

Abstract
Subcellular localization influences the nature of Ras/extracellular signal-regulated kinase (ERK) signals by unknown mechanisms. Herein, we demonstrate that the microenvironment from which Ras signals emanate determines which substrates will be preferentially phosphorylated by the activated ERK1/2. We show that the phosphorylation of epidermal growth factor receptor (EGFr) and cytosolic phospholipase A2 (cPLA2) is most prominent when ERK1/2 are activated from lipid rafts, whereas RSK1 is mainly activated by Ras signals from the disordered membrane. We present evidence indicating that the underlying mechanism of this substrate selectivity is governed by the participation of different scaffold proteins that distinctively couple ERK1/2, activated at defined microlocalizations, to specific substrates. As such, we show that for cPLA2 activation, ERK1/2 activated at lipid rafts interact with KSR1, whereas ERK1/2 activated at the endoplasmic reticulum utilize Sef-1. To phosphorylate the EGFr, ERK1/2 activated at lipid rafts require the participation of IQGAP1. Furthermore, we demonstrate that scaffold usage markedly influences the biological outcome of Ras site-specific signals. These results disclose an unprecedented spatial regulation of ERK1/2 substrate specificity, dictated by the microlocalization from which Ras signals originate and by the selection of specific scaffold proteins.
Ras GTPases operate as key molecular switches that convey extracellular signals from surface receptors to the interior of the cell, thereby regulating essential processes, including proliferation, differentiation, and survival (10). It has been long known that Ras proteins must be attached to the inner leaflet of the plasma membrane (PM) to be functional, but only recently has it been established that Ras isoforms are distinctively segregated in different PM microdomains: H-Ras can be found in the bulk membrane and in lipid rafts, both caveolar and noncaveolar. K-Ras is exclusively located in the bulk membrane (27, 28), while N-Ras is mainly detected in noncaveolar lipid rafts (20). Moreover, Ras proteins are present and functional in endomembranes, such as endosomes, the endoplasmic reticulum (ER), and the Golgi complex (9, 26, 32). However, how these distinct microenvironments affect the signals generated by Ras is just beginning to be unfolded. In this respect, recent findings support the notion that the Ras signals emanating from these different sublocalizations are quantitatively and qualitatively different, in the effector routes that they engage in and in the resulting transcriptomal and biological outcomes (1, 9, 15, 21).
The pathway leading to the activation of extracellular signal-regulated kinase 1 and 2 (ERK1/2) mitogen-activated protein kinases is a key component of Ras signals (29). To date, evidence is mounting indicating that the cellular microenvironment can play an essential role in the regulation of ERK1/2 functions. ERK1/2 can be regulated by processes strictly dependent on subcellular localization (33), and ERK1/2 themselves regulate some biochemical processes in a compartment-dependent fashion (14). Furthermore, the balance among different compartment-specific components of ERK1/2 signals seems determinant for the biological outcomes resulting from ERK1/2 activation (3, 30). ERK1/2 phosphorylate a great variety of substrates distributed in different subcellular localizations and microenvironments (40). However, whether, depending on the activating stimuli, ERK1/2 display variable selectivity toward these different pools of substrates and the extent to which the subcellular atmosphere can regulate ERK1/2 interactions with their cognate targets are largely unanswered issues.
ERK1/2 signals are modulated by different types of regulatory proteins. Scaffold proteins serve such a purpose by connecting the different components of the cascade into an enzymatic macrostructure by which the amplitude and duration of ERK1/2 signals are fine-tuned, while providing signal fidelity by isolating the signaling complex from undesired, molecular interferences (12, 17, 23). Furthermore, scaffolds can also serve a role in the spatial control of ERK1/2 signals by regulating their activity in a sublocalization-specific fashion. In this respect, KSR1 acts preferentially on ERK1/2 signals emanating from PM cholesterol-rich domains (22). MP-1 regulates ERK1/2 at endosomes (35), Sef regulates ERK1/2 at the Golgi complex (37), and paxillin regulates ERK1/2 at focal adhesions (16). But the precise mechanisms whereby scaffolds can achieve spatial specificity for ERK1/2 signals are largely unknown.
Herein, we have taken a look at these questions. We demonstrate that the signaling platform from which the activating Ras signals spring dictates ERK1/2 selectivity toward different substrates. In this process, we unveil the participation of scaffold proteins that distinctively couple ERK1/2 activated at defined microlocalizations to specific substrates. Our results provide the first evidence pointing both to the activating signal microenvironment and to a differential scaffold usage, as regulators of ERK1/2 substrate specificity.
MATERIALS AND METHODS
Plasmids.
The expression vectors encoding the localization-targeted H-RasV12 and the wild type (5, 21) have been previously described. Myc-Sef was a gift from Z. Chang (38). UO126, SB203580, and epidermal growth factor (EGF) were from Calbiochem. Lysophosphatidic acid (LPA) and tetradecanoyl phorbol acetate (TPA) were from Sigma.
Cell culture and transfection.
HEK293T cells and wild-type, H-Ras−/− N-Ras−/− (H/N-Ras−/−) and Ksr−/− mouse embryonic fibroblasts (MEFs) were grown in Dulbecco's modified Eagle's medium-10% fetal calf serum, and NIH 3T3 cells were grown in 10% calf serum. Subconfluent cells were transfected by Lipofectamine. Small interfering RNAs (siRNAs) were from Dharmacon; a pool of four different siRNAs was used in each particular case. Sequences are available upon request. siRNAs were transfected by following the manufacturer's guidelines.
Immunoblotting and immunoprecipitations.
Immunoblotting and immunoprecipitations were performed exactly as described previously. Immunoprecipitations were performed for lysates normalized by total protein contents (33). Mouse monoclonal antihemagglutinin (anti-HA), -Myc, and -AU5 were from Covance. The mouse monoclonal antibodies anti-ERK1/2 and -phospho-ERK; the rabbit polyclonals anti-KSR1, -RSK1, -caveolin, and -phospho-RSK1; and the goat polyclonal anti-cytosolic phospholipase A2 (anti-cPLA2) were from Santa Cruz. The mouse monoclonal antibodies anti-Elk1 and -phospho-Elk1 were from Cell Signaling. The mouse monoclonal antibodies anti-IQGAP1 and -transferrin receptor were from BD Transduction. The rabbit polyclonal anti-EGF receptor (anti-EGFr) was a gift from Atanasio Pandiella (Salamanca).
Nucleocytoplasmic fractionations.
Nucleocytoplasmic fractionations were performed as described previously (3).
S100/P100 separations.
S100/P100 separations were performed as described previously (4).
Sucrose gradients.
Cells were resuspended in 25 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, and 0.25% Triton X-100 with rocking at 4°C for 1 h. The lysates were set to a sucrose concentration of 41%. Layers of 8.5 ml of 35% sucrose (in 10 mM Tris [pH 7.4]) and of 2.5 ml of 16% sucrose were sequentially overlaid and centrifuged in a Beckman SW41 rotor for 18 h at 35,000 rpm. Ten to 12 1-ml fractions were collected, precipitated in 6.5% trichloroacetic acid, resuspended in loading buffer, and fractionated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels.
cPLA2 activation assays.
