) plasmid, and pLuc-PRD(III-I)3 or pLuc-IFN-β with BGLF4-targeted or control small interfering RNA (siRNA) using Lipofectamine 2000. At 18 h or 24 h posttransfection, luciferase activities were determined and normalized to GFP activity. The BGLF4-targeted siRNAs, siBGLF4-1 (5′-UUCUCCGGCAGCUUUACAUAGAGGG-3′) and siBGLF4-2 (5′-UAAUCAGUCAGGACCAGCCUACCCA-3′), and a control siRNA with comparable GC content, siCtrl (5′-UUCAGUGGCCCGACAUUUAUACGGG-3′), were purchased from Invitrogen.
RESULTS
Interaction of BGLF4 kinase with a cellular IRF3 variant.
EBV BGLF4 is comprised of 429 amino acids and is a member of the herpesviral UL kinases, for which very few identified cellular substrates have been identified. To explore possible biological functions of BGLF4 in cellular signaling, a HeLa cell cDNA library was screened with GAL4DBD-BGLF4(1-293) or GAL4DBD-BGLF4(201-429), which does not contain kinase activity, as bait in a yeast two-hybrid system. One clone that interacts with GAL4DBD-BGLF4(1-293) was identified (Fig. 1A). This two-hybrid isolate contains a cDNA fragment matching codons 282 to 452 of a human IRF3 splicing variant (GenBank accession no. AAH00660) (Fig. 1B). The putative protein encoded by this splicing variant is designated IRF3′ hereafter. The splicing variant IRF3′ contains an additional 16 nucleotides at codon 326 of wild-type IRF3, which results in a frameshift after aa 326 and the addition of another 126 residues to the protein. Compared to wild-type IRF3 (GenBank accession no. NP_001562), IRF3′ lacks part of the IAD (aa 327 to 375) and the transactivation domain (aa 327 to 394) of the IRF3 protein. To confirm the interaction between BGLF4 and IRF3′, 293T cells were cotransfected with E1/BGLF4(1-293) and HA-IRF3′(282-452) expression constructs and subjected to coimmunoprecipitation with E1 tag-specific MAb 5C11 (13). HA-IRF3′(282-452) was coimmunoprecipitated from cell extracts specifically with E1/BGLF4(1-293) (Fig. 1C).
FIG. 1.

BGLF4 interacts with a splicing variant and authentic form of IRF3. (A) GAL4DBD-BGLF4(1-293) and a two-hybrid isolate, GAL4AD-IRF3′(282-452), from a HeLa cDNA library were retransformed into PJ69-4A yeast cells and grown on nutrient selective plates (lacking Ura and Leu; lacking Ura, Leu, and His; or lacking Ura, Leu, and Ade). Negative controls (NC) are GAL4AD and GAL4DBD-BGLF4(1-293). Positive controls (PC) are the known interaction partners (Nijmegen breakage syndrome 1 [NBS1] and importin-α) (68). (B) Summary of IRF3, alternative splicing variant IRF3′ (GenBank accession number AAH00660), and the two-hybrid isolate truncated IRF3′ (amino acid 282 to 452). Functional domains of IRF3 including the N-terminal DBD, nuclear export signal (NES), proline-rich region (Pro), IAD, response domain (RD), and transactivation domain are indicated (3, 17, 47, 64, 73). (C) 293T cells were transiently transfected with plasmids expressing E1/BGLF4(1-293) and HA-IRF3′(282-452) separately or together for 24 h. The tagged proteins were then immunoprecipitated (IP) and immunoblotted (IB) with the indicated antibodies. GST antibody was used as a control antibody (Ctrl. Ab) for immunoprecipitation. (D) 293T cells were transiently transfected with plasmids expressing E1/BGLF4(1-293) and HA-IRF3(282-427) separately or together for 24 h. The tagged proteins were then immunoprecipitated and immunoblotted with the antibodies indicated. (E) HeLa cells were cotransfected with vector control (V) or plasmids expressing wild-type BGLF4 (K) or kinase-dead mutant KD (K102I) with IRF3-Flag or IRF3(5D)-Flag for 24 h. The tagged proteins were then immunoprecipitated by anti-Flag antibody and immunoblotted with the antibodies indicated. Whole-cell extract (WCE) was also applied as an input control. (F) HeLa cells were cotransfected with vector control (V) or plasmids expressing wild-type BGLF4 (K) or the kinase-dead mutant KD (K102I) with IRF3-Flag or IRF3(5D)-Flag for 24 h. Cell lysates were then immunoprecipitated by anti-BGLF4 antibody and immunoblotted with the antibodies indicated. (G) In vitro-translated [35S]methionine-labeled HA-IRF3(282-427) was examined for its ability to interact with GST, GST-BGLF4, or GST-K102I as described in Materials and Methods. (H) In vitro-translated [35S]methionine-labeled BGLF4 was examined for its ability to interact with GST, GST-IRF3(282-427), or GST-IRF3.
BGLF4 interacts with IRF3.
According to AceView at the NCBI website, the sequence of the IRF3 gene is defined by 576 GenBank entries from 540 cDNA clones, which were derived from 26 different mRNA species, including 25 alternatively spliced variants and 1 unspliced form, and these transcripts presumably encode 23 different isoforms of the IRF3 protein. Among them, the wild-type IRF3 protein was supported by 150 cDNA clones, and the splicing variant IRF3′ was supported by 84 cDNA clones. Because the expression pattern and biological function of the IRF3′ protein have never been reported, and the two-hybrid isolate contains a portion of the IAD of IRF3, we then determined whether BGLF4 can also interact with IRF3. The coimmunoprecipitation of E1/BGLF4(1-293) and HA-IRF3(282-427) was demonstrated in 293T cells using E1 tag-specific MAb 5C11 (Fig. 1D). It was reported previously that the activation status of IRF3 determines the cellular localization of IRF3 (42), and BGLF4 is expressed predominantly in the nucleus. We then determined whether the activation status of IRF3, or the kinase activity of BGLF4, is required for the appropriate interaction of the full-length proteins. HeLa cells were transfected with BGLF4 or the kinase-dead mutant K102I (KD) in combination with IRF3-Flag or constitutively active mutant IRF3(5D)-Flag, and the lysates were subjected to coimmunoprecipitation assays. Our results demonstrated that BGLF4 interacts with IRF3 independently of its activity (Fig. 1E, lanes 4 to 6). Compared to IRF3-Flag, IRF3(5D)-Flag had a strong ability to pull down both BGLF4 and K102I (Fig. 1E, lanes 5, 6, 8, and 9, and F, lanes 5, 6, 8, and 9), suggesting that the activation and nuclear translocation of IRF3 may promote its interaction with BGLF4. To ensure the specificity of the interaction of IRF3 and BGLF4, we overexpressed BGLF4 or K102I and GFP in HeLa cells and demonstrated that GFP was not coimmunoprecipitated by BGLF4 or K102I (see Fig. S1 in the supplemental material). Furthermore, bacterially expressed GST-BGLF4 and GST-K102I, but not GST, pulled down in vitro-translated and [35S]methionine-labeled HA-IRF3(282-427) (Fig. 1G). Consistently, in vitro-translated BGLF4 bound to bacterially expressed GST-IRF3 and GST-IRF3(282-427) (Fig. 1H). Taken together, these results suggest that BGLF4 interacts physically with IRF3 in addition to IRF3′.
