

Abstract
The Prep1 homeodomain transcription factor is essential in embryonic development. Prep1 hypomorphic mutant mouse (Prep1i/i) embryos (embryonic day 9.5) display an increased terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling reaction compared to wild-type (WT) littermates. Prep1i/i mouse embryo fibroblasts (MEFs) show an increased basal level of annexin V binding activity, reduction of the mitochondrial-membrane potential, and increased caspase 9 and 3 activation, indicating increased apoptosis. Prep1i/i MEFs also respond faster than WT MEFs to genotoxic stress, indicating increased activation of the intrinsic apoptotic pathways. We did not observe an increase in p53 or an abnormal p53 response to apoptotic stimuli. However, hypomorphic MEFs have decreased endogenous levels of antiapoptotic Bcl-XL mRNA and protein, and Bcl-x overexpression rescues the defect of Prep1i/i MEFs. Using transient transfections and chromatin immunoprecipitation, we identified the Bcl-x promoter as a novel target of Prep1. Thus, Prep1 directly controls mitochondrial homeostasis (and the apoptotic potential) by modulating Bcl-x gene expression.
Prep1 transcription factor DNA independently heterodimerizes with Pbx family members (4, 8, 22), which allows its nuclear localization and activity (1, 2). Prep1 is essential for embryonic development: in particular, Prep1 null embryos die before gastrulation (L. C. Fernandez-Diaz and F. Blasi, unpublished data), while Prep1 hypomorphic mutant mouse (Prep1i/i) embryos, which express 2 to 3% of Prep1 mRNA and up to 10% of the protein, show a leaky embryonic-lethal phenotype and defects in angiogenesis, hematopoiesis, and eye development. The molecular basis of the Prep1i/i phenotype depends, at least in part, on reduction of the Pbx protein level (13, 15, 29).
Programmed cell death may be triggered by external signals mediated by specific cell surface receptors (17) or by damage or stress-generated intrinsic signals mediated by mitochondria (12). Pathway-specific caspase cascade activation, in turn, induces characteristic biochemical and morphological changes and, ultimately, cell death (27). Proteins of the Bcl-2 family are central regulators of apoptosis, with antiapoptotic (Bcl-2-like survival factors: Bcl-2, Bcl-XL, Bcl-w, and Mcl1), as well as proapoptotic, members. These, in turn, can be distinguished as follows: Bax-like death factors (Bax, Bak, and Bcl-XS) and BH3-only death factors (BimL, Bad, Bid, Noxa, Puma, and Bik) (18). Proapoptotic members of the Bcl-2 family increase the permeability of the outer mitochondrial membrane, whereas antiapoptotic members inhibit their action and maintain mitochondrial homeostasis (12). Thus, the balance of pro- and antiapoptotic proteins at the mitochondrial outer membrane determines a cell's fate (5). The p53 tumor suppressor regulates the balance between these proteins and thus controls the apoptotic destiny of a cell. Upon genotoxic stress, p53 accumulates in the nucleus (26, 32) and transcriptionally activates genes that promote apoptosis, in particular, the proapoptotic members of the Bcl-2 family, such as Bax (16, 19). The increased ratio of proapoptotic to antiapoptotic Bcl-2 proteins at the outer mitochondrial membrane favors the release of apoptogenic proteins and the activation of caspases, ultimately tipping the balance toward cell death.
Here, we report that hypomorphic Prep1i/i embryos display substantial generalized apoptosis and that Prep1i/i mouse embryo fibroblasts (MEFs) have increased basal apoptosis compared to those of wild-type (WT) littermates and respond faster than the WT to intrinsic, but not extrinsic, apoptotic stimuli. Endogenous p53 mRNA and protein levels are only marginally affected, as is the genotoxic-stress-induced p53 response. However, Prep1i/i MEFs have decreased levels of endogenous Bcl-XL protein, a regulator of mitochondrial-membrane permeability (35). Transient-transfection and chromatin immunoprecipitation (ChIP) analyses showed that Bcl-x is a direct target of Prep1 and that restoring Bcl-XL levels rescues the apoptotic phenotype of Prep1i/i MEFs. Therefore, Prep1 affects apoptosis by directly modulating mitochondrial homeostasis through the control of Bcl-x gene expression.
MATERIALS AND METHODS
Cell culture.
Cos7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 5 mM sodium pyruvate, 2 mM glutamine, and streptomycin-penicillin at 37°C in a humidified incubator with 5% CO2.
Genotyping of mice, MEF extraction, and culture.
Animals were maintained in a specific-pathogen-free mouse facility. Genotyping was performed as described previously (15). Primary MEFs were obtained from embryonic day 14.5 (E14.5) embryos after mating Prep1+/i heterozygous animals (15). Each embryo was dissected and treated with 0.25% trypsin, 0.02% EDTA in phosphate-buffered saline (PBS) for 30 min on ice. Trypsinization was blocked by the addition of complete DMEM. After mechanical dissociation, the embryo fragments were cultured in 6-cm dishes containing complete DMEM and were incubated at 37°C with 5% CO2. MEFs were used for experiments between passages 2 and 5.
TUNEL assay.
E9.5 WT and Prep1i/i embryos were fixed in 4% paraformaldehyde, pH 7.4, at 4°C overnight. The embryos were incubated in PBS-20% sucrose solution at 4°C for 12 h, embedded in Killik (Bio-Optics, Italy), and frozen at −80°C until they were used. Each frozen embryo was cut with a cryotome into 8-μm-thick sagittal sections. A terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay was performed following the manufacturer's instructions with an Apoptag fluorescein in situ apoptosis detection kit (Chemicon International). The slides were incubated with Hoechst stain (1:1,000 in PBS) for 5 min and finally mounted under a glass coverslip. Sections were analyzed with a DeltaVision microscope (Olympus IX70; Applied Precision) using a 20× objective, and paneling of the acquired images was done using SoftWoRx software.
Apoptotic treatments.
MEFs (3 × 105; third passage) were plated in 6-cm dishes for fluorescence-activated cell sorter (FACS) analysis. After 24 h, the MEFs were exposed to UV light using a UV lamp (Vilber Lourmat; VL-115.C; UV C; 254 nm; 1,000 J/m2) or a UV Stratalinker 1800 (Stratagene, La Jolla, CA; UV C; 254 nm; 60 J/m2). Alternatively, the MEFs were treated with etoposide (Sigma, St. Louis, MO) or tumor necrosis factor alpha (TNF-α) (Roche) as indicated in the figure legends. For biochemical analyses, 1 × 106 cells (plated in 10-cm dishes and treated as described above) were used.