The cPLA2 activation assays were performed as described previously (33). Briefly, cells were labeled with [3H]arachidonic acid (1 μCi per well in a 24-well plate) for 18 h. The cells were then washed twice with phosphate-buffered saline (PBS), and new medium (Dulbecco's modified Eagle's medium-1% fatty acid-free bovine serum albumin) was added. Where indicated, the cells were stimulated with EGF (100 ng/ml) for 30 min. The medium was removed and centrifuged at 2,000 × g, and free, labeled arachidonic acid released into the medium was measured by liquid scintillation counting.
EGFr phosphorylation assays.
32P metabolic labeling was performed as described previously (33). After 18 h of starvation and upon treatment with mitogens or inhibitors where appropriate, the cells were lysed and the lysates were immunoprecipitated with anti-EGFr antibody. The immunoprecipitates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and EGFr phosphorylation was made evident by prolonged exposition of the dried gel.
Focus formation and senescence assays in MEFs.
The focus formation and senescence assays were performed exactly as described previously (18).
Luciferase assays.
Luciferase assays were performed as described previously (33), using the reporter pGE51-luc and a vector encoding the transactivation domain of Elk-1 (amino acids 307 to 428) fused to the GAL4 DNA-binding domain. The luciferase activities were determined using a commercial kit (Promega, Madison, WI) and normalized by the β-galactosidase activity.
Immunofluorescence.
Cells were grown on glass slides, washed twice in PBS, fixed with 3.7% formaldehyde in PBS for 10 min at room temperature, and permeabilized, when necessary, with 0.5% Triton X-100-PBS for 20 min. The preparations were sequentially incubated with 0.5% Triton X-100-PBS for 15 min and 0.1 M glycine-PBS for 30 min and blocked with 1% bovine serum albumin, 0.01% Tween 20 in PBS for 5 min. The preparations were then rinsed in PBS-0.05% Tween 20, incubated for 1 h with the primary antibody, washed, and incubated for 45 min with the appropriate secondary antibodies. Coverslips were mounted in VectaShield and sealed with nail polish. Confocal microscopy was performed with a Bio-Rad MRC-1024 microscope, using excitation wavelengths of 488 nm.
RESULTS
ERK1/2 cytoplasmic substrates are differentially activated depending on Ras localization.
To take an initial glance into how Ras sublocalization affected ERK1/2 functions, we looked at the activation of several bona fide ERK1/2 substrates, which are known to be present in different subcellular compartments. To this end, we utilized the previously described H-RasV12 constructs selectively tethered to defined microlocalizations by specific localization signals: the ER (avian infectious bronchitis virus M1 protein), lipid rafts (LCK myristoylation signal), the disordered membrane (CD8α transmembrane domain), and the Golgi complex (KDEL receptor N193D) (21), transiently expressed in 293T cells. With the exception of the Golgi signal, which evoked little ERK activation, endogenous ERK1/2 were activated to similar levels independently of Ras localization and of Ras expression levels (Fig. 1A). The expression levels of the different site-specific Ras constructs were determined by the amount of Ras that can fit into each of the sublocalizations, rather than on the intrinsic expression capacity of the constructs themselves, as extensively discussed before (21). This pattern of activation was almost identical to that previously described for NIH 3T3 cells stably expressing these targeted RasV12 constructs (21).
FIG. 1.

Ras sublocalization defines ERK1/2 substrate specificity. (A) Phosphorylation of ERK1/2 resulting from compartmentalized Ras activation. 293T cells were transfected with H-RasV12 (0.5 μg), untargeted (V12) or specifically tethered to the ER (M1), lipid rafts (LCK), the disordered membrane (CD8), or the Golgi complex (KDELr). Phosphorylated and total ERK1/2 levels were determined in total lysates by immunoblotting. Anti-HA Western blotting (WB) reveals the expression levels of the Ras constructs. C, control. (B) Phosphorylation of EGFr resulting from compartmentalized Ras activity. Top panel, EGFr phosphorylation was determined by immunoprecipitation (IP) in 32P-labeled 293T cells transfected with the Ras constructs. Bottom panel, EGFr phosphorylation is ERK dependent. 293T cells transfected with the targeted Ras constructs and incubated with 2 μM U0126 for 2 h (+) were analyzed for EGFr phosphorylation. (C) Phosphorylation of RSK1 induced by compartmentalized Ras activation. Total and phosphorylated RSK1 levels were determined in starved NIH 3T3 and 293T cells expressing the Ras constructs. Right panel, RSK1 phosphorylation is ERK dependent. 293T cells transfected with H-RasV12 and incubated with U0126 for 2 h (+) were analyzed for RSK1 phosphorylation. (D) Regulation of cPLA2 activation by Ras sublocalization. 3H-labeled arachidonic acid release was measured in NIH 3T3 cells. (E) ERK1/2 substrate specificity is unaffected by targeted Ras expression levels. Cells were transfected with increasing concentrations (0.5 to 5 μg) of LCK- or CD8-RasV12 and RSK1 phosphorylation and cPLA2 activation were examined. (D and E) Shown are the averages ± standard errors of the means of the results from three independent experiments relative to the levels in the control cells. α, anti.
First, we explored whether the platform in which Ras-activating signals were generated could determine if the activated ERK1/2 preferentially selected substrates localized at distinct sites. We assayed the phosphorylation of the EGFr, an ERK substrate localized at the PM (24). The 293T cells were transfected with the targeted RasV12 constructs and metabolically labeled with orthophosphate in order to disclose the phosphorylation of EGFr. It was found that EGFr was intensively phosphorylated in cells expressing RasV12 at lipid rafts and, to a lesser extent, at the disordered membrane. On the other hand, RasV12 at endomembranes, whether ER or Golgi, was not capable of inducing a substantial phosphorylation of the EGFr (Fig. 1B, top panels). To validate that the RasV12 signals inducing the phosphorylation of EGFr were mediated by ERK1/2, we used the MEK inhibitor UO126, which was shown to completely inhibit the phosphorylation of EGFr induced by Ras, irrespective of its localization (Fig. 1B, bottom panels). In NIH 3T3 cells expressing ectopic EGFr, LCK-RasV12 could evoke the phosphorylation of a protein of the same size as the one detected in 293T cells, whereas in parental NIH 3T3 cells that do not express EGFr, this band was not apparent (data not shown), thereby validating EGFr identity. Since protein kinase D can phosphorylate EGFr (6), we investigated the possible participation of this kinase by suppressing its expression using siRNA interference. It was found that the downregulation of protein kinase D had no effects on EGFr phosphorylation induced by RasV12 (data not shown).