BGLF4 suppresses IRF3-dependent transcriptional activation.
As mentioned above, the function of IRF3 but not IRF3′ is well studied. We decided, therefore, to determine whether BGLF4 affects IRF3 function. IRF3 plays a vital role in antiviral defense by inducing IFN-β production. As depicted in Fig. 2A, IRF3 regulates IFN-β promoter activity by binding to the IRF3-responsive element PRDIII-I of the IFN-β promoter region (70). To investigate the possible BGLF4-mediated regulation of IRF3 transactivation activity, poly(I:C) was used to trigger IRF3 activation together with a luciferase reporter plasmid containing the PRDIII-I or IFN-β promoter sequence. Various amounts of wild-type or kinase-dead BGLF4 expression plasmids were then transfected with the reporter plasmid into HeLa cells. Indeed, BGLF4 suppressed poly(I:C)-triggered IRF3 transactivation activity in a dose- and kinase activity-dependent manner not only in the minimal IRF3-responsive element-based reporter (PRDIII-I) but also in the context of the IFN-β promoter (Fig. 2B and C). We then determined whether similar effects can be observed in 293-TLR3 cells, which express larger amounts of TLR3 and can be stimulated by adding poly(I:C) to the medium. The expression of BGLF4 also suppressed IRF3-dependent transcriptional activation on the PRDIII-I-based reporter in 293-TLR3 cells (Fig. 2D).
FIG. 2.

BGLF4 suppresses IRF3 transactivation activity in transient reporter assay. (A) Schematic diagram of DNA sequence and transcription factor binding sites of the IFN-β promoter (70). Reporter assays were performed by cotransfecting increasing amounts of plasmids expressing BGLF4 or K102I with pCMV-R. reniformis luciferase and reporter plasmid pLuc-PRD(III-I)3 into HeLa cells (B), pLuc-IFN-β into HeLa cells (C), or pLuc-PRD(III-I)3 into 293-TLR3 cells (D). At 24 h posttransfection, cells were stimulated with or without poly(I:C) for 8 h, and luciferase activities were measured and normalized to R. reniformis luciferase activities. Open bars indicate poly(I:C) treatment, and black bars indicate mock poly(I:C) treatment. (E) The reporter assay was performed by cotransfecting plasmids expressing constitutively active IRF3(5D) and increasing amounts of BGLF4 or KD (K102I) with GFP and reporter plasmid pLuc-PRD(III-I)3 into HeLa cells. At 24 h posttransfection, luciferase activities were measured and normalized to GFP activity. In all experiments, results are means ± standard deviations (SD) from two separate transfections. Data are representative of two independent experiments. IB, immunoblot.
To clarify whether BGLF4 affects IRF3 function directly, rather than through other poly(I:C)-stimulated pathways, constitutively active IRF3(5D) was used to mimic the activation status of IRF3 in the reporter system. As expected, BGLF4 suppressed IRF3(5D)-dependent transcriptional activation on the PRDIII-I-based reporter (Fig. 2E). Together, these results demonstrated that BGLF4 suppresses IRF3 transactivation, mainly in a kinase activity-dependent manner, in both HeLa and 293-TLR3 cells.
BGLF4 suppresses the poly(I:C)-triggered endogenous IRF3 signaling pathway.
To demonstrate the effects of BGLF4 on endogenous IRF3 signaling, we examined the poly(I:C)-stimulated expression of IFN-β mRNA and the IFN-β-transduced STAT1 phosphorylation at Tyr701. HeLa cells were transfected with plasmids expressing BGLF4, K102I, or the vector control and stimulated with poly(I:C). The relative ratio of expression of IFN-β to GAPDH mRNA was monitored by quantitative real-time RT-PCR at various time points, and STAT1 phosphorylation at Tyr701 was detected with a specific antibody. In vector-transfected HeLa cells, poly(I:C)-triggered IFN-β mRNA expression was elevated slightly at 2 h poststimulation and peaked at 6 h poststimulation (Fig. 3A). STAT1 phosphorylation (Tyr701) was observed at 4 h poststimulation and declined at 8 h poststimulation (Fig. 3B), which is in accordance with previously reported observations (15). Because it is known that the phosphorylated STATs can induce IRF7 expression and turn on IFN positive-feedback regulation through the binding of the IRF3/IRF7 complex to the IFN-β promoter (31), we speculate that the IRF3-dependent activation of the IFN-β promoter takes place predominantly before 4 h post-poly(I:C) treatment, whereas IFN-dependent amplification of the IFN-β promoter may occur after 4 h of poly(I:C) treatment. In the presence of BGLF4, the expression of IFN-β was repressed significantly after poly(I:C) stimulation (Fig. 3A), and the phosphorylation signal of STAT1 was barely detectable at 4 h and decreased rapidly at 8 h post-poly(I:C) stimulation (Fig. 3B). This indicates that the expression of BGLF4 suppresses poly(I:C)-triggered endogenous IRF3-mediated expression of IFN-β. As for K102I-expressing HeLa cells, IFN-β mRNA was expressed at a level similar to that of the vector control at 4 h post-poly(I:C) stimulation, while the reduction in IFN-β expression was observed at 6 h poststimulation (Fig. 3A). The phosphorylation intensities of STAT1 in the presence of K102I were similar to those of the vector control at 4 h and 6 h post-poly(I:C) stimulation (Fig. 3B). This indicates that the expression of K102I does not repress the IRF3-dependent activation of the IFN-β promoter during the early phase after poly(I:C) stimulation.
FIG. 3.

BGLF4 suppresses the endogenous poly(I:C)-triggered signaling pathway. HeLa cells were transiently transfected with vector control or the expression plasmids encoding BGLF4 or K102I for 24 h and stimulated with or without 1 μg/ml poly(I:C) for the indicated periods of time. (A) Total RNA was extracted and subjected to quantitative real-time RT-PCR analysis using primers specific for IFN-β. The relative abundance of IFN-β mRNA normalized to its own GAPDH mRNA is shown. (B) The cell lysates were collected and immunoblotted with antibodies against phospho-STAT1(Tyr701), STAT1, IRF3, BGLF4, or GAPDH. The relative abundance of phospho-STAT1(Tyr701) normalized to the amount of STAT1 protein is shown for the indicated periods of time post-poly(I:C) treatment. hpt, hours posttreatment.
BGLF4 does not suppress poly(I:C)-triggered IRF3 dimerization, translocation, and CBP recruitment in the IRF3 activation pathway.