Flow cytometry analysis.
Apoptosis was measured with the Annexin V-FITC Apoptosis Detection KIT II (BD Pharmingen, San Diego, CA) and analyzed by flow cytometry (FACScan; Becton Dickinson,). For the JC-1 experiment, cells were stained using 10 μM JC-1 dye (Molecular Probes, Invitrogen; United Kingdom) for 30 min at 37°C.
Protein extraction and immunoblotting.
Total extracts were prepared in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% Na-deoxycholate) containing a protease inhibitor cocktail (Roche, Basel, Switzerland) and clarified by centrifugation. The collected supernatants were quantitated by the Bradford assay (Bio-Rad, Richmond, CA). Nuclear-protein extraction was performed as described previously (25). Protein extracts were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted to polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were incubated with primary antibodies for 1 h at room temperature and incubated with a peroxidase-conjugated secondary antibody for 1 h at room temperature in milk-PBS-Tween 20 solution (dry milk, 5%; Tween 20, 0.1%). The peroxidase activity was measured with an enhanced chemiluminescence kit (Pierce, Rockford, IL) following the manufacturer's instructions. The membranes were exposed to a Hyperfilm (Amersham, Arlington Heights, IL), and the resulting bands were quantitated by densitometric analysis (Personal Densitometer; Molecular Dynamics). The following antibodies were used for immunoblot decoration: p53 (monoclonal antibody; JM-3036-100; MBL, Woburn, MA); Bcl-XL (monoclonal antibody; sc-8392; Santa Cruz Biotechnology, CA); caspase 3 (polyclonal antibodies; JM-3138-100; MBL); caspase 8 (polyclonal antibodies; JM-3020-100; MBL); caspase 9 (monoclonal antibody; M054-3; MBL); caspase 3 (polyclonal antibodies; Cell Signaling Technology, Danvers, MA); β-actin (polyclonal antibodies; sc-1616; Santa Cruz Biotechnology); tubulin-α (monoclonal antibody; N356; Amersham Lifescience, United Kingdom); and Prep1 (Meis 4.1; monoclonal antibody; 05-766; Upstate Biotechnology, Upstate House, Dundee, United Kingdom).
RNA extraction and quantitative RT-PCR.
RNA extraction was performed with the RNeasy mini kit (Qiagen, Germany) following the manufacturer's instructions. Reverse transcription (RT) on total RNA was performed with the Superscript First-Strand Synthesis System for RT-PCR kit (Invitrogen, United Kingdom). Triplicate real-time PCRs were carried out in an ABI/Prism 7900 HT Sequence Detector System (Applied Biosystems, Foster City, CA) using 5 ng of the RT reaction mixture. Primers were designed based on the sequences corresponding to accession numbers NM_011640 (p53) and NM_009743.3 (Bcl-XL).
Confocal microscopy.
Untreated and treated MEFs were stained with 10 μM (final concentration) JC-1 (Molecular Probes). Images were acquired with a TCS SP2 confocal laser scanning microscope (Leica), using a 63× Plan-Apochromat oil immersion objective, numerical aperture 1.4, equipped with 488- and 543-nm laser lines, using a pinhole equivalent to 1 Airy disk, at the Advanced Light and Electron Microscopy BioImaging Center, S. Raffaele Scientific Institute, Milan, Italy.
Retroviral infection.
pBabe-puro-Prep1 was generated by amplifying the cDNA for Prep1 with the primers FW-SnaBI (5′-AGCTTTTACGTAATGATGGCTACACAGACATTAAG-3′) and RW-SalI (5′-TTCCGCGGCCGCTATGGCCGACGTCGACCTACTGCAGGGAGTCACCTGTTCG-3′) and was inserted into the SnaBI and SalI sites of pBabe-Puro. The Bcl-XL vector was provided by D. Green (9). The vectors were transfected into Phoenix packaging cells by the calcium phosphate method. Viral supernatants were supplemented with 5 μg Polybrene, used to infect MEFs (at passage 2), and used after four rounds of infection.
Luciferase assay.
Cos7 cells were transfected (in triplicate) with plasmids (see Fig. 6) using the Lipofectamine 2000 reagent (Invitrogen, United Kingdom) following the manufacturer's instructions. The plasmids used for transfection were Bcl-x promoter (−198), kindly provided by Maurice Bondurant (Veterans Affairs Medical Center, Nashville, TN); Prep1 and Pbx1b expression vectors (4); cytomegalovirus β-galactosidase plasmid (Invitrogen); and pBluescript SK II(+), used to normalize the DNA content (Stratagene, La Jolla, CA). The mutated form of the −198 plasmid was obtained using the QuickChange II XL Site-Directed Mutagenesis kit (Stratagene). The mutagenic primers, designed following the provided instructions, were Fwd1 (5′-GGAATATTGTAAGGTAAATTCCATGCATATTAAATTTTTC-3′), Rew1 (5′-GAAAAATTTAATATGCATGGAATTTACCTTACAATATTCC-3′), Fwd2 (5′-GGAATATTGTAAGGTAAATTCACTGCATATTAAATTTTTC-3′), and Rew2 (5′-GAAAAATTTAATATGCAGTGAATTTACCTTACAATATTCC-3′).
FIG. 6.