We then determined if ERK1/2 cytoplasmic substrates were also subject to differential regulation depending on the Ras activation site. To do so, we monitored RSK1, a substrate phosphorylated by ERK1/2 primarily at the cytoplasm (13). Both in NIH 3T3 and in 293T cells, RSK1 phosphorylation was most prominent when induced by Ras signals originated at the disordered membrane. UO126 completely abrogated RSK1 phosphorylation induced by RasV12 (Fig. 1C), demonstrating that ERK1/2 were the sole mediators in this event. It was important to know whether other ERK cytoplasmic substrates responded similarly or if this behavior was unique for RSK1. As such, we analyzed a second cytoplasmic substrate, cPLA2 (19). Upon examination of radiolabeled arachidonic acid release in cells expressing the tethered Ras constructs, it was found that cPLA2 was activated mostly by Ras at lipid rafts and, to a lesser extent, by signals elicited from the ER (Fig. 1D). These results demonstrated that ERK1/2 activated from different Ras signaling platforms selectively targeted distinct cytoplasmic substrates. To ascertain that ERK1/2 substrate selectivity was not altered by the levels of the targeted Ras proteins, we analyzed cPLA2 and RSK1 activation in response to increasing concentrations of LCK- or CD8-RasV12. As shown in Fig. 1E, even the highest levels of active Ras at the disordered membrane failed to affect cPLA2 activation. Likewise, Ras targeted to lipid rafts could not elicit any significant RSK1 phosphorylation, irrespective of its expression levels.
Ras localization does not affect ERK1/2 nuclear substrates.
Next, we focused on ERK1/2 nuclear substrates. Noticeably, in NIH 3T3 cells expressing the Ras constructs, the phosphorylation of Elk1 transcription factor, induced by untargeted RasV12, was remarkably higher than that generated by the site-specific RasV12 constructs, among which there were no major differences. Only Ras signals emanating from the Golgi complex were slightly less effective. Similar results were observed in 293T cells (Fig. 2A). To further substantiate this observation, we looked at how Ras sublocalization affected phosphorylated ERK1/2 nuclear influx. To this end, nuclear and cytoplasmic fractions were purified from 293T cells expressing the targeted Ras constructs. It was found that Ras sublocalization was not a factor for determining ERK1/2 nucleocytoplasmic distribution, again except with Golgi Ras signals, which were less effective for eliciting ERK1/2 nuclear entry (Fig. 2B).
FIG. 2.

Effects of Ras sublocalization on the activation of ERK1/2 nuclear substrates. (A) Phosphorylation of Elk1 by compartmentalized Ras signals. Total and phosphorylated Elk1 levels were determined by immunoblotting in NIH 3T3 and 293T cells expressing the targeted Ras constructs. C, control. (B) Effects of compartmentalized Ras signals on ERK1/2 nucleocytoplasmic distribution. Total and phosphorylated ERK1/2 levels were examined in nuclear (N) and cytoplasmic (C) fractions from 293T cells expressing the targeted Ras constructs. Bottom panel, the purity of the fractions was ascertained by blotting against Rho-GDP dissociation inhibitor (αGDI) and Elk1. (C) Elk1 activation dependence on Ras compartmentalized signals. Left panel, Elk1 transactivation was assayed in NIH 3T3 cells transfected with GAL4-Elk1 and the targeted Ras constructs plus siRNAs against ERK1 and -2 or JNK1 and -2 (10 ng), as shown in the key. C, control. Shown are the averages ± standard errors of the means of the results from three independent experiments relative to the levels in the control cells. Right panel, diminished expression of the endogenous mitogen-activated protein kinases resulting from siRNA treatments. V12, untargeted H-RasV12; M1, H-RasV12 specifically tethered to the ER; LCK, H-RasV12 specifically tethered to lipid rafts; CD8, H-RasV12 specifically tethered to the disordered membrane; KDELr, H-RasV12 specifically tethered to the Golgi complex.
Since Elk1 is also a substrate for other MAPKs, such as p38 and JNK, we investigated the contribution of each of these MAPKs to its phosphorylation, in response to compartmentalized Ras signals. For this, we utilized a GAL4-Elk1-dependent luciferase reporter. Emulating the results obtained by assaying Elk1 phosphorylation, untargeted RasV12 induced the greatest transactivation of the Elk1 reporter, whereas few differences were found among the site-specific Ras constructs (Fig. 2C, left panel). The depletion of ERK1/2 levels by siRNA interference (Fig. 2C, right panel) resulted in pronounced drops in Elk1 transactivation in all cases. Noticeably, Elk1 activation elicited by Golgi Ras signals was the least responsive to ERK1/2 downregulation but was the most affected by JNK1/2 decline. On the other hand, p38 inhibition by treatment with SB203580 was ineffective in all cases (data not shown).
It was essential to validate the above findings in a physiologically relevant context. For this purpose, we resorted to fibroblasts obtained from H-Ras/N-Ras knockout mice, in which lipid rafts are completely devoid of Ras (21), since K-Ras is exclusively located in disordered membrane (27). We reasoned that in these cells, the phosphorylation of ERK substrates regulated by Ras signals from lipid rafts, but not of those regulated from disordered membrane, should be compromised. In these MEFs, ERK1/2 activation resulting from EGF stimulation was significantly reduced compared to that in wild-type MEFs (Fig. 3A). When we evaluated the activation of cPLA2 in H/N-Ras−/− MEFs, the response to EGF was reduced by nearly 70% in comparison to that of the wild-type MEFs (Fig. 3B). The same situation occurred when we analyzed EGFr phosphorylation induced by LPA stimulation (Fig. 3C). Conversely, EGF-induced RSK1 and Elk1 phosphorylations were similar to those encountered in wild-type MEFs (Fig. 3D), thereby corroborating our hypothesis. To further ascertain our case, we restored Ras at the lipid rafts of H/N-Ras−/− MEFs by transfecting them with the LCK-H-Ras wild type. The renewed presence of Ras in lipid rafts markedly rescued cPLA2 activation upon EGF stimulation (Fig. 3E), whereas in cells transfected with H-Ras tethered to the ER, the rescue of cPLA2 activation was only modest and insignificant in those cells harboring disordered membrane- or Golgi-tethered Ras. Conversely, the restoration of H-Ras at lipid rafts had no effects in the phosphorylation of RSK1 induced by EGF, which was slightly augmented in those cells transfected with disordered membrane-tethered Ras (Fig. 3F). These results demonstrated that, both in physiological contexts and in ectopic expression systems, the Ras signaling platform regulated ERK1/2 selectivity toward different substrates.
FIG. 3.