IRF3 is activated by phosphorylation, which leads to its dimerization, nuclear translocation, and CBP recruitment to the promoter region of target genes (29). To examine the possible effects of BGLF4 on individual steps of IRF3 activation, BGLF4-expressing HeLa cells were stimulated with poly(I:C) and examined at various time points. IRF3 dimerization was monitored by native PAGE, and no difference was observed among cells expressing BGLF4, the vector control, or the kinase-dead mutant K102I (Fig. 4A), suggesting that BGLF4 does not block poly(I:C)-triggered IRF3 dimerization. Next, an immunofluorescence assay was performed to investigate whether BGLF4 affects poly(I:C)-triggered IRF3 nuclear translocation. As shown in Fig. 4B, poly(I:C) promoted the nuclear accumulation of IRF3 in HeLa cells independent of the presence or absence of BGLF4. These results demonstrated that poly(I:C) stimulation activates IRF3 dimer formation and nuclear translocation without resultant gene transcription in the presence of BGLF4. The reason was investigated further by examining the association of IRF3 with the coactivator CBP. Coimmunoprecipitation experiments revealed that the affinity of the poly(I:C)-induced association of IRF3 with CBP was similar in the presence of BGLF4, vector control, or K102I at various time points (Fig. 4C). Of note, both BGLF4 and K102I were coimmunoprecipitated with CBP (Fig. 4C). These data suggest that BGLF4 does not block the poly(I:C)-induced association of IRF3 and CBP, and BGLF4 itself forms complexes with CBP or the activated IRF3/CBP.
FIG. 4.

BGLF4 does not suppress poly(I:C)-triggered IRF3 dimerization, translocation, or CBP recruitment. (A) HeLa cells were transiently transfected with vector control (V) or expression plasmids encoding BGLF4 (K) or K102I (KD) for 24 h and stimulated with or without 1 μg/ml poly(I:C) for the indicated periods of time. IRF3 dimerization was analyzed by native gel electrophoresis and immunoblotted with IRF3 antibody. The cell lysates were separated by SDS-PAGE and immunoblotted with antibody against BGLF4 or GAPDH. (B) HeLa cells were transiently transfected with vector control or the expression plasmids encoding BGLF4 or KD (K102I) for 24 h and stimulated with or without 1 μg/ml poly(I:C) for 3 h. The subcellular location of IRF3 was analyzed with IRF3 antibody; DNA was stained with Hoechst 33258 stain; BGLF4 was detected with MAb 2224. Scale bar, 10 μm. (C) HeLa cells were transiently transfected with vector control (V) or expression plasmids encoding BGLF4 (K) or K102I (KD) for 24 h and stimulated with 1 μg/ml poly(I:C) for the indicated periods of time. CBP was immunoprecipitated (IP) from these cell lysates with specific antibody, and the immunocomplexes were immunoblotted (IB) with the antibodies indicated. Whole-cell extract (WCE) also was immunoblotted with the antibodies indicated. hpt, hours posttreatment.
BGLF4 prevents the binding of poly(I:C)-triggered IRF3 to the IRF3-responsive promoter.
Once activated, IRF3 forms complexes with CBP and binds to IRF3-responsive elements containing sequences such as in IFN-β promoter regions to activate gene transcription (67, 70). To test the possibility that BGLF4 suppresses activated IRF3 binding to its cognate promoter, the occupancy of the IRF3-responsive elements in the IFN-β promoter by IRF3 was examined by ChIP assay. HeLa cells were transfected with plasmids expressing BGLF4, K102I, or the vector control and stimulated with poly(I:C). At 3 h poststimulation, the cross-linked chromatin fragments were immunoprecipitated by specific antibody, and DNA from antibody-bound chromatin was amplified by specific primers for the IFN-β promoter region by PCR. In vector-transfected cells, poly(I:C) increased the amounts of IRF3 associated with the IFN-β promoter region at 3 h poststimulation, whereas the stimulation was not observed in the presence of BGLF4 (Fig. 5A and B). In K102I-expressing cells, poly(I:C) stimulation still promotes IRF3 binding to the IFN-β promoter. Data here suggest that the failure of activated IRF3 to bind the IRF3-responsive promoter region is probably the determinant step of the BGLF4-mediated repression of IRF3 signaling, and the kinase activity of BGLF4 is important for effective repression.
FIG. 5.
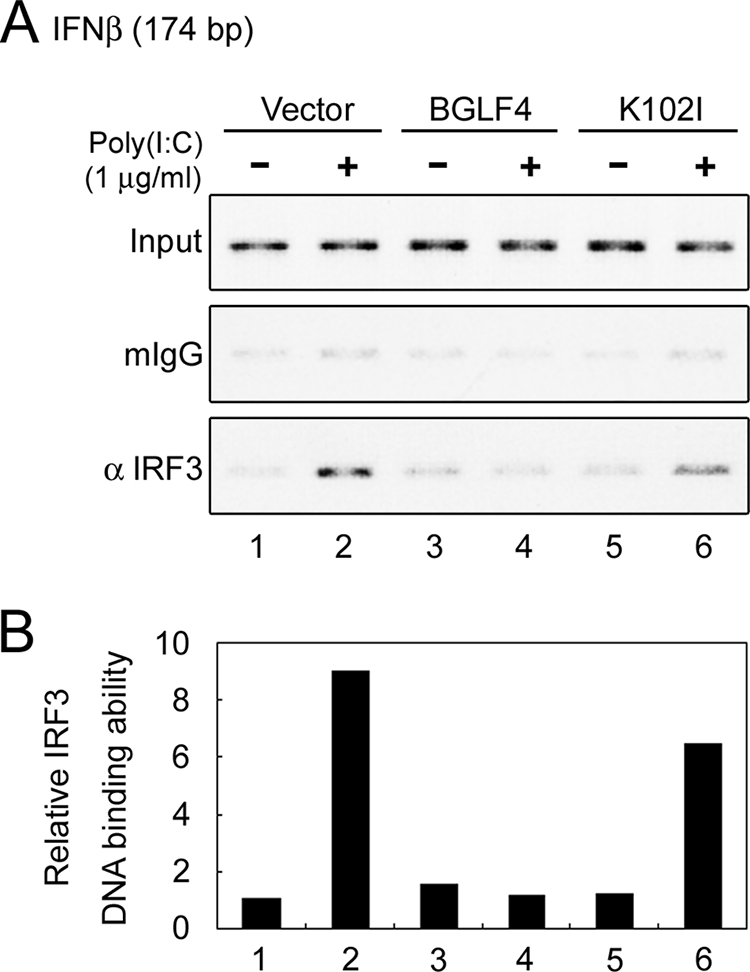
BGLF4 suppresses poly(I:C)-triggered IRF3 binding to PRDIII-I of the IFN-β promoter. HeLa cells were transiently transfected with vector control (V) or the expression plasmids encoding BGLF4 (K) or KD (K102I) for 24 h and then stimulated with 1 μg/ml poly(I:C) or not for 3 h. ChIP assays were performed using antibodies for immunoprecipitation as described in Materials and Methods. Mouse IgG (mIgG) was used as the antibody control in the ChIP assay. ChIP assays were performed on the IFN-β promoter. (B) The data in A were quantified by ImageQuant software and normalized with respect to the total DNA inputs. The designations of the lanes are shown above (A).