Bcl-x expression is controlled by the Prep1-Pbx1 complex. (A) Total RNA was extracted from untreated MEFs obtained from Prep1+/+ (+/+) and Prep1i/i (i/i) (n = 4 each) embryos, retrotranscribed, and quantitated for Bcl-XL mRNA by real-time PCR (see Materials and Methods). Data from individual reactions were normalized to 18S rRNA and averaged. A value of 1 was arbitrarily given to the Bcl-XL mRNA amount of each Prep1+/+ MEF. The error bars indicate standard deviations. A.U., arbitrary units. (B) Total extracts from Prep1+/+, Prep+/i (+/i), and Prep1i/i MEFs were fractionated, transferred to a PVDF membrane as described in the text, immunoblotted with anti-Bcl-XL antibody, normalized to β-actin levels, and analyzed by densitometry. A value of 100 was arbitrarily given to Prep1+/+ MEFs. n, number of embryos analyzed. (C) Total extracts from Prep1+/+ and Prep1i/i MEFs infected with the pBabe empty vector and from Prep1i/i MEFs infected with the pBabe-Bcl-XL vector were immunoblotted with monoclonal anti-Bcl-XL and α-tubulin antibodies and quantitated by densitometry. A value of 100 was arbitrarily given to the endogenous Bcl-XL protein in Prep1+/+ MEFs infected with the empty vector. The number of early apoptotic cells was determined by flow cytometry in MEFs infected with pBabe or pBabe-Bcl-XL vector (untreated), 24 h after treatment with 200 μM etoposide, or 12 h after treatment with UV-C light (254 nm; 1,000 J/m2). Empty-vector-infected WT MEFs were used as controls, and a value of 1 was arbitrarily given. Representative results from one of two independent experiments performed using MEFs from two and three different WT and Prep1i/i embryos, respectively, are shown. U, untreated. (D) Total extracts from Prep1i/i MEFs infected with the empty and Prep1-expressing retroviral pBabe vectors were immunoblotted with anti-Prep1, anti-Bcl-XL, and anti-α-tubulin (for normalization) antibodies and quantitated by densitometry. Empty-vector-infected WT MEFs were used as controls. A value of 100 was arbitrarily given to Bcl-XL protein levels in Prep1+/+ MEFs infected with the pBabe empty vector. The number of spontaneous apoptotic cells was determined by flow cytometry using annexin V binding in MEFs infected with the pBabe or pBabe-Prep1 vector. Empty-vector-infected WT MEFs were used as controls and were arbitrarily given a value of 1. Representative results from one of three independent experiments are shown. (E) Transient transfections of Cos7 cells with the WT (−198) and mutated (−198**) Bcl-x promoters and with equimolar amounts of the indicated expression plasmids. β-Galactosidase-normalized luciferase activities of total cell extracts (see Materials and Methods) are shown in the graph. (F) ChIP assay of the Bcl-x (P1) promoter with the indicated antibodies. DNA was amplified by real-time PCR using primers (see Fig. S2 in the supplemental material) spanning the region used for luciferase assays containing the Prep1/Pbx binding site. The data are expressed as enrichment of the immunoprecipitated DNA relative to the input DNA.
Thirty hours after transfection, the cells were washed with PBS and lysed with Reporter Lysis Buffer 1× Solution (Promega Corporation, WI) according to the manufacturer's protocol; 5 μl of each lysate was analyzed using the Luciferase Assay System (Promega Corporation) with a Mithras LB 940 luminometer (Berthold Technologies) equipped with Mikro Win 2000 software. The colorimetric β-galactosidase reaction was measured with Versa max (Molecular Devices), equipped with SOFT max PRO 4.3 software, at a wavelength of 420 nm and used to normalize the luciferase assay data.
ChIP and quantitative PCR.
Cross-linked chromatin was prepared from confluent MEFs (at passage 4) as described previously (14), and 20-μg aliquots were then incubated overnight with the appropriate antibodies (see below) or without antibodies (mock controls) in a total volume of 1 ml and immunoprecipitated. The resulting material was processed as described previously (14) and resuspended in 250 ml of distilled water. Triplicate samples of 4 μl of immunoprecipitated genomic DNA were amplified in a Light Cycler apparatus (Roche) and quantitated using a FastStart DNA mix SYBR green I kit (Roche). The PCR conditions were as follows: (i) denaturation and DNA polymerase activation at 95°C for 10 min; (ii) denaturation (95°C; 15 s), annealing (60°C; 6 s), and extension (72°C; 20 s) for 55 cycles; and (iii) melting curve (99°C for 5 s; cooling at 30°C for 5 s). The values are expressed as nanograms of enrichment with respect to input DNA. The primer sequences were as follows: Bcl-xFW, 5′-CGGACTCAGACCTTCATAAGAGCC-3′, and Bcl-xREV, 5′-CCAAAACACCTGCTCACTTACTGG-3′.
The antibodies for ChIP were as follows: anti-Pbx1b (15) (kindly provided by Micheal Cleary, Department of Pathology, Stanford University School of Medicine, Stanford, CA); anti-Prep1 (polyclonal) (4); and uPAR (polyclonal) (31).
RESULTS
Prep1i/i embryos display generalized apoptosis.
TUNEL immunofluorescence on sections of WT and Prep1i/i embryos at E9.5 (Fig. 1 is representative of many serial sections analyzed from a total of three embryos) showed that hypomorphic embryos display many more areas of intense fluorescence than WT embryos. Apoptosis was more evident in the central nervous system, the roof of the hindbrain, and intersomitic, as well as more ventral and caudal, regions. Similar observations were made with E11.5 Prep1i/i embryos (not shown). These results are in general agreement with the massive apoptosis observed in the central nervous system of Prep1.1 morpholino-injected early zebrafish embryos (11).
FIG. 1.

TUNEL analysis of Prep1i/i embryos. Sections of Killik-embedded E9.5 WT (+/+) and Prep1i/i (i/i) embryos were stained by TUNEL to detect apoptosis, and nuclei were counterstained with Hoechst stain (see Materials and Methods). The white arrowheads indicate neural and intersomitic regions; the empty arrowheads indicate other nonneural tissues.
Prep1i/i MEFs have a higher apoptotic potential than their WT counterparts.
We compared MEFs generated from E14.5 WT and Prep1i/i (hypomorphic) littermate embryos. Figure 2A shows Prep1 immunoblots from nuclear cell extracts of WT (Prep1+/+), heterozygous (Prep1+/i), and two Prep1i/i homozygous MEFs. Prep1i/i MEFs displayed very low residual Prep1 levels, as expected (15), while heterozygous Prep1+/i MEFs showed about 50% of the WT expression. We then analyzed the basal apoptotic potentials of these cells plated at low confluence and cultured for 24 h (when they reached 80% confluence). Figure 2B shows the average results obtained with MEFs from several different embryos. Annexin V binding (determined by flow cytometry) was two to threefold higher in Prep1i/i than in WT MEFs. This was not observed in Prep1+/i MEFs (data not shown). Similar results were obtained with late apoptotic cells by measuring the number of annexin V-binding and propidium iodide-incorporating cells (not shown). Thus, Prep1i/i MEFs have high spontaneous apoptosis.
FIG. 2.