cPLA2 but not RSK1 activation is impaired in MEFs devoid of Ras at lipid rafts. (A) ERK1/2 activation in H/N-Ras−/− MEFs. Total and phosphorylated ERK1/2 levels were analyzed in wild type (wt) and H/N-Ras−/− MEFs after starvation (−) and stimulation with 10 ng/ml EGF for 5 min (+). Figures show ERK1/2 phosphorylation levels relative to those found in unstimulated, wild-type MEFs. (B) cPLA2 activation is impaired in H/N-Ras−/− MEFs. 3H-labeled arachidonic acid release was measured in wild-type and H/N-Ras−/− MEFs after stimulation (+) with EGF. Shown are the averages ± standard errors of the means of the results from three experiments relative to the levels in unstimulated cells. ***, P < 0.001, with a 95% confidence interval. (C) EGFr phosphorylation is impaired in H/N-Ras−/− MEFs. EGFr phosphorylation was assayed in wild-type and H/N-Ras−/− MEFs after stimulation with LPA (5 μM) for 5 min (+). (D) Phosphorylation of RSK1 and Elk1 is unaffected in H/N-Ras−/− MEFs. Total and phosphorylated RSK1/Elk1 levels were determined by immunoblotting in wild-type and H/N-Ras−/− MEFs after stimulation with EGF. (E) The presence of Ras at lipid rafts in H/N-Ras−/− MEFs restores PLA2 activation. 3H-labeled arachidonic acid release was measured in H/N-Ras−/− MEFs transfected with wild-type H-Ras targeted to its different microlocalizations (1 μg) after stimulation with EGF. Shown are the averages ± standard errors of the means of the results from two experiments relative to the levels detected in unstimulated cells transfected with the respective site-specific, wild-type H-Ras constructs. *, P < 0.05, with a 95% confidence interval. (F) The restoration of Ras at lipid rafts in H/N-Ras−/− MEFs does not affect RSK1 activation. Total and phosphorylated RSK1 levels were determined for H/N-Ras−/− MEFs transfected with LCK- or CD8-tethered wild-type H-Ras (1 μg) after stimulation with EGF (+). Targeted H-Ras protein expression levels were determined by anti-HA immunoblotting. α, anti; c, control.
Sublocalization dictates ERK1/2 substrate selectivity under physiological stimulation.
Subsequently, we asked whether for external stimuli acting upstream of Ras, ERK1/2 also exhibited a localization-dependent substrate selectivity. To this end, we engineered AU5-tagged ERK2 constructs specifically tethered to disordered membrane and to lipid rafts (Fig. 4A), from which the activations of RSK1 and EGFr were, respectively, regulated. Such constructs would enable us to monitor ERK2 activation in the immediate proximity of these microenvironments, upon the receipt of external stimuli. To verify the response specificity of these constructs to signals emanating from lipid rafts or disordered membrane, they were cotransfected with CD8- and LCK-RasV12. As shown in Fig. 4B, CD8-RasV12 only induced the phosphorylation of the disordered membrane-tethered ERK2, whereas LCK-RasV12 just affected lipid raft-bound ERK2, thereby corroborating their spatial specificity.
FIG. 4.

Sublocalization dictates ERK1/2 substrate selectivity under physiological stimulation. (A) Subcellular fractionation of targeted ERK2 proteins. 293T cells expressing AU5-tagged ERK2 specifically tethered to lipid rafts (LCK) or to the disordered membrane (CD8) (1 μg) were solubilized in 0.25% Triton X-100 and partitioned in a sucrose gradient. The presence of the targeted ERK2 proteins in the different fractions was analyzed by anti-AU5 immunoblotting. Anti-caveolin-1 (α Cav) identifies the lipid raft fractions, and anti-transferrin receptor (α Tfr) identifies the disordered membrane fractions. (B) Targeted ERK2 proteins recognize signals in site-specific fashion. Cells were transfected with LCK- or CD8-AU5-tagged ERK2 in addition to LCK-RasV12 or CD8-RasV12 (1 μg each) as shown. Total and phosphorylated ERK levels were examined in anti-AU5 immunoprecipitates. (C) Time-dependent activation profiles of the targeted ERK2 proteins. 293T cells were transfected with the tethered ERK2 constructs and their kinetics of activation were compared when stimulated with EGF (10 ng/ml), LPA (5 μM), or TPA (100 nM) for the indicated times (in minutes) by monitoring total and phosphorylated ERK levels in anti-AU5 immunoprecipitates. For comparison, the phosphorylation kinetics of endogenous ERK1/2 are shown (top two panels). (D) EGFr phosphorylation correlates with the activation of ERK2 at lipid rafts. 32P-labeled 293T cells were stimulated, after starvation, with LPA for the indicated times and phosphorylation of the EGFr was analyzed. Notice that the kinetics of EGFr phosphorylation match those of LCK-ERK2 stimulated with LPA. (E) RSK1 phosphorylation correlates with the activation of ERK2 at the disordered membrane and varies accordingly depending on the stimulus. 293T cells were stimulated, after starvation, with EGF or TPA for the indicated times, and the phosphorylation of RSK1 was analyzed. Notice that the kinetics of RSK1 activation match those of CD8-ERK2 stimulated with EGF or with TPA.
These constructs were transfected in 293T cells that were then treated with different agonists. Interestingly, the activation of ERK2 at these microdomains followed different kinetics depending on the stimulus. At lipid rafts, upon treatment with EGF, the phosphorylation of ERK2 reached a rapid maximum after just 2 min and faded progressively afterwards. Conversely, at the disordered membrane, ERK2 exhibited a slower phosphorylation course, with a late peak 10 min after stimulation that was still apparent after 30 min (Fig. 4C). Stimulation with LPA evoked a similar pattern of activation: the activation of ERK2 at the disordered membrane lagged behind that one taking place at lipid rafts. Interestingly, when cells were treated with the phorbol ester TPA, ERK2 followed similar patterns of activation at both localizations, with robust peaks 5 min after stimulation that were sustained after 30 min.
We then examined if the kinetics of ERK2 activation at lipid rafts and disordered membrane correlated with the activation patterns exhibited by those substrates specifically regulated from each of these localizations, namely, EGFr and RSK1. In a physiological setting, such as parental 293T cells stimulated with LPA, the phosphorylation of EGFr displayed a maximum after 5 min, decreasing thereafter (Fig. 4D). Noticeably, this activation course was similar to the one exhibited by ERK2 at lipid rafts upon LPA stimulation (Fig. 4C). In the same fashion, the phosphorylation of RSK1 induced by EGF displayed a late onset (Fig. 4D). Remarkably, RSK1 activation paralleled EGF-induced ERK2 activation at the disordered membrane. More interestingly, the RSK1 activation pattern completely changed when the activating stimulus was TPA (Fig. 4D). In this case, its kinetics resembled that of TPA-induced ERK2 activation at disordered membrane, with an early peak and sustained activation. These results added further support to the notion that, regardless of the activating stimulus, the microenvironment in which ERK1/2 are activated dictates their substrate selectivity.
cPLA2 and RSK1 occupy distinct subcytoplasmic domains.
We wished to understand how two substrates that, supposedly, shared the same subcellular compartment were distinctively selected by ERK1/2 activated at different localizations. We hypothesized that even though RSK1 and cPLA2 both reside at the cytoplasm, they could be present at different microdomains. To test this possibility, we explored the localizations of RSK1 and cPLA2 by immunofluorescence. Interestingly, even though these two proteins exhibited clear cytoplasmic staining, hardly any colocalization was apparent, suggesting that they could be occupying different sites within the cytoplasm (Fig. 5A, profile A). Next, we examined the colocalization of these two substrates with activated ERK1/2 in cells in which the activating Ras signal originated at different sites. In cells expressing RasV12 at lipid rafts, phosphorylated ERK1/2 markedly colocalized at the cytoplasm with cPLA2, in particular in the perinuclear region and at the cellular periphery (Fig. 5B, profile B), but very little colocalization with RSK1 was evident (Fig. 5C). Conversely, in cells expressing RasV12 at the disordered membrane, a profuse colocalization of phosphorylated ERK1/2 with RSK1 but not with cPLA2 was apparent (Fig. 5D and E). We studied the microlocalization of these substrates in further detail by immunogold electron microscopy, in which cPLA2 and RSK1 were labeled with gold particles of different diameters. Interestingly, instead of appearing intermixed and randomly scattered throughout the cytoplasm, both cPLA2 and RSK1 formed clusters at specific sites, clearly separated from each other (Fig. 6 and Table 1). This was not a general feature for all cytoplasm residents, since both cPLA2 and RSK1 were intermixed with another cytoplasmic protein such as ubiquitin (data not shown). These results suggested that depending on the membrane localization from which the Ras signal emanated, activated ERK1/2 were directed to defined subcytoplasmic compartments where different substrates resided.