BGLF4 phosphorylates IRF3 in vitro but does not adapt a Pin1-mediated IRF3 degradation mechanism to block IRF3 signaling.
Given that BGLF4 prevents IRF3 from binding to its responsive promoter mainly in a kinase activity-dependent manner upon poly(I:C) treatment, we then determined whether IRF3 is a substrate of BGLF4. The immunoprecipitation kinase assay was performed with BGLF4 or K102I expressed in HeLa cells and bacterially expressed GST-IRF3. A specific phosphorylation signal was observed in the presence of BGLF4 but not K102I (Fig. 6A), suggesting that BGLF4 may modulate IRF3 function directly through phosphorylation.
FIG. 6.
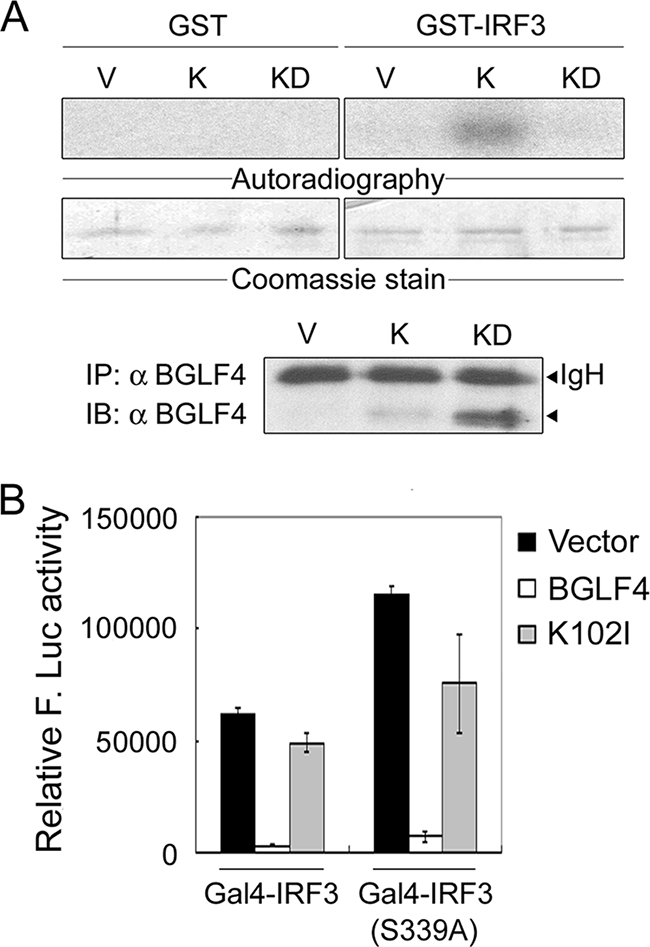
BGLF4 phosphorylates IRF3 in vitro but does not adapt a Pin1-mediated mechanism to block IRF3 activation. (A) 293T cell lysate containing BGLF4 (K) or KD (K102I) was precipitated with antibody against BGLF4. The immunoprecipitation (IP) kinase assay was performed as described in Materials and Methods. IB, immunoblot. (B) Luciferase reporter assays were performed by cotransfecting pGal4-IRF3 or pGal4-IRF3(S339A) and the vector control or plasmids expressing BGLF4 or KD (K102I) with 4× Gal4-Luc (pTK-MH100x4-Luc) and pCMV-R. reniformis luciferase into HeLa cells. At 24 h posttransfection, luciferase activities were measured and normalized with R. reniformis luciferase activities. In all experiments, results are means ± SD from two separate transfections. Data are representative of two independent experiments.
Phosphorylation-dependent posttranslational modifications of IRF3 are crucial for regulating the function of IRF3. Most of these phosphorylation modifications contribute to IRF3 activation (19, 37, 53, 61, 63, 65). However, the phosphorylation of IRF3 at the Ser339-Pro340 motif by an unidentified putative “proline-directed kinase” leads to its interaction with peptidyl-prolyl isomerase Pin1, and the resultant conformational change of IRF3 facilitates its polyubiquitination and subsequent proteasome-dependent degradation (60). Because BGLF4 is a proline-directed kinase, the Gal4-IRF3 fusion protein (wild type or S339A), which contains aa 57 to 427 of IRF3, and the Gal4-luciferase reporter system (60) were used to determine whether BGLF4 adapts a Pin1-mediated mechanism to degrade IRF3. As shown in Fig. 6B, Gal4-IRF3(S339A) showed an enhanced transactivation ability because of its escape from Pin1-mediated degradation as reported previously (60). Compared to the vector control, BGLF4 suppressed the transactivation of Gal4-IRF3 and Gal4-IRF3(S339A) to similar extents, indicating that BGLF4 does not suppress IRF3 transactivation through a Pin1-mediated degradation mechanism.
Ser123, Ser173, and Thr180 of IRF3 contribute additively to BGLF4-mediated suppression of IRF3(5D).
Because BGLF4 phosphorylates TP or SP, and Thr3 and Ser339 of IRF3 were excluded for the BGLF4-mediated repression of transactivation, we decided to use constitutively active IRF3(5D) for analyzing whether Ser123, Ser173, and Thr180 of IRF3 are involved in this regulation (Fig. 7A). A mutant with Ser123, Ser173, and Thr180 all mutated into Ala was generated as IRF3(5D)3A. In a cotransfection experiment, BGLF4 suppressed IRF3(5D), but not IRF3(5D)3A, transactivation activities on a PRDIII-I-based reporter in a dose-dependent manner (Fig. 7B). Among these three residues, Ser173 and Thr180 were located within the proline-rich region of IRF3, whereas Ser123 sits between the DNA binding and nuclear export signals of IRF3 (Fig. 7A). To further dissect which residue(s) is critical for BGLF4-mediated repression, single or double mutations were generated and examined for their transactivation activities in the absence or presence of BGLF4. Interestingly, all the mutants showed slightly enhanced transactivation activities (Fig. 7C). Except for IFR3(5D)3A, all the mutants were sensitive to BGLF4-mediated suppression (Fig. 7C). Data here suggest that Ser123, Ser173, and Thr180 contribute additively to the BGLF4-mediated repression of the IRF3 transactivation activity.
FIG. 7.