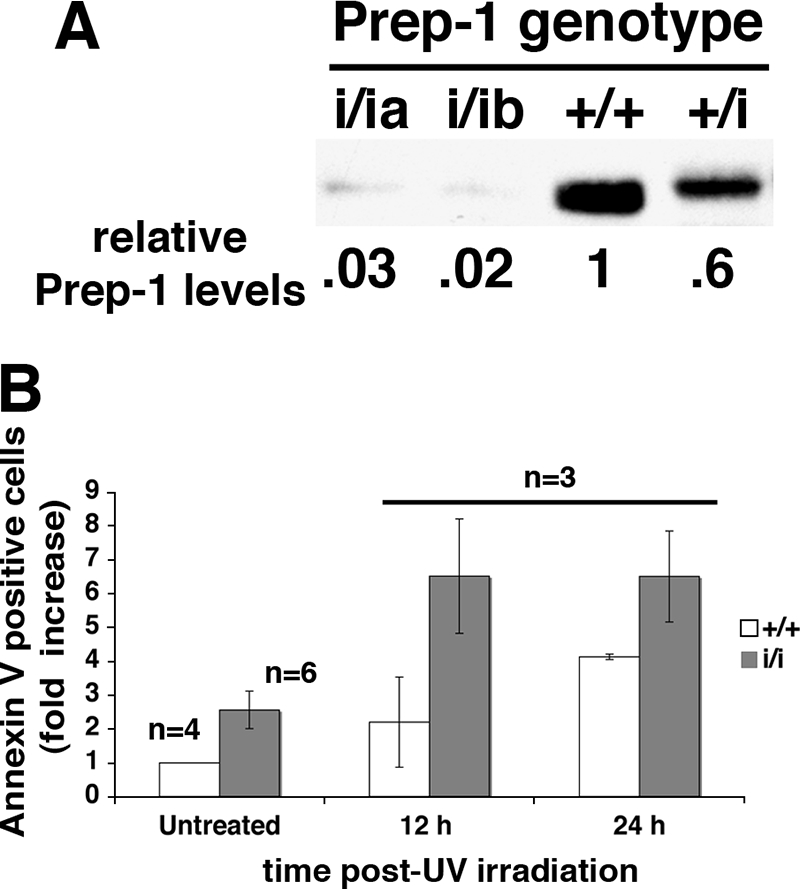
MEFs from Prep1i/i embryos are more apoptotic and more sensitive to UV irradiation than MEFs from WT embryos. (A) SDS-8% PAGE immunoblots of nuclear extracts of MEFs from E14.5 Prep1+/+ (+/+), Prep1+/i (+/i), and Prep1i/i (i/ia and i/ib) littermate embryos. Actin-normalized densitometric quantitation is shown under each lane. The Prep1 content of Prep1+/+ MEFs was arbitrarily set to 1. (B) Prep1+/+ and Prep1i/i MEFs were irradiated with UV light (UV-C; 254 nm; 1,000 J/m2). The number of early apoptotic cells was determined by flow cytometry using annexin V binding. WT untreated cells were given an arbitrary value of 1. n, number of embryos whose MEFs were analyzed. The error bars indicate standard deviations.
Prep1 affects the intrinsic apoptotic pathway.
We compared the responses of WT and Prep1i/i MEFs to stimuli that trigger mitochondrial responses. The number of early apoptotic MEFs, 12 h after high UV irradiation (1,000 J/m2), was about threefold higher in Prep1i/i than in WT MEFs, as determined by flow cytometry using annexin V binding (Fig. 2B), and remained higher at later time points (24 h). In the WT, this number increased at 24 h postirradiation, but it remained substantially lower than in Prep1i/i MEFs.
Immunoblot analysis with anti-initiator caspase 9 antibodies showed no signal in untreated cells, a very faint signal in WT MEFs 8 h postirradiation, and a stronger and earlier signal in Prep1i/i MEFs, reaching a maximum at 12 h (Fig. 3A and B). Analogous behavior was observed for the executioner caspase 3 (Fig. 3A and B). Likewise, at lower UV doses (60 J/m2) or after etoposide treatment, active caspase 9 was increased three- to fivefold in Prep1i/i MEFs 24 h after irradiation or etoposide treatment (Fig. 3C and D). Thus, Prep1i/i MEFs have higher basic and genotoxic stress-stimulated apoptosis.
FIG. 3.

The intrinsic apoptotic pathway is affected by decreased levels of Prep1. (A) Total extracts from E14.5 Prep1+/+ (+/+) and Prep1i/i (i/i) MEFs, untreated (U) or 4, 8, or 12 h after UV irradiation (UV-C; 254 nm; 1,000 J/m2), were fractionated in 12% SDS-PAGE, transferred to PVDF membranes, and immunoblotted with active caspase 9 and 3 monoclonal and polyclonal antibodies, respectively. β-Actin was used for normalization. Representative results from one of three independent experiments are shown. (B) Densitometric analysis of the β-actin-normalized immunoblots in panel A. A.U., arbitrary units. (C) Twenty-four hours after treatment, total extracts were prepared from untreated (U), 200 μM etoposide-treated, or UV-C light (254 nm; 60 J/m2)-irradiated MEFs from WT and Prep1i/i littermate embryos; fractionated in SDS-PAGE; transferred to PVDF membranes; and immunoblotted with active caspase 9 and β-actin antibodies. Representative results from one of three independent experiments are shown. (D) β-Actin-normalized densitometric analysis of the immunoblots shown in panel C. (E) MEFs from WT and hypomorphic embryos were treated (or not) with actinomycin D (1 μg/ml) for 30 min and then with 0.1, 1, and 10 ng/ml of TNF-α. Total extracts were prepared 6 h after treatment, fractionated, and transferred, and active caspase 8 protein levels were probed with specific polyclonal antibodies. β-Actin was used for normalization. Representative results from one of three independent experiments are shown. U, untreated. (F) β-Actin-normalized densitometric analysis of the immunoblot shown in panel E. (G) MEFs from WT, Prep+/i (+/i), and Prep1i/i littermates were either treated (or not) with 200 μM etoposide for 24 h, irradiated with UV-C light (254 nm; 60 J/m2), or treated with actinomycin D and 1 ng/ml of TNF-α for 6 h (see above). Immunoblotting was performed with anti-active caspase 3 and anti-α-tubulin polyclonal and monoclonal antibodies, respectively. Representative results from one of three independent experiments are shown. U, untreated. (H) α-Tubulin-normalized densitometric analysis of the immunoblot shown in panel G.
We also tested TNF-α, which triggers the extrinsic pathway (20). The annexin V binding differences between WT and Prep1i/i MEFs observed 12 and 24 h after the treatment were not as prominent as for genotoxic agents (not shown). TNF-α at 1 and/or 10 ng/ml induced caspase 8 activation within 6 h, and the effect was somewhat more pronounced in Prep1i/i than in WT MEFs (Fig. 3E and F).