FIG. 5.

RSK and cPLA2 are located in distinct subcytoplasmic microlocalizations. (A) RSK and cPLA2 do not colocalize. Growing NIH 3T3 cells were costained with anti-RSK and anti-cPLA2 antibodies and visualized by immunofluorescence and confocal microscopy. (B and C) cPLA2 but not RSK1 colocalizes with phospho-ERK (p-ERK) in NIH 3T3 cells stably expressing LCK-RasV12. Inset, detail of the sublocalization at the cytoplasm (thick arrow). (D and E) RSK1 but not cPLA2 colocalizes with phospho-ERK in NIH 3T3 cells stably expressing CD8-RasV12. To highlight the cytoplasmic signals, the cells were not permeabilized (except in panel B), and the nuclear signals are quenched. Pseudocolors were red and green and as indicated; yellow indicates a merge. Scale bar = 10 μM. Profile A, pseudocolor intensity profile, across the section shown in panel A. Notice that the colocalization of peaks is minimal. Profile B, pseudocolor intensity profile at a PM localization (thin arrow) shown in panel B.
FIG. 6.

RSK1 and cPLA2 occupy distinct cytoplasmic microdomains. Immunogold electron microscopy was performed on NIH 3T3 cells. The secondary antibodies disclosing RSK1 (asterisks) and cPLA2 (arrows) were labeled with 15-nm and 10-nm gold particles, respectively. Scale bar = 100 nm.
TABLE 1.
RSK and cPLA2 are located in distinct subcytoplasmic microlocalizationsa
| Particles | Distance between gold particles (nm) | P value |
|---|---|---|
| cPLA2-cPLA2 | 8.21 ± 1.21 | >0.5 |
| RSK1-RSK1 | 8.02 ± 1.88 | >0.5 |
| cPLA2-RSK1 | 160.82 ± 13.08 | <0.005 |
Distance between RSK1 and cPLA2 particles. Immunogold electron microscopy was performed with NIH 3T3 cells. The secondary antibodies disclosing RSK1 and cPLA2 were labeled with 15-nm and 10-nm gold particles, respectively. Results show the distances between gold particles as the averages ± standard errors of the means of six sections. Student's t test highlights the significance of the distance between RSK1 and cPLA2.
ERK1/2 site-specific substrate selectivity entails distinct usage of scaffold proteins.
It was important to understand the molecular mechanisms underlying ERK1/2 site-dependent substrate specificity. Since some scaffold proteins have been shown to regulate ERK1/2 signals in a localization-regulated fashion (17), we investigated their implication in substrate specificity. To this end, we first analyzed the activation of several ERK substrates when the expression of KSR1, a scaffold involved in ERK1/2 signaling orchestrated from lipid rafts (22), was downregulated by siRNA interference in 293T cells. Whereas the reduction of KSR1 levels forestalled EGF-induced cPLA2 activation by more than 75%, RSK1 phosphorylation was completely unaltered (Fig. 7A). Furthermore, gradually increasing the amount of KSR1 that was transfected resulted in a biphasic cPLA2 activation pattern in response to EGF stimulation, typical of scaffold proteins, whereas RSK1 activation was, again, unaffected (Fig. 7B). These results suggested that KSR1 was the scaffold utilized by ERK1/2 for activating cPLA2 but not RSK1.
FIG. 7.

KSR1 couples Ras signals from lipid rafts to cPLA2 activation. (A) Depletion of KSR1 prevents cPLA2 but not RSK1 activation. 293T cells were transfected with (+) or without (-) a siRNA (10 nM) for KSR1 and stimulated with EGF (10 ng/ml, 5 min) after starvation. Endogenous cPLA2 activation was determined by [3H]arachidonic acid release. Bottom panels show total and phosphorylated RSK1 (p-RSK1), and KSR1 levels were determined by immunoblotting. (B) Biphasic activation of cPLA2 resulting from increasing KSR1 levels. Cells were transfected with increasing concentrations of FLAG-KSR1 and stimulated with EGF after starvation. Bottom panels show total and phosphorylated RSK1, and KSR1 (FLAG) levels were determined by immunoblotting. (A and B) Shown are the averages ± standard errors of the means of the results from three independent experiments relative to the levels in starved (C) cells. (C) ERK1/2-KSR1 association is enhanced by Ras signals from lipid rafts but not the disordered membrane. Endogenous ERK1/2, total and phosphorylated, present in native KSR1 immunoprecipitates was determined in cells transfected with RasV12 (0.5 μg) tethered to lipid rafts (LCK) or to the disordered membrane (CD8) or stimulated with EGF. (D) ERK1/2-KSR1 association is enhanced in wild-type but not in H/N-Ras−/− MEFs. ERK1/2, total and phosphorylated, present in KSR1 immunoprecipitates were analyzed in MEFs, starved (C) or EGF stimulated. (E) The association of KSR1 and endogenous cPLA2 is enhanced by EGF stimulation and by Ras signals from lipid rafts and is inhibited by UO126 (2 μM, 25 min prior to stimulation). Bottom panels, RSK1 does not associate with KSR1 in response to Ras signals coming from lipid rafts or to EGF stimulation. (F) The association of KSR1 and endogenous cPLA2 in response to EGF is diminished in H/N-Ras−/− MEFs compared to wild-type fibroblasts. TL, total lysate; IP, immunoprecipitations performed with a specific antibody; PI, immunoprecipitations performed with preimmune serum; α, anti.
Following this argument, we examined whether KSR1 coupled ERK and cPLA2 activations in a site-dependent fashion. To do so, we analyzed the composition of the KSR1 complex upon the receipt of Ras signals originated at different localizations. It was found that the levels of total and phosphorylated ERK1/2 that assembled to endogenous KSR1 markedly increased in cells stimulated with EGF and in those transfected with lipid raft-associated RasV12 but not in those expressing RasV12 tethered to the disordered membrane (Fig. 7C). These results were ascertained in the entirely physiological background offered by the H/N-Ras null MEFs and under ectopic stimulation. Whereas in wild-type MEFs EGF induced an increase in the total and phosphorylated ERK1/2 bound to endogenous KSR1, in H/N-Ras−/− MEFs, devoid of Ras at lipid rafts, the levels of total and activated ERK1/2 which associated with KSR1 upon EGF stimulation where unchanged (Fig. 7D). Most interestingly, endogenous cPLA2 was completely absent from KSR1 immunoprecipitates under resting conditions, but significant proportions of cPLA2 were found to associate with KSR1 in cells stimulated with EGF or transfected with lipid raft-associated RasV12. As we had previously demonstrated (8), the interaction between cPLA2 and KSR1 was completely dependent on ERK1/2 activation, since preincubation with UO126 completely inhibited cPLA2 binding to KSR1 (Fig. 7E). On the other hand, RSK1 was found not to associate with KSR1, either in cells transfected with H-RasV12 or in response to EGF stimulation (Fig. 7E). In a similar fashion, in wild-type MEFs, EGF induced a marked incorporation of cPLA2 to the KSR1 complex, whereas in H/N-Ras−/− MEFs, the levels of cPLA2 which associated with KSR1 upon EGF stimulation where minimal (Fig. 7F).