Ser123, Ser173, and Thr180 of IRF3 contribute additively to BGLF4-mediated suppression of IRF3(5D) in a transient reporter assay. (A) Summary of currently identified phosphorylation sites on IRF3 and IRF3(5D)-based mutants used in this study. Thr135 is the phosphorylation site of DNA-PK (37), and the seven phosphorylation sites in the response domain (RD) can be phosphorylated by TBK1 or IKKi (14, 19, 53, 63, 65). The proline-dependent phosphorylation sites are Thr3, Ser123, Ser173, Thr180, and Ser339. Ser339 is phosphorylated by unidentified cellular kinase and contributes to Pin-mediated IRF3 degradation (60). Mutants generated for mapping residues responsible for BGLF4-mediated suppression are listed. NES, nuclear export signal. (B) A reporter assay was performed by cotransfecting plasmids expressing constitutively active IRF3(5D) or IRF3(5D)3A and increasing amounts of BGLF4 with reporter plasmid pLuc-PRD(III-I)3 and the GFP control into HeLa cells. At 24 h posttransfection, luciferase activities were measured and normalized to GFP activity. IB, immunoblot. (C) A reporter assay was performed by cotransfecting IRF3(5D), IRF3(5D)S123A, IRF3(5D)S173A, IRF3(5D)T180A, IRF3(5D)S123A S173A, IRF3(5D)S173A T180A, or IRF3(5D)S123A S173A T180A [IRF3(5D)3A] in the presence of BGLF4 or vector with the reporter plasmid into HeLa cells and assayed as described above (B). In all experiments, results are means ± SD from two separate transfections. Data are representative of two independent experiments.
Knockdown of BGLF4 enhances IRF3-responsive reporter activity in EBV-reactivated cells.
Two EBV-positive cell lines, EREV8, which harbors EBV Akata and a doxycycline-inducible Rta, and NA, which is derived from an EBV Akata-infected NPC cell line, were adapted to investigate the interaction between BGLF4 and IRF3. We demonstrate that IRF3 can be specifically coimmunoprecipitated with BGLF4 in doxycycline-induced EREV8 cells (Fig. 8A). Additionally, the IRF3-responsive reporter system was used to indicate the IRF3-dependent transactivation activity in Rta-transduced EBV-positive NA cells (Fig. 8B and C). Using an siRNA approach, we demonstrate that the knockdown of BGLF4 indeed enhanced IRF3 activities monitored at 18 h and 24 h posttreatment not only in the minimal IRF3-responsive element-based reporter (PRDIII-I) but also in the context of the IFN-β promoter (Fig. 8B and C). Together, these data suggest that BGLF4 interacts with IRF3 and suppresses IRF3 transactivation in reactivated EBV-positive cells.
FIG. 8.

BGLF4 interacts with IRF3 in EBV-reactivated EBV-positive epithelial cells, and the knockdown of BGLF4 enhances IRF3-responsive reporter activity in EBV-reactivated NPC cells. (A) EREV8 cells were induced by 100 ng/ml doxycycline (Dox) for the expression of Rta and EBV lytic replication or mock treated. At 48 h posttreatment, cell lysates were collected and immunoblotted with antibodies (Ab) against Rta, BMRF1, BGLF4, IRF3, or GAPDH. The cell lysates were then immunoprecipitated (IP) by anti-BGLF4 MAb 2224 and immunoblotted with the antibodies against IRF3 or BGLF4 (2616). Whole-cell extract was applied as an input control. GST antibody was used as a control antibody (Ctrl. Ab) in immunoprecipitations. Reporter assays were performed by cotransfecting Rta-expressing plasmid or vector, GFP plasmid, and BGLF4-targeted siRNAs (siBGLF4-1 or siBGLF4-2) or a control siRNA (siCtrl.) with pLuc-PRD(III-I)3 (B) or pLuc-IFN-β (C) into NA cells. At 18 h or 24 h posttransfection, luciferase activities were measured and normalized to GFP activities. Data shown are from triplicate samples and are representative of three independent experiments. The statistical significance of differences between BGLF4-targeted and control siRNA is indicated at the top of the bars. “*,” P < 0.05; “**,” P < 0.01 (Student's t test). The cell lysates of individual samples were immunoblotted for the expression of BGLF4, Rta, BMRF1, IRF3, and GAPDH (bottom).
DISCUSSION
The innate immune response is the first line of defense against invading viruses. The attachment or entry of herpesviruses has been shown to initiate the IFN signaling pathway, including the activation of IRFs and the expression of ISGs. Various viral strategies counteract these signaling pathways and are important for efficient replication. In this study, we demonstrate that BGLF4 kinase interacts physically with and phosphorylates IRF3, which is the initial activator of transcription in the innate immune response (Fig. 1 and 6A). In a transient reporter assay, BGLF4 suppresses not only poly(I:C)-stimulated activation in the context of the IFN-β promoter or minimal IRF3-responsive element but also the transactivation activities of constitutively active IRF3(5D) (Fig. 2B to E). Correspondingly, the poly(I:C)-stimulated expression of IFN-β mRNA and subsequent IFN-β-dependent STAT1 phosphorylation were repressed in the presence of BGLF4 (Fig. 3). The suppression effects in reporter assays are predominantly BGLF4 kinase activity dependent, whereas K102I appeared to repress the endogenous poly(I:C)-induced signaling moderately at 6 h post-poly(I:C) stimulation (Fig. 3). We suspect that K102I may affect enhanceosome assembly following IRF3-dependent expression of IFN and the subsequent IFN-dependent gene expression. Analysis of the mechanism involved in BGLF4-mediated repression revealed that poly(I:C) induced dimerization, translocation, and CBP recruitment of IRF3 were not affected by BGLF4. However, the binding of active IRF3 to its cognate DNA sequences on the IFN-β promoter was diminished in the presence of BGLF4 (Fig. 5). By transfecting BGLF4 siRNA into EBV-positive NA cells, IRF3-dependent transactivation activity is enhanced in a minimal IRF3-responsive element-based reporter (PRDIII-I) and in the context of the IFN-β promoter (Fig. 8B and C), suggesting that BGLF4 may downregulate the IFN signaling pathway through inhibiting IRF3 function.
BGLF4 is a proline-directed Ser/Thr kinase that phosphorylates several cellular and viral substrates at Cdk1 target sites (22). According to the amino acid sequence of IRF3, it contains five Ser/Thr-Pro motifs (Thr3, Ser123, Ser173, Thr180, and Ser339). Because BGLF4 represses the transactivation of Gal4-IRF3 (Fig. 6B), which contains aa 57 to 427 of IRF3, in a Gal4-IRF3 reporter system, the involvement of Thr3 in BGLF4 kinase activity-dependent repression may be excluded (Fig. 6B). Additionally, Ser339 of IRF3, which is responsible for Pin1-mediated degradation, was also excluded for BGLF4-mediated IRF3 suppression (Fig. 6B). Notably, our results reveal that Ser123, Ser173, and Thr180 of IRF3 contribute additively to BGLF4-mediated suppression (Fig. 7). All these three residues are located in a linker-like domain between the well-studied DNA binding and IRF association domains. Recently, the conformational changes of full-length wild-type and mutant IRF3 upon binding to DNA were monitored by the fluorescence spectrum. The data indicate that the dimerization of IRF3 is the basis of its strong binding to PRDIII-PRDI sites, which leads to a ∼100° twist of DNA (17). Therefore, our most favorable postulation is that the phosphorylation of the proline-rich region may affect the flexibility required for the stable binding of the IRF3 dimer on its DNA target sequence as proposed previously by Dragan et al. (17), although it cannot be excluded that BGLF4-mediated phosphorylation may interfere with its ability to interact with other components of the enhanceosome complex. A similar phosphorylation-mediated regulation of transactivator activities was also reported for the androgen receptor (AR) and tumor suppressor p53. The AR also contains a hinge region, which has three proline-directed phosphorylation sites at Ser81, Ser94, and Ser650, in between its DNA and steroid binding domains. It was demonstrated previously that phosphorylation at Ser650 contributes to the optimal transactivation activity of AR (75). Ser81-Pro82 in the proline-rich region of p53 is responsible for DNA damage-induced p53-Chk2 (checkpoint kinase 2) interaction, which is required for the subsequent regulation of p53/Mdm2 loop (4). A recent study further indicates that Pin1-mediated regulation at phosphorylated Ser81 of p53 stimulates its binding to p300 and acetylation (50). Thus, it would be interesting to examine whether Pin1 is also involved in the regulation of other transactivators at their proline-directed phosphorylation sites.