We compared the levels of active caspase 3 in WT, Prep1+/i (heterozygous), and Prep1i/i MEFs after 24 h of treatment with etoposide or UV irradiation (60 J/m2) and 6 h after TNF-α treatment at 1 ng/ml. In all cases, caspase 3 increased more in Prep1i/i than in Prep1+/i MEFs, and even less in WT cells (Fig. 3G and H). However, etoposide and UV had greater effects than TNF-α. Interestingly, a stronger caspase 3 response was observed in heterozygous than in WT MEFs, indicating a direct relationship between Prep1 levels and the apoptotic response. We also observed the same correlation upon high UV irradiation (see Table S1 in the supplemental material).
In conclusion, Prep1i/i MEFs are more sensitive than WT MEFs to apoptotic stimuli, and in particular, to genotoxic stress. The TNF-α pathway was not analyzed further.
Alteration of the mitochondrial membrane potential in Prep1i/i MEFs.
The increased response of Prep1i/i MEFs to genotoxic stress suggests an altered mitochondrial function, since a decrease in the mitochondrial-membrane potential releases mitochondrial apoptogenic factors and induces caspase 9 activation (23). We investigated mitochondrial function by assessing the mitochondrial transmembrane potential (ΔΨm) in viable WT and Prep1i/i MEFs using the fluorescent JC-1 probe (30) and confocal microscopy. JC-1 is a positively charged carbocyanine dye that accumulates in the inner mitochondrial membrane and fluoresces in green at low and in red at high membrane potentials. During apoptosis, the mitochondrial potential collapses and JC-1 no longer accumulates in the mitochondria, inducing a diffuse green cytoplasmic fluorescence. Confocal microscopy showed that JC-1-stained, untreated WT MEFs displayed healthy mitochondria (i.e., rod-like green and red fluorescent figures), whereas in many Prep1i/i MEFs, JC-1 fluorescence was mostly green and diffuse in the cytoplasm (Fig. 4A). Etoposide or UV at low (60-J/m2) or high (1,000-J/m2) dose induced no major alteration in JC-1 staining of WT MEFs, consistent with the presence of active mitochondria, even if some cells displayed a partial shift of JC-1 emission from red to yellow-green. Genotoxic-stress-treated Prep1i/i MEFs, on the other hand, showed a decrease in the active mitochondrial staining and a diffuse green or yellow-green JC-1 fluorescence.
FIG. 4.

Prep1i/i MEFs have a lower mitochondrial-membrane potential (ΔΨm), which is further decreased upon genotoxic stress. (A) Untreated, 200 μM etoposide-treated or UV-C light (254 nm; 60 or 1,000 J/m2)-irradiated MEFs from Prep1+/+ (+/+) and Prep1i/i (i/i) embryos were stained with JC-1 (10 μM) for 30 min at 37°C and analyzed by confocal microscopy 12 h after each treatment. Some cells displayed a partial shift of JC-1 emission from red to yellow-green (white arrows). The results shown are representative of two independent experiments with at least 20 fields observed. (B) FACScan analysis of JC-1-stained (see above) Prep1+/+ and Prep1i/i MEFs (from a different litter than for panel A). The cells were analyzed before and 12 h after 200 μM etoposide or UV-C light (254 nm; 60 J/m2) treatment.
Flow cytometry of JC-1 red versus green fluorescence in genotoxic-stress-treated or untreated WT and Prep1i/i MEFs showed a twofold-higher green emission (low mitochondrial membrane potential) in untreated Prep1i/i MEFs than in WT MEFs, indicating altered mitochondrial homeostasis. Mitochondrion-deficient cells further increased in Prep1i/i, but not in WT MEFs, after etoposide or UV treatment (Fig. 4B).
The observation that Prep1i/i MEFs have compromised mitochondria provides a rationale for the high basal and genotoxic-stress-induced apoptosis in these cells.
p53 and its response is only marginally affected in Prep1i/i MEFs.
Mitochondrial deficiency and the anomalous response to genotoxic stress may depend on increased nuclear levels of p53 and the consequent upregulation of proapoptotic and downregulation of antiapoptotic genes (16, 24). We measured endogenous p53 levels in MEFs from several Prep1i/i and WT embryos by quantitative RT-PCR and immunoblotting. In untreated MEFs, p53 mRNA and protein were lower in Prep1i/i than WT MEFs (Fig. 5A and B), indicating that increased p53 could not be the determinant of the higher apoptotic potential of Prep1i/i MEFs. After etoposide or low UV irradiation, p53 increased in both Prep1i/i and WT MEFs, although less in Prep1i/i cells (Fig. 5C). This showed that the p53 response is not impaired in Prep1i/i MEFs. Indeed, genotoxic treatments equally increased the mRNA level of the p53 target Bax gene in WT and Prep1i/i MEFs (data not shown). Thus, we can exclude the possibility that the higher apoptotic potential of untreated Prep1i/i MEFs is caused by an increase in p53.
FIG. 5.

The p53-dependent response to genotoxic stress is slightly affected in Prep1i/i MEFs. (A) Total RNA extracted from untreated MEFs was obtained from Prep1+/+ (+/+) (n = 4) and Prep1i/i (i/i) (n = 4) embryos and retrotranscribed, and quantitative RT-PCR was performed for p53 mRNA in triplicate (see Materials and Methods). Data from individual reactions were normalized to 18S rRNA values obtained with the same sample in a parallel reaction, averaged, and plotted. The p53 mRNA level detected in WT MEFs was arbitrarily set to 100. The error bars indicate standard deviations. A.U., arbitrary units. (B) Total extracts of MEFs isolated from different Prep1+/+ (n = 3) and Prep1i/i (n = 4) embryos were fractionated, and the levels of endogenous p53 were analyzed by immunoblotting, quantitated, and normalized to α-tubulin by densitometry. A value of 100 was arbitrarily given to p53 protein levels in Prep1+/+ MEFs. (C) MEFs were treated (or not) with 200 μM etoposide or irradiated with UV-C light (254 nm; 60 J/m2). Crude extracts were prepared after 24 h and resolved on 12% SDS-PAGE, the levels of p53 were analyzed by immunoblotting, and the quantitation by α-tubulin-normalized densitometry was plotted. Note that in this experiment the p53 signal was not detected in total extracts from untreated cells, unlike in Fig. 5A where the immunoblot was exposed longer. Representative results from one of three independent experiments are shown.