These results were further substantiated by immunofluorescence studies, in which, in wild-type MEFs, cPLA2 was shown to profusely colocalize with KSR1 at PM localizations in response to EGF stimulation (Fig. 8A and B, arrows), while such interaction was lost in H/N-Ras−/− MEFs (Fig. 8C and D). An identical situation was observed in biochemical studies evaluating EGF-induced KSR1 and cPLA2 association with the PM, extracted from these two types of MEFs (Fig. 9). Overall, these results supported the notion that KSR1 coupled activated ERK1/2 selectively to its substrate, cPLA2, in response to signals generated at lipid rafts.
FIG. 8.

KSR1 and cPLA2 colocalize at the PM of wild-type (wt) but not H/N-Ras−/− (−/−) MEFs. Endogenous RSK and cPLA2 were costained with anti-RSK and anti-cPLA2 antibodies and visualized by immunofluorescence and confocal microscopy. The wild-type and H/N-Ras−/− MEFs were starved (A and C) or stimulated with EGF (10 ng/ml, 5 min) (B and D). Pseudocolors were red and green as indicated; yellow indicates a merge. Scale bar = 10 μM.
FIG. 9.
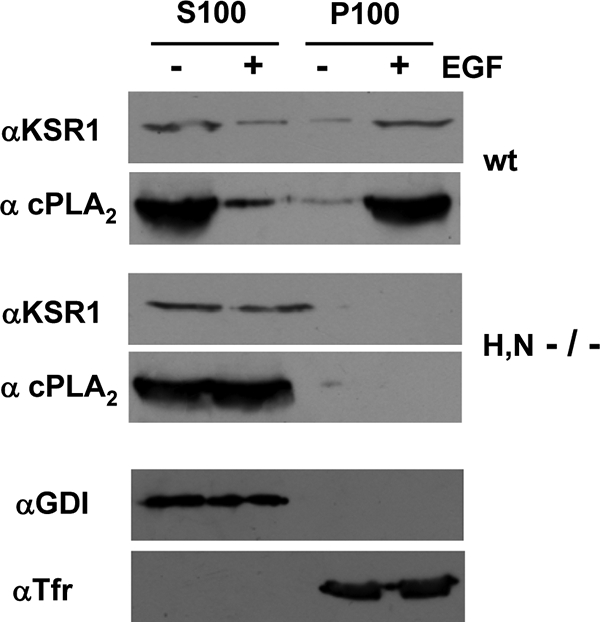
KSR1 and cPLA2 localize at the PM of wild-type (wt) but not H/N-Ras−/− MEFs upon stimulation. Soluble (S100) and particulate (P100) fractions were isolated from wild-type and H/N-Ras−/− MEFs, starved (−) or stimulated (+) with EGF (10 ng/ml, 5 min). The presence of KSR1 and cPLA2 was examined by immunoblotting. The purity of the fractions was ascertained by blotting against Rho-GDP dissociation inhibitor (αGDI) and transferrin (Tfr) receptor in lysates from wild-type MEFs.
In light of our data demonstrating that cPLA2 could be activated from distinct Ras platforms, though to different extents, we then investigated whether KSR1 would couple ERK1/2 to the activation of cPLA2, irrespective of the site where the activating Ras signal was generated. For this, we evaluated cPLA2 activation in 293T cells transfected with RasV12, tethered to lipid rafts, the ER, and the disordered membrane, plus a siRNA against KSR1. It was found that the downregulation of KSR1 expression affected only cPLA2 activation elicited from lipid rafts and not that resulting from Ras activation at the ER or at the disordered membrane (Fig. 10A), thereby demonstrating that KSR1 exhibited both substrate and localization specificity.
FIG. 10.

Sef couples Ras signals from the ER to cPLA2 activation. (A) Depletion of KSR1 inhibits cPLA2 activation by Ras signals from lipid rafts but not from the ER or disordered membrane. 293T cells were transfected with RasV12 (0.5 μg) tethered to lipid rafts, the ER, and the disordered membrane with (+) or without (−) a siRNA (10 nM) for KSR1. (B) Effects of depleting the levels of different scaffold proteins on cPLA2 activation by Ras signals from the ER. Cells were transfected with ER-targeted RasV12 (M1) (+) plus siRNAs (10 nM) for the shown scaffold proteins. DGLY., dystroglycan; β ARR. 2, β-arrestin 2; PAXILL., paxillin. (C) cPLA2 activation by Ras signals from the ER exhibits a biphasic response to increasing Sef concentrations. Cells were transfected with ER-targeted RasV12 (M1) (+), in addition to increasing concentrations of Myc-Sef as shown. (D) Sef does not intervene in cPLA2 activation elicited from lipid rafts. Cells were transfected with RasV12 tethered to lipid rafts with (+) or without (-) a siRNA against Sef. (A to D) cPLA2 activation was determined by [3H]arachidonic acid release after 18 h of starvation. Shown are the averages ± standard errors of the means of the results from three independent experiments relative to the levels in starved (C) cells. (E) Sef/cPLA2 association is enhanced by Ras signals from the ER but not from lipid rafts. Endogenous cPLA2, present in anti-Myc (α Myc) immunoprecipitates, was determined in cells transfected with Myc-Sef (1 μg) plus RasV12 tethered to lipid rafts (LCK) or to the ER (M1). The dependence on ERK was determined by incubation with UO126 (2 μM, 25 min prior to stimulation). TL, total lysate; IP, immunoprecipitations performed with a specific antibody; PI, immunoprecipitations performed with preimmune serum.
It was of interest to identify the scaffold protein that coupled ERK to cPLA2 activation when the Ras signal was coming from the ER. To do so, we analyzed the activation of cPLA2 under conditions where the availability of different scaffold proteins was reduced. The 293T cells were transfected with ER-targeted RasV12 and siRNAs against a set of well-known ERK1/2 scaffolds, namely, dystroglycan, IQGAP1, KSR1, β-arrestin, MORG, MP-1, paxillin, and Sef. These siRNAs efficiently downregulated the expression of all the scaffold proteins tested (8), but only the drop in Sef expression levels had a negative impact on cPLA2 activation induced by ER Ras (Fig. 10B). Moreover, increasing concentrations of ectopically expressed Sef brought about a biphasic response for cPLA2 activation (Fig. 10C), pointing to Sef having a scaffold function in this process. We next asked if Sef would also intervene in cPLA2 activation as regulated from other sublocalizations. In this respect, cPLA2 activation induced by lipid raft-targeted RasV12 was completely unaffected by the siRNA-induced drop in Sef expression (Fig. 10D). Furthermore, when we analyzed the composition of Sef complexes upon the receipt of Ras signals originated at different localizations, cPLA2 associated with Sef in an activated ERK-dependent fashion in cells transfected with ER-bound RasV12 but not in those expressing lipid raft-associated RasV12 (Fig. 10E). In conclusion, these results indicated that Sef was the scaffold protein specifically regulating cPLA2 activation by ERK1/2 responding to Ras signals coming from the ER.