Because IRF3 plays a central role in the innate immune response, it is not surprising that many viral proteins have been reported to disrupt IRF3 activation. For example, the P protein of rabies virus is involved in the negative regulation of IRF3 by inhibiting IRF3 phosphorylation (7). The leader protein of mengovirus prevents IRF3 dimerization (27), while the leader protein of Theiler's virus reduces IRF3 translocation to the nucleus (16). vIRF1 of Kaposi's sarcoma-associated herpesvirus interferes with the interaction between IRF3 and CBP (46). K-bZIP of Kaposi's sarcoma-associated herpesvirus competes for IRF3 DNA binding sites to inhibit IRF3 DNA binding (45). bICP0 of bovine herpesvirus reduces IRF3 protein levels through the proteolytic degradation of IRF3 (59). Our study thus demonstrates that EBV evolved with a different mechanism to block IRF3 function.
The BGLF4 kinase-mediated suppression of the IFN signaling pathway also provides a possible interpretation of data from a previous study by Shibaki et al. (66), using the UL13 gene-deleted mutant of HSV-1 (VRΔ13). Those authors demonstrated that VRΔ13, but not wild-type HSV-1 or a UL13 revertant (VRΔ13R), was cleared in the early period of intraperitoneal infection of BALB/c mice, coupled with a higher level of IFN induction. It was suggested that UL13 may regulate viral genes such as ICP0 or ICP22 to mediate the IFN suppression function, while the involvement of IRF3 was not revealed.
Given that IRF3 serves as the initial transactivator of IFN signaling, the abundant virion glycoprotein pp65 of human cytomegalovirus was found to block IFN by subverting IRF3 function upon infection (1, 6). Because BGLF4 is also a virion-associated protein kinase (2, 36, 69), we suggest that BGLF4 may either block IRF3 signaling during reactivation or function upon infection of new host cells. BGLF4 may cooperate with viral IRF7 suppressors, BZLF1 or the recently identified tegument protein LF2, to prevent the activation of the IFN-β promoter (24, 71) and to facilitate maximal viral replication.
Taken together, we demonstrate that BGLF4 interacts with and suppresses IRF3 transactivation activities through a novel mechanism dependent on its kinase activity. Because IRF3-initiated ISGs and the expression of IFNs may lead to an inhibition of transcription and translation and induction of apoptosis to suppress viral replication (28, 62), we suggest that BGLF4-mediated suppression of IRF3 function may enhance viral lytic progression by subverting these antiviral mechanisms. Additionally, IRF3 also is involved in the regulation of cell cycle progression and tumor suppression (18, 40, 56); therefore, the possible contribution of BGLF4 to EBV pathogenesis and oncogenesis needs to be explored in the future.
Supplementary Material
Acknowledgments
We thank Katherine A. Fitzgerald (Division of Infectious Diseases and Immunology, University of Massachusetts Medical School, Worcester, MA) for providing reporter plasmids pLuc-IFN-β and pLuc-PRD(III-I)3 and 293-TLR3 cells and Shoji Yamaoka (Department of Molecular Virology, Graduate School of Medicine, Tokyo Medical and Dental University, Tokyo, Japan) for providing plasmids pGal4-IRF3 and pGal4-IRF3(S339A). We appreciate Yi-Ling Lin (Institute of Biomedical Sciences, Academia Sinica, ROC) for providing plasmids pCR3.1-huIRF3, pCR3.1-huIRF3(5D), pIRF3-Flag, and pIRF3(5D)-Flag and helpful discussion and Hsiu-Ming Shih (Institute of Biomedical Sciences, Academia Sinica, ROC) for plasmid pTK-MH100x4-Luc. We are grateful to Chien-Kuo Lee (Graduate Institute of Immunology, College of Medicine, National Taiwan University, ROC) for helpful assistance and discussion with quantitative real-time RT-PCR analysis. We also show appreciation to Tim J. Harrison of the Royal Free and University College Medical School (University College London) for critical reading and editing of the manuscript.
This study was supported by the National Health Research Institutes, Taiwan (grants NHRI-EX96-9609BI and NHRI-EX97-9609BI), and the National Science Council, Taiwan (grant NSC95-2320-B002-087-MY3).