Bcl-x is a target of the Prep1/Pbx complex, and Bcl-XL downregulation accounts for the anomalous apoptotic response of Prep1i/i MEFs.
The balance between pro- and antiapoptotic mitochondrial-membrane proteins maintains the organelle's homeostasis. Bcl-XL is involved in maintaining the mitochondrial-membrane potential (35), and its reduction in Prep1i/i MEFs may be at least in part responsible for increased apoptosis. Quantitative RT-PCR and immunoblotting showed 40% and 60% decreases in endogenous Bcl-XL mRNA and protein, respectively, in untreated Prep1i/i MEFs (Fig. 6A and B), while in Prep1+/i MEFs, Bcl-XL protein appeared only slightly reduced (Fig. 6B). To determine the generality of this finding, we also analyzed the Bcl-XL protein in whole extracts of E14.5 WT and Prep1i/i fetal livers, which also contained less Bcl-XL protein than the WT (see Fig. S1 in the supplemental material). Therefore, the decrease in Bcl-XL appears to be a general phenomenon and might account for the anomalous apoptosis of Prep1i/i MEFs and embryos.
To demonstrate this point, we infected Prep1i/i MEFs with a retrovirus expressing Bcl-XL (9) and measured the degrees of spontaneous and genotoxic-stress-induced apoptosis. Indeed, overexpression of Bcl-XL (Fig. 6C) reduced the apoptotic phenotype in both untreated and UV (1,000-J/m2)- or etoposide-treated MEFs, as shown by flow cytometry analysis of annexin V binding (Fig. 6C).
Moreover, overexpression of Prep1 in Prep1i/i MEFs restored high levels of Bcl-XL, confirming the dependence of the latter on Prep1 levels (Fig. 6D). Accordingly, the apoptotic phenotype of Prep1-infected Prep1i/i MEFs was restored (Fig. 6D).
We also transiently transfected Cos7 cells with a luciferase reporter plasmid driven by the murine Bcl-x regulatory region (−198 bp) (28, 34), which contains at least one potential Prep1/Pbx binding TGACAT site (see Fig. S2 in the supplemental material). Cotransfection of the reporter plasmid with both Prep1 and Pbx1b expression vectors increased luciferase activity (Fig. 6E), in agreement with the known fact that Prep1 activates transcription in complex with Pbx1 (3, 4). A reporter plasmid mutated in the Prep1/Pbx binding site responded much more weakly. The expression of only Prep1 or Pbx1 had no effect (not shown).
Finally, we immunoprecipitated cross-linked, sonicated chromatin with anti-Prep1 and -Pbx1b antibodies and analyzed the precipitated DNA by real-time PCR with a set of primers within the same Bcl-x P1 promoter (28) used for the transfection assays and including the Prep1/Pbx binding site. Indeed, Prep1 and Pbx1b antibodies immunoprecipitated this DNA region in WT, but not in Prep1i/i[r], MEFs (Fig. 6F). Therefore, Prep1 and Pbx1b are absent from the Bcl-x regulatory region in Prep1i/i MEFs. The absence of Pbx1 was expected, since one of the major phenotypes of the Prep1i/i embryos is an overall decrease in the Pbx proteins (15, 29).
Thus, the data indicate that the Prep1/Pbx heterodimer stimulates transcription of the murine Bcl-x gene. In the absence of Prep1, Bcl-x is downregulated, and the decreased Bcl-XL alters mitochondrial homeostasis and increases spontaneous and induced apoptosis.
DISCUSSION
Prep1i/i hypomorphic mouse embryos are smaller than their WT littermates, showing retarded growth and organ hypoplasia (13, 15). This phenotype might depend on a deficient general cellular function, such as, for instance, apoptosis. This possibility is supported by our TUNEL analysis (Fig. 1), showing widespread apoptosis in Prep1i/i embryos; agrees with the increased brain apoptosis observed in Prep1.1-downregulated zebrafish (11); and suggests that Prep1 has a conserved, important function in cell death regulation.
Hypomorphic Prep1i/i MEFs display a very low level of Prep1 (Fig. 1) and of its partners Pbx1 and Pbx2 (data not shown). In fact, they show higher levels of spontaneous apoptosis than the WT (Fig. 2), confirming the TUNEL analysis. The depletion of Prep1 affects mainly the intrinsic apoptotic pathway. In fact, Prep1i/i MEFs were much more sensitive to stimuli that trigger the intrinsic (UV or etoposide) than the extrinsic (TNF-α) apoptosis pathway (Fig. 2 and 3), both in terms of the number of cells binding annexin V and in the timing and extent of activation of “initiator” caspase 9 and “excutioner” caspase 3. Interestingly, we observed a direct relationship between the Prep1 level and caspase 3 activation, since an intermediate caspase 3 activation was observed in heterozygous versus WT or homozygous Prep1i/i MEFs upon each apoptotic treatment. Moreover, the different Prep1 residual levels inversely correlated with caspase 3 activation (Fig. 3; see Table S1 in the supplemental material).
Surprisingly, we did not observe an increase in p53, a major regulator of the intrinsic pathway (16) and highly sensitive to genotoxic stress, in Prep1i/i MEFs. In fact, our data, if anything, show a decrease in p53 mRNA and protein, a normal increase of p53 upon genotoxic stress (Fig. 5), and no difference in the regulation of a p53 target gene, Bax, in Prep1i/i MEFs upon genotoxic stress (data not shown). On this basis, p53 does not appear to be a mediator of the apoptotic phenotype of Prep1i/i cells.
The lack of Prep1 interferes with mitochondrial homeostasis, as shown by confocal-microscopy analysis, which demonstrated a sizable population of Prep1i/i MEFs with low mitochondrial-membrane potential (Fig. 4). Thus, the mitochondrial amplification of the etoposide- or UV-generated p53-dependent apoptotic response is already operative in these cells, indicating that the lack of Prep1 acts downstream of p53.
The balance of pro- and antiapoptotic proteins regulates mitochondrial permeability (23). Since Bcl-XL is an antiapoptotic member of the Bcl-2 family and is involved in maintaining the mitochondrial-membrane potential (23), the lower levels of Bcl-XL mRNA and protein in Prep1i/i MEFs might tilt the balance toward apoptosis, explaining why Prep1i/i MEFs undergo spontaneous apoptosis and faster and stronger apoptosis after genotoxic stress. Indeed, overexpression of Bcl-XL in Prep1i/i MEFs brought the levels of spontaneous and genotoxic-stress-induced apoptosis back to WT levels (Fig. 6), indicating that Bcl-XL is a key molecule in the Prep1i/i MEFs' apoptotic phenotype. The levels of Bcl-XL in Prep1i/i MEFs depend on Prep1; in fact, overexpression of Prep1 in these cells restores high levels of Bcl-XL and rescues their apoptotic phenotype.