Our results had demonstrated that Ras signals from lipid rafts regulated the phosphorylation of two ERK substrates, cPLA2 and EGFr, and that KSR1 was the ERK scaffold mediating in cPLA2 activation. As such, we questioned whether KSR1 also participated in ERK1/2 signals directed to EGFr, thus behaving as a “general” lipid raft scaffold. Remarkably, EGFr phosphorylation resulting from Ras activation at lipid rafts was unaffected by the downregulation of KSR1 levels (Fig. 11A), speaking against KSR1 having a scaffold role in the activation of this substrate. Accordingly, we looked for the scaffold mediating in this event by assaying the phosphorylation of EGFr, as induced by lipid raft-tethered RasV12, under limiting levels of scaffold proteins, brought about by the utilization of the siRNA battery mentioned before. In this case, it was found that a drop in IQGAP1 expression evoked a marked reduction in EGFr phosphorylation (Fig. 11B). Further, in cells transfected with lipid raft-tethered RasV12, IQGAP1 was found in association with the EGFr (Fig. 11C), thereby pointing to IQGAP1 as the scaffold participating in the lipid raft-induced ERK signal, directed to this particular substrate. Since Ras signals derived from lipid rafts also regulated the activation of cPLA2, we tested whether IQGAP1 could also intervene in this process. It was found that the siRNA-mediated depletion of IQGAP1 levels did not affect whatsoever cPLA2 activation as induced by RasV12 acting at lipid rafts (Fig. 11D).
FIG. 11.

IQGAP1 couples Ras signals from lipid rafts to EGFr phosphorylation. (A) Depletion of KSR1 does not affect the phosphorylation of EGFr elicited by Ras signals from lipid rafts. 293T cells were transfected with lipid raft-targeted RasV12 (LCK) (0.5 μg) plus a siRNA (10 nM) (+) for KSR1. EGFr phosphorylation was determined by immunoprecipitation (IP) in 32P-labeled cells. (B) Effects of downregulating the levels of different scaffold proteins on EGFr phosphorylation induced by Ras signals from lipid rafts. Cells were transfected with lipid raft-targeted RasV12, plus siRNAs for the shown scaffold proteins. DGLY., dystroglycan; β ARR. 2, β-arrestin 2; PAXILL., paxillin. (C) EGFr/IQGAP1 association is enhanced by Ras signals from lipid rafts. Endogenous IQGAP1, present in anti-EGFr immunoprecipitates, was measured in cells transfected with RasV12, untargeted or tethered to lipid rafts (LCK). TL, total lysate; IP, immunoprecipitations performed with a specific antibody; PI, immunoprecipitations performed with preimmune serum. (D) IQGAP1 does not intervene in cPLA2 activation elicited from lipid rafts. Cells were transfected with RasV12 tethered to lipid rafts with (+) or without (−) a siRNA against IQGAP1. cPLA2 activation was determined by [3H]arachidonic acid release after starvation. (E) RSK1 phosphorylation by Ras signals from the disordered membrane is unaffected by the depletion of scaffold proteins. Cells were transfected with disordered membrane-targeted RasV12 (CD8), plus siRNAs (10 ng) for the shown scaffold proteins. Total and phosphorylated RSK1 levels were detected by immunoblotting. α, anti.
Subsequently, we set out to find the scaffold protein mediating the phosphorylation of RSK1, as induced by Ras signals flowing from the disordered membrane. With this aim, once again, we utilized the set of siRNAs previously described in cells expressing RasV12 tethered to this specific sublocalization. Interestingly, the levels of RSK1 phosphorylation remained unaltered in all cases (Fig. 11E), suggesting that none of the tested scaffold proteins participated in the disordered membrane-evoked ERK signal targeting RSK1. Overall, these results supported the notion that Ras localization within different membrane microdomains could dictate ERK1/2 substrate specificity and that such an effect was possible due to the selective participation of defined scaffold proteins.
Finally, we examined the extent to which scaffold usage could influence the biological output of Ras site-specific signals. It has been shown that KSR1 is an essential mediator in Ras-induced replicative senescence (18). As such, we tested how site-specific Ras signals coupled to KSR1, such as those emanating from lipid rafts, could induce replicative senescence compared to those generated at the disordered membrane, which we have shown not to utilize KSR1. To this end, wild-type MEFs (+/+) were transfected with the constructs expressing RasV12 at lipid rafts and the disordered membrane. In these cells, both site-specific signals were capable of promoting transformation, slightly more prominently in the case of those signals generated at the disordered membrane (Fig. 12A). However, the situation was completely reversed when we monitored their ability to induce senescence: whereas the amount of β-galactosidase-positive +/+ MEFs resulting from the expression of lipid raft-tethered RasV12 was similar to that encountered in cells transfected with “total” RasV12, their numbers were reduced by more than 50% in MEFs transfected with RasV12 bound to the disordered membrane (Fig. 12B). To test the participation of KSR1 in this process, we repeated the same experiment with MEFs devoid of KSR1 (−/−) (18). It was found that the absence of this scaffold dramatically reduced the ability of the different Ras constructs to induce senescence in all cases, though less pronouncedly in the case of disordered membrane-targeted CD8-RasV12. Likewise, when the levels of KSR1 were reconstituted by ectopic expression, the regained capacity of the Ras signals emanating from the disordered membrane to induce senescence was far lower than the one exhibited by Ras signals coming from lipid rafts (Fig. 12B). In agreement with our biochemical data, these results demonstrated that the biological responses caused by Ras signaling from the disordered membrane were mostly independent of KSR1, whereas those resulting from Ras signals originating at lipid rafts were strongly dependent on the presence of this scaffold protein.
FIG. 12.

KSR1 affects the ability of site-specific Ras signals to induce senescence. (A) Transforming potential of Ras site-specific signals. MEFs were transfected with vectors encoding H-RasV12 constructs as shown in the inset key (1 μg each) in addition to c-Myc (1 μg). +/+, wild-type MEFs. (B) Site-specific Ras signals require KSR1 to induce senescence. MEFs, wild-type (+/+) or deficient for KSR1 (−/−), were transfected with vectors encoding the H-RasV12 constructs as shown in the inset key in panel A (1 μg each). Where indicated, ectopic KSR1 (1 μg) was also transfected. After 3 weeks, the cells were stained and either the number of foci or the number of senescent cells was scored. Shown are the averages ± standard errors of the means of the results from three independent experiments. +ve, positive.