Footnotes
Published ahead of print on 3 December 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Abate, D. A., S. Watanabe, and E. S. Mocarski. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 7810995-11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asai, R., A. Kato, K. Kato, M. Kanamori-Koyama, K. Sugimoto, T. Sairenji, Y. Nishiyama, and Y. Kawaguchi. 2006. Epstein-Barr virus protein kinase BGLF4 is a virion tegument protein that dissociates from virions in a phosphorylation-dependent process and phosphorylates the viral immediate-early protein BZLF1. J. Virol. 805125-5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Au, W. C., P. A. Moore, W. Lowther, Y. T. Juang, and P. M. Pitha. 1995. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 9211657-11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger, M., N. Stahl, G. Del Sal, and Y. Haupt. 2005. Mutations in proline 82 of p53 impair its activation by Pin1 and Chk2 in response to DNA damage. Mol. Cell. Biol. 255380-5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boehme, K. W., J. Singh, S. T. Perry, and T. Compton. 2004. Human cytomegalovirus elicits a coordinated cellular antiviral response via envelope glycoprotein B. J. Virol. 781202-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browne, E. P., and T. Shenk. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. USA 10011439-11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzozka, K., S. Finke, and K. K. Conzelmann. 2005. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J. Virol. 797673-7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, T. H., C. L. Liao, and Y. L. Lin. 2006. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 8157-171. [DOI] [PubMed] [Google Scholar]
- 9.Chang, Y., C. H. Tung, Y. T. Huang, J. Lu, J. Y. Chen, and C. H. Tsai. 1999. Requirement for cell-to-cell contact in Epstein-Barr virus infection of nasopharyngeal carcinoma cells and keratinocytes. J. Virol. 738857-8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chee, A. V., and B. Roizman. 2004. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J. Virol. 784185-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, C., and H. Okayama. 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 72745-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, M. R., S. J. Chang, H. Huang, and J. Y. Chen. 2000. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 743093-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, M. R., C. H. Tsai, F. F. Wu, S. H. Kan, C. S. Yang, and J. Y. Chen. 1999. The major immunogenic epitopes of Epstein-Barr virus (EBV) nuclear antigen 1 are encoded by sequence domains which vary among nasopharyngeal carcinoma biopsies and EBV-associated cell lines. J. Gen. Virol. 80(Pt. 2)447-455. [DOI] [PubMed] [Google Scholar]
- 14.Clément, J. F., A. Bibeau-Poirier, S.-P. Gravel, N. Grandvaux, É. Bonneil, P. Thibault, S. Meloche, and M. J. Servant. 2008. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. J. Virol. 833984-3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai, X., K. Sayama, K. Yamasaki, M. Tohyama, Y. Shirakata, Y. Hanakawa, S. Tokumaru, Y. Yahata, L. Yang, A. Yoshimura, and K. Hashimoto. 2006. SOCS1-negative feedback of STAT1 activation is a key pathway in the dsRNA-induced innate immune response of human keratinocytes. J. Investig. Dermatol. 1261574-1581. [DOI] [PubMed] [Google Scholar]
- 16.Delhaye, S., V. van Pesch, and T. Michiels. 2004. The leader protein of Theiler's virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 784357-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dragan, A. I., V. V. Hargreaves, E. N. Makeyeva, and P. L. Privalov. 2007. Mechanisms of activation of interferon regulator factor 3: the role of C-terminal domain phosphorylation in IRF-3 dimerization and DNA binding. Nucleic Acids Res. 353525-3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duguay, D., F. Mercier, J. Stagg, D. Martineau, J. Bramson, M. Servant, R. Lin, J. Galipeau, and J. Hiscott. 2002. In vivo interferon regulatory factor 3 tumor suppressor activity in B16 melanoma tumors. Cancer Res. 625148-5152. [PubMed] [Google Scholar]
- 19.Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4491-496. [DOI] [PubMed] [Google Scholar]
- 20.Fuld, S., C. Cunningham, K. Klucher, A. J. Davison, and D. J. Blackbourn. 2006. Inhibition of interferon signaling by the Kaposi's sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 803092-3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao, S. J., C. Boshoff, S. Jayachandra, R. A. Weiss, Y. Chang, and P. S. Moore. 1997. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 151979-1985. [DOI] [PubMed] [Google Scholar]
- 22.Gershburg, E., and J. S. Pagano. 2008. Conserved herpesvirus protein kinases. Biochim. Biophys. Acta 1784203-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gershburg, E., S. Raffa, M. R. Torrisi, and J. S. Pagano. 2007. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J. Virol. 815407-5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hahn, A. M., L. E. Huye, S. Ning, J. Webster-Cyriaque, and J. S. Pagano. 2005. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. J. Virol. 7910040-10052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hannon, G. J., D. Demetrick, and D. Beach. 1993. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev. 72378-2391. [DOI] [PubMed] [Google Scholar]
- 26.Harmon, M. A., M. F. Boehm, R. A. Heyman, and D. J. Mangelsdorf. 1995. Activation of mammalian retinoid X receptors by the insect growth regulator methoprene. Proc. Natl. Acad. Sci. USA 926157-6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hato, S. V., C. Ricour, B. M. Schulte, K. H. Lanke, M. de Bruijni, J. Zoll, W. J. Melchers, T. Michiels, and F. J. van Kuppeveld. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 92921-2930. [DOI] [PubMed] [Google Scholar]
- 28.Heylbroeck, C., S. Balachandran, M. J. Servant, C. DeLuca, G. N. Barber, R. Lin, and J. Hiscott. 2000. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 743781-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hiscott, J. 2007. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 28215325-15329. [DOI] [PubMed] [Google Scholar]
- 30.Honda, K., A. Takaoka, and T. Taniguchi. 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25349-360. [DOI] [PubMed] [Google Scholar]
- 31.Honda, K., and T. Taniguchi. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6644-658. [DOI] [PubMed] [Google Scholar]
- 32.Hsu, T. Y., Y. Chang, P. W. Wang, M. Y. Liu, M. R. Chen, J. Y. Chen, and C. H. Tsai. 2005. Reactivation of Epstein-Barr virus can be triggered by an Rta protein mutated at the nuclear localization signal. J. Gen. Virol. 86317-322. [DOI] [PubMed] [Google Scholar]
- 33.Iwamura, T., M. Yoneyama, K. Yamaguchi, W. Suhara, W. Mori, K. Shiota, Y. Okabe, H. Namiki, and T. Fujita. 2001. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells 6375-388. [DOI] [PubMed] [Google Scholar]
- 34.James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 1441425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaworska, J., A. Gravel, K. Fink, N. Grandvaux, and L. Flamand. 2007. Inhibition of transcription of the beta interferon gene by the human herpesvirus 6 immediate-early 1 protein. J. Virol. 815737-5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johannsen, E., M. Luftig, M. R. Chase, S. Weicksel, E. Cahir-McFarland, D. Illanes, D. Sarracino, and E. Kieff. 2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 10116286-16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karpova, A. Y., M. Trost, J. M. Murray, L. C. Cantley, and P. M. Howley. 2002. Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. USA 992818-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato, K., Y. Kawaguchi, M. Tanaka, M. Igarashi, A. Yokoyama, G. Matsuda, M. Kanamori, K. Nakajima, Y. Nishimura, M. Shimojima, H. T. Phung, E. Takahashi, and K. Hirai. 2001. Epstein-Barr virus-encoded protein kinase BGLF4 mediates hyperphosphorylation of cellular elongation factor 1delta (EF-1delta): EF-1delta is universally modified by conserved protein kinases of herpesviruses in mammalian cells. J. Gen. Virol. 821457-1463. [DOI] [PubMed] [Google Scholar]