Finally, the decrease in Bcl-XL in Prep1i/i MEFs appears to be due to the lack of direct transcriptional regulation of the Bcl-x gene by Prep1 (Fig. 6). Indeed, the regulatory region of the Bcl-x gene containing one Prep1/Pbx binding site in the first 198 bp (see Fig. S2 in the supplemental material) and several Prep1/Pbx binding sites upstream (not shown) responds to the ectopic expression of a Prep1-Pbx1b complex in cotransfection experiments in vitro. The single Prep1/Pbx binding site appears degenerate (TGACATG and not TGACAG), resulting in a low-affinity binding site. However, ChIP experiments with anti-Prep1 and anti-Pbx1b showed that this complex is present in vivo ion the Bcl-x regulatory region in WT MEFs and that not only Prep1, but also Pbx1b, is absent in Prep1i/i MEFs. This shows that Bcl-x is a target of the Prep1-Pbx1 complex and not just of Prep1 and agrees with the absence of Pbx in Prep1i/i MEFs and embryos (data not shown) (15, 29). Prep1 exerts hierarchical control over the levels of its Pbx partners mainly at the protein stability level (references 15, 25 and data not shown). However, Pbx1 and Pbx2 knockout mice and Pbx4 lazarus knockdown zebrafish did not generate an apoptotic phenotype (6, 11, 33), while Prep1.1 depletion did (11). This is probably due to the fact that the absence of one Pbx gene can be compensated for by other members of the family (7, 33), whereas the absence of Prep1 totally depletes all Pbx isoforms (33). Therefore, the apoptotic phenotype is due to the absence of Prep1, which represents a novelty in the mechanism of action and in the function of Prep1.
The relevance of our results is twofold: (i) we identified Bcl-x as a transcriptional target of Prep1 and (ii) we showed that by controlling Bcl-XL, Prep1 modulates mitochondrial homeostasis and therefore apoptosis. The 60% decrease in Bcl-XL in Prep1i/i MEFs is not marginal, since Bcl-x +/− heterozygous male germ cells are apoptotic (21) and since cell lines in which the Bcl-XL level is downregulated undergo apoptosis because of an altered mitochondrial homeostasis (10, 36).
In conclusion, we have identified Bcl-x as a transcriptional target of Prep1 and showed that by controlling Bcl-XL, Prep1 modulates mitochondrial homeostasis and therefore apoptosis. This provides a possible explanation for the general organ hypoplasia and the small size of Prep1i/i embryos and mice (13, 15, 29) and may explain other aspects of this phenotype.
Supplementary Material
Acknowledgments
We are grateful to Gerry Melino (Università di Roma Tor Vergata) for discussion and advice, Daniela Talarico (S. Raffaele Scientific Institute) and Elena Longobardi (IFOM) for critical discussions and technical help, Silvia Mori (S. Raffaele Scientific Institute) for the pBabe-Prep1 retroviral vector, and Marco Giorgio and Raluca Marcu (IFOM) for their help in JC-1 staining experiments.
This work was supported by grants from the Italian Association for Cancer Research (AIRC) (number 4/2005) Telethon and the Italian Ministry of University and Scientific Research to F.B. and from AIRC (number 63/2006) and the Istituto Superiore di Sanità to M.P.C.
Footnotes
Published ahead of print on 22 December 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Berthelsen, J., C. Kilstrup-Nielsen, F. Blasi, F. Mavilio, and V. Zappavigna. 1999. The subcellular localization of PBX1 and EXD proteins depends on nuclear import and export signals and is modulated by association with PREP1 and HTH. Genes Dev. 13946-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berthelsen, J., L. Viggiano, H. Schulz, E. Ferretti, G. G. Consalez, M. Rocchi, and F. Blasi. 1998. PKNOX1, a gene encoding PREP1, a new regulator of Pbx activity, maps on human chromosome 21q22.3 and murine chromosome 17B/C. Genomics 47323-324. [DOI] [PubMed] [Google Scholar]
- 3.Berthelsen, J., V. Zappavigna, E. Ferretti, F. Mavilio, and F. Blasi. 1998. The novel homeoprotein Prep1 modulates Pbx-Hox protein cooperativity. EMBO J. 171434-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berthelsen, J., V. Zappavigna, F. Mavilio, and F. Blasi. 1998. Prep1, a novel functional partner of Pbx proteins. EMBO J. 171423-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borner, C. 2003. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol. 39615-647. [DOI] [PubMed] [Google Scholar]
- 6.Brendolan, A., E. Ferretti, V. Salsi, K. Moses, S. Quaggin, F. Blasi, M. L. Cleary, and L. Selleri. 2005. A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development 1323113-3126. [DOI] [PubMed] [Google Scholar]
- 7.Capellini, T. D., G. Di Giacomo, V. Salsi, A. Brendolan, E. Ferretti, D. Srivastava, V. Zappavigna, and L. Selleri. 2006. Pbx1/Pbx2 requirement for distal limb patterning is mediated by the hierarchical control of Hox gene spatial distribution and Shh expression. Development 1332263-2273. [DOI] [PubMed] [Google Scholar]
- 8.Chang, C. P., Y. Jacobs, T. Nakamura, N. A. Jenkins, N. G. Copeland, and M. L. Cleary. 1997. Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol. Cell. Biol. 175679-5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chipuk, J. E., L. Bouchier-Hayes, T. Kuwana, D. D. Newmeyer, and D. R. Green. 2005. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 3091732-1735. [DOI] [PubMed] [Google Scholar]
- 10.Coluccia, A. M., S. Perego, L. Cleris, R. H. Gunby, L. Passoni, E. Marchesi, F. Formelli, and C. Gambacorti-Passerini. 2004. Bcl-XL down-regulation suppresses the tumorigenic potential of NPM/ALK in vitro and in vivo. Blood 1032787-2794. [DOI] [PubMed] [Google Scholar]