- 39.Kato, K., A. Yokoyama, Y. Tohya, H. Akashi, Y. Nishiyama, and Y. Kawaguchi. 2003. Identification of protein kinases responsible for phosphorylation of Epstein-Barr virus nuclear antigen leader protein at serine-35, which regulates its coactivator function. J. Gen. Virol. 843381-3392. [DOI] [PubMed] [Google Scholar]
- 40.Kim, T. K., J. S. Lee, S. Y. Oh, X. Jin, Y. J. Choi, T. H. Lee, E. Lee, Y. K. Choi, S. You, Y. G. Chung, J. B. Lee, R. A. DePinho, L. Chin, and H. Kim. 2007. Direct transcriptional activation of promyelocytic leukemia protein by IFN regulatory factor 3 induces the p53-dependent growth inhibition of cancer cells. Cancer Res. 6711133-11140. [DOI] [PubMed] [Google Scholar]
- 41.Kudoh, A., T. Daikoku, Y. Ishimi, Y. Kawaguchi, N. Shirata, S. Iwahori, H. Isomura, and T. Tsurumi. 2006. Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J. Virol. 8010064-10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar, K. P., K. M. McBride, B. K. Weaver, C. Dingwall, and N. C. Reich. 2000. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol. 204159-4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee, C. P., J. Y. Chen, J. T. Wang, K. Kimura, A. Takemoto, C. C. Lu, and M. R. Chen. 2007. Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. J. Virol. 815166-5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee, C. P., Y. H. Huang, S. F. Lin, Y. Chang, Y. H. Chang, K. Takada, and M. R. Chen. 2008. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 8211913-11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lefort, S., A. Soucy-Faulkner, N. Grandvaux, and L. Flamand. 2007. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 8110950-10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin, R., P. Genin, Y. Mamane, M. Sgarbanti, A. Battistini, W. J. Harrington, Jr., G. N. Barber, and J. Hiscott. 2001. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 20800-811. [DOI] [PubMed] [Google Scholar]
- 47.Lin, R., Y. Mamane, and J. Hiscott. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 192465-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu, C. C., Y. C. Chen, J. T. Wang, P. W. Yang, and M. R. Chen. 2007. Xeroderma pigmentosum C is involved in Epstein Barr virus DNA replication. J. Gen. Virol. 883234-3243. [DOI] [PubMed] [Google Scholar]
- 49.Makarova, O., E. Kamberov, and B. Margolis. 2000. Generation of deletion and point mutations with one primer in a single cloning step. BioTechniques 29970-972. [DOI] [PubMed] [Google Scholar]
- 50.Mantovani, F., F. Tocco, J. Girardini, P. Smith, M. Gasco, X. Lu, T. Crook, and G. Del Sal. 2007. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat. Struct. Mol. Biol. 14912-920. [DOI] [PubMed] [Google Scholar]
- 51.Melroe, G. T., N. A. DeLuca, and D. M. Knipe. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 788411-8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller, D. M., Y. Zhang, B. M. Rahill, W. J. Waldman, and D. D. Sedmak. 1999. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J. Immunol. 1626107-6113. [PubMed] [Google Scholar]
- 53.Mori, M., M. Yoneyama, T. Ito, K. Takahashi, F. Inagaki, and T. Fujita. 2004. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 2799698-9702. [DOI] [PubMed] [Google Scholar]
- 54.Morin, P., J. Braganca, M. T. Bandu, R. Lin, J. Hiscott, J. Doly, and A. Civas. 2002. Preferential binding sites for interferon regulatory factors 3 and 7 involved in interferon-A gene transcription. J. Mol. Biol. 3161009-1022. [DOI] [PubMed] [Google Scholar]
- 55.Netterwald, J. R., T. R. Jones, W. J. Britt, S. J. Yang, I. P. McCrone, and H. Zhu. 2004. Postattachment events associated with viral entry are necessary for induction of interferon-stimulated genes by human cytomegalovirus. J. Virol. 786688-6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parekh, B. S., and T. Maniatis. 1999. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-beta promoter. Mol. Cell 3125-129. [DOI] [PubMed] [Google Scholar]
- 57.Preston, C. M., A. N. Harman, and M. J. Nicholl. 2001. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J. Virol. 758909-8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rickinson, A. B., and E. D. Kieff. 2007. Epstein-Barr virus, p. 2656-2700. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 59.Saira, K., Y. Zhou, and C. Jones. 2007. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J. Virol. 813077-3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saitoh, T., A. Tun-Kyi, A. Ryo, M. Yamamoto, G. Finn, T. Fujita, S. Akira, N. Yamamoto, K. P. Lu, and S. Yamaoka. 2006. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat. Immunol. 7598-605. [DOI] [PubMed] [Google Scholar]
- 61.Sarkar, S. N., K. L. Peters, C. P. Elco, S. Sakamoto, S. Pal, and G. C. Sen. 2004. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol. 111060-1067. [DOI] [PubMed] [Google Scholar]
- 62.Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55255-281. [DOI] [PubMed] [Google Scholar]
- 63.Servant, M. J., N. Grandvaux, B. R. tenOever, D. Duguay, R. Lin, and J. Hiscott. 2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem. 2789441-9447. [DOI] [PubMed] [Google Scholar]
- 64.Servant, M. J., B. ten Oever, C. LePage, L. Conti, S. Gessani, I. Julkunen, R. Lin, and J. Hiscott. 2001. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276355-363. [DOI] [PubMed] [Google Scholar]
- 65.Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 3001148-1151. [DOI] [PubMed] [Google Scholar]
- 66.Shibaki, T., T. Suzutani, I. Yoshida, M. Ogasawara, and M. Azuma. 2001. Participation of type I interferon in the decreased virulence of the UL13 gene-deleted mutant of herpes simplex virus type 1. J. Interf. Cytok. Res. 21279-285. [DOI] [PubMed] [Google Scholar]
- 67.Suhara, W., M. Yoneyama, I. Kitabayashi, and T. Fujita. 2002. Direct involvement of CREB-binding protein/p300 in sequence-specific DNA binding of virus-activated interferon regulatory factor-3 holocomplex. J. Biol. Chem. 27722304-22313. [DOI] [PubMed] [Google Scholar]
- 68.Tseng, S. F., C. Y. Chang, K. J. Wu, and S. C. Teng. 2005. Importin KPNA2 is required for proper nuclear localization and multiple functions of NBS1. J. Biol. Chem. 28039594-39600. [DOI] [PubMed] [Google Scholar]
- 69.Wang, J. T., P. W. Yang, C. P. Lee, C. H. Han, C. H. Tsai, and M. R. Chen. 2005. Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J. Gen. Virol. 863215-3225. [DOI] [PubMed] [Google Scholar]
- 70.Wathelet, M. G., C. H. Lin, B. S. Parekh, L. V. Ronco, P. M. Howley, and T. Maniatis. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1507-518. [DOI] [PubMed] [Google Scholar]
- 71.Wu, L., E. Fossum, C. H. Joo, K. Lee, Y. C. Shin, E. Johannsen, L. M. Hutt-Fletcher, J. Hass, and J. U. Jung. 5 November 2008. Epstein-Barr virus LF2: an antagonist to type I interferon. J. Virol. doi: 10.1128/JVI.00602-08. [DOI] [PMC free article] [PubMed]
- 72.Yang, P. W., S. S. Chang, C. H. Tsai, Y. H. Chao, and M. R. Chen. 2008. Effect of phosphorylation on the transactivation activity of Epstein-Barr virus BMRF1, a major target of the viral BGLF4 kinase. J. Gen. Virol. 89884-895. [DOI] [PubMed] [Google Scholar]
- 73.Yoneyama, M., W. Suhara, Y. Fukuhara, M. Fukuda, E. Nishida, and T. Fujita. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 171087-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yue, W., E. Gershburg, and J. S. Pagano. 2005. Hyperphosphorylation of EBNA2 by Epstein-Barr virus protein kinase suppresses transactivation of the LMP1 promoter. J. Virol. 795880-5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou, Z. X., J. A. Kemppainen, and E. M. Wilson. 1995. Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol. Endocrinol. 9605-615. [DOI] [PubMed] [Google Scholar]
- 76.Zhu, F. X., S. M. King, E. J. Smith, D. E. Levy, and Y. Yuan. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 995573-5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.