- 11.Deflorian, G., N. Tiso, E. Ferretti, D. Meyer, F. Blasi, M. Bortolussi, and F. Argenton. 2004. Prep1.1 has essential genetic functions in hindbrain development and cranial neural crest cell differentiation. Development 131613-627. [DOI] [PubMed] [Google Scholar]
- 12.Desagher, S., and J. C. Martinou. 2000. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 10369-377. [DOI] [PubMed] [Google Scholar]
- 13.Di Rosa, P., J. C. Villaescusa, E. Longobardi, G. Iotti, E. Ferretti, V. M. Diaz, A. Miccio, G. Ferrari, and F. Blasi. 2007. The homeodomain transcription factor Prep1 (pKnox1) is required for hematopoietic stem and progenitor cell activity. Dev. Biol. 311324-334. [DOI] [PubMed] [Google Scholar]
- 14.Ferrai, C., D. Munari, P. Luraghi, L. Pecciarini, M. G. Cangi, C. Doglioni, F. Blasi, and M. P. Crippa. 2007. A transcription-dependent micrococcal nuclease-resistant fragment of the urokinase-type plasminogen activator promoter interacts with the enhancer. J. Biol. Chem. 28212537-12546. [DOI] [PubMed] [Google Scholar]
- 15.Ferretti, E., J. C. Villaescusa, P. Di Rosa, L. C. Fernandez-Diaz, E. Longobardi, R. Mazzieri, A. Miccio, N. Micali, L. Selleri, G. Ferrari, and F. Blasi. 2006. Hypomorphic mutation of the TALE gene Prep1 (pKnox1) causes a major reduction of Pbx and Meis proteins and a pleiotropic embryonic phenotype. Mol. Cell. Biol. 265650-5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridman, J. S., and S. W. Lowe. 2003. Control of apoptosis by p53. Oncogene 229030-9040. [DOI] [PubMed] [Google Scholar]
- 17.Fulda, S., and K. M. Debatin. 2006. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 254798-4811. [DOI] [PubMed] [Google Scholar]
- 18.Gross, A., J. M. McDonnell, and S. J. Korsmeyer. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 131899-1911. [DOI] [PubMed] [Google Scholar]
- 19.Hemann, M. T., and S. W. Lowe. 2006. The p53-Bcl-2 connection. Cell Death Differ. 131256-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Igney, F. H., and P. H. Krammer. 2002. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer. 2277-288. [DOI] [PubMed] [Google Scholar]
- 21.Kasai, S., S. Chuma, N. Motoyama, and N. Nakatsuji. 2003. Haploinsufficiency of Bcl-x leads to male-specific defects in fetal germ cells: differential regulation of germ cell apoptosis between the sexes. Dev. Biol. 264202-216. [DOI] [PubMed] [Google Scholar]
- 22.Knoepfler, P. S., and M. P. Kamps. 1997. The highest affinity DNA element bound by Pbx complexes in t(1;19) leukemic cells fails to mediate cooperative DNA-binding or cooperative transactivation by E2a-Pbx1 and class I Hox proteins—evidence for selective targeting of E2a-Pbx1 to a subset of Pbx-recognition elements. Oncogene 142521-2531. [DOI] [PubMed] [Google Scholar]
- 23.Kroemer, G., N. Zamzami, and S. A. Susin. 1997. Mitochondrial control of apoptosis. Immunol. Today 1844-51. [DOI] [PubMed] [Google Scholar]
- 24.Lakin, N. D., and S. P. Jackson. 1999. Regulation of p53 in response to DNA damage. Oncogene 187644-7655. [DOI] [PubMed] [Google Scholar]
- 25.Longobardi, E., and F. Blasi. 2003. Overexpression of PREP-1 in F9 teratocarcinoma cells leads to a functionally relevant increase of PBX-2 by preventing its degradation. J. Biol. Chem. 27839235-39241. [DOI] [PubMed] [Google Scholar]
- 26.Meek, D. W. 1998. Multisite phosphorylation and the integration of stress signals at p53. Cell Signal. 10159-166. [DOI] [PubMed] [Google Scholar]
- 27.Nicholson, D. W. 1999. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 61028-1042. [DOI] [PubMed] [Google Scholar]
- 28.Pecci, A., L. R. Viegas, J. L. Baranao, and M. Beato. 2001. Promoter choice influences alternative splicing and determines the balance of isoforms expressed from the mouse bcl-X gene. J. Biol. Chem. 27621062-21069. [DOI] [PubMed] [Google Scholar]
- 29.Penkov, D., P. Di Rosa, L. Fernandez Diaz, V. Basso, E. Ferretti, F. Grassi, A. Mondino, and F. Blasi. 2005. Involvement of Prep1 in the alphabeta T-cell receptor T-lymphocytic potential of hematopoietic precursors. Mol. Cell. Biol. 2510768-10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reers, M., S. T. Smiley, C. Mottola-Hartshorn, A. Chen, M. Lin, and L. B. Chen. 1995. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 260406-417. [DOI] [PubMed] [Google Scholar]
- 31.Resnati, M., I. Pallavicini, R. Daverio, N. Sidenius, P. Bonini, and F. Blasi. 2006. Specific immunofluorimetric assay detecting the chemotactic epitope of the urokinase receptor (uPAR). J. Immunol. Methods 308192-202. [DOI] [PubMed] [Google Scholar]
- 32.Sakaguchi, K., J. E. Herrera, S. Saito, T. Miki, M. Bustin, A. Vassilev, C. W. Anderson, and E. Appella. 1998. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 122831-2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Selleri, L., J. DiMartino, J. van Deursen, A. Brendolan, M. Sanyal, E. Boon, T. Capellini, K. S. Smith, J. Rhee, H. Popperl, G. Grosveld, and M. L. Cleary. 2004. The TALE homeodomain protein Pbx2 is not essential for development and long-term survival. Mol. Cell. Biol. 245324-5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian, C., P. Gregoli, and M. Bondurant. 2003. The function of the bcl-x promoter in erythroid progenitor cells. Blood 1012235-2242. [DOI] [PubMed] [Google Scholar]
- 35.Vander Heiden, M. G., N. S. Chandel, E. K. Williamson, P. T. Schumacker, and C. B. Thompson. 1997. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 91627-637. [DOI] [PubMed] [Google Scholar]
- 36.Wang, D., H. Masutani, S. Oka, T. Tanaka, Y. Yamaguchi-Iwai, H. Nakamura, and J. Yodoi. 2006. Control of mitochondrial outer membrane permeabilization and Bcl-xL levels by thioredoxin 2 in DT40 cells. J. Biol. Chem. 2817384-7391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.