DISCUSSION
An essential requirement for an efficient signal transduction system is a fast response upon the receipt of a stimulus. This can be accomplished by triggering immediate associations and functional interactions among its constituents. However, considering that the environment in which most of these reactions take place, the cytoplasm, is densely crowded and mostly occupied by organelles and macromolecular complexes, it seems unlikely that such instantaneous reactions could be achieved if the signaling intermediaries were randomly scattered. To overcome this problem, one possibility would be that signaling molecules resided in defined subcytoplasmic confinements, in a preassembled mode, so as to guarantee a swift wiring, connecting those sites at which the signal is received to the desired effectors at particular cellular locations. The results presented herein support this notion.
We show that the Ras signaling platform from which the activating signals originate dictates ERK1/2 substrate specificity. Our results indicate that the phosphorylation of PM substrates, like EGFr, is preferentially regulated from lipid rafts. This is consistent with the EGFr being highly concentrated in this type of membrane (39). Regarding ERK1/2 cytoplasmic substrates, our results point to variations in the spatial regulation of their activation. As such, cPLA2 is primarily regulated by lipid raft-generated Ras signals, once again consistent with the physical and functional association of cPLA2 with lipid rafts, caveolar in particular (2, 25). On the other hand, the activation of RSK1 is regulated from the disordered membrane. An explanation for this divergent regulation for two substrates that a priori reside within the same compartment is that, within the cytoplasm, they occupy distinct microlocalizations. In support of this notion, our electron microscopy data demonstrate that these two cytoplasmic proteins are clustered in clearly separated cytoplasmic microdomains. Thus, it can be envisioned that upon the activation of Ras at lipid rafts or at the disordered membrane, activated ERK1/2 would be, respectively, directed to cPLA2- or to RSK1-enriched cytoplasmic confinements. Indeed, we demonstrate that Ras activity at lipid rafts induces the colocalization of phosphorylated ERK1/2 with cPLA2 but not with RSK1, whereas RSK1 but not cPLA2 colocalizes with activated ERK1/2 when the activating Ras signals emanate from the disordered membrane.
Importantly, we demonstrate that the coupling of ERK1/2 to specific substrates depending on the activating signaling platform is not solely restricted to oncogenic Ras-generated signals but also applies to physiological agonists, like EGF and LPA, and to drugs like TPA. Indeed, in an entirely physiological context, we show that ERK activation kinetics at defined localizations resembles those of its cognate substrates and that both fluctuate in parallel depending on the stimulus. As such, EGFr activation kinetics are similar to those for ERK2 at lipid rafts, whereas the time course of RSK1 activity mirrors that of ERK2 at the disordered membrane.
With respect to ERK1/2 nuclear substrates, we have examined the activation of the transcription factor Elk1. We have found that its activation is not due solely to ERK1/2 activity but also to JNK1/2, but not to p38. Interestingly, the JNK component displays a clear spatial regulation, being the Ras signals emanating from endomembranes, in particular from the Golgi complex, the most sensible to JNK1/2 depletion. This result is in agreement with our previous findings, in which Ras at these localizations exhibited the most prominent JNK activation (21). Regarding the ERK1/2 component, unlike membrane and cytoplasmic substrates, our results indicate that the activation of nuclear substrates is mostly independent of the Ras sublocalization from which ERK1/2 are switched on. Interestingly, we show that untargeted RasV12 elicits a much stronger Elk1 activation than the site-specific RasV12 constructs. It must be noticed that our site-specific Ras proteins are fixed to their compartments, whereas untargeted Ras is free to translocate between localizations and the cytoplasm (31). This could cause differences in signal intensities and therefore impact the resulting biological outcomes. Also, another possibility is that a more-efficient Elk1 transactivation takes place under the input from several Ras sublocalizations, attainable by untethered Ras molecules free to move, while, as elicited by the site-specific, single signals generated by our targeted Ras proteins, Elk1 activation may be less effective.
Herein we demonstrate that ERK1/2 selectivity toward different substrates, dependent on Ras sublocalization, is attained through the participation of specific scaffold proteins, which mediate such an interaction. We show that ERK1/2 activated by signals originating at lipid rafts are coupled to cPLA2 by the participation of KSR1. Interestingly, KSR1 is not the only scaffold utilized in ERK1/2-mediated signaling routes emanating from lipid rafts, as we demonstrate that the ERK-induced phosphorylation of EGFr is regulated not by KSR1 but by IQGAP1. Another concept unveiled by our present results is that targeting a defined substrate is not exclusive for a given scaffold protein. In this respect, we demonstrate that cPLA2 activation is regulated by KSR1 when ERK1/2 are activated from lipid rafts, whereas Sef is the mediator if the ERK1/2-activating signal evolves from the ER. While the association of IQGAP1 with EGFr has been previously reported (7), the interactions of cPLA2 with KSR1 or with Sef were unknown before our studies (8). Noticeably, we have observed that the activation of the cytoplasmic kinase RSK1 by signals coming from the disordered membrane is unaffected by the deprivation of the best-known scaffold proteins. Even though we cannot ultimately discard the idea that such a reaction takes place in an unscaffolded fashion, our previous results indicating that the depletion of ERK dimers affects RSK1 phosphorylation (8) suggest the participation of some scaffold. It is possible that the limited tactic that we have utilized herein has not allowed us to identify the scaffold mediating in this pathway and that an unbiased, genome-wide approach is necessary.
Having acquired the notion of scaffold proteins playing an essential role in the spatial regulation of Ras sublocalization-directed ERK1/2 substrate specificity, our data showing that the activation of ERK1/2 nuclear substrates is not dependent on the Ras signaling platform are further endorsed by previous reports demonstrating that scaffold proteins such as KSR1 (34), β-arrestin (11, 36), and Sef (37) are mostly involved in cytoplasmic events and that their effects on ERK-mediated nuclear effects are limited. In line with these findings, our recent results demonstrate that scaffolds act as dimerizing platforms for ERK1/2 and, subsequently, serve in the activation of cytoplasmic substrates, undertaken by ERK1/2 dimers, whereas they are dispensable for the activation of nuclear substrates exerted by ERK1/2 monomers (8).
As of today, the mechanisms whereby ERK1/2 scaffold complexes respond to localization-defined Ras signals and how they can be directed to different subcytoplasmic microenvironments, to target different substrates, are completely unknown and under investigation. One possibility is the existence of microlocalization-specific determinants within the structure of scaffold proteins, which could orchestrate ERK1/2 signals in a spatially dependent fashion. This possibility is supported by the fact that some scaffold proteins regulate ERK1/2 signals specifically at localizations like the Golgi complex (37), endosomes (35), and focal adhesions (16). Overall, our results disclose a novel mechanism whereby space can regulate the outcome of a signal by a selective wiring connecting the sites at which such a signal emanates for the activation of defined substrates through the specific intervention of molecular intermediaries, which are, in this particular case, scaffold proteins.
Acknowledgments
We are indebted to A. Pandiella, T. Iglesias, J. Lozano, and Z. Chang for providing reagents.
A.P. and L.A.-I. are Spanish Ministry of Education predoctoral fellows. P.C.'s lab is supported by grants BFU2005-00777 and GEN2003-20239-C06-03 from the Spanish Ministry of Education, the EU Sixth Framework Program under the GROWTHSTOP (LSHC CT-2006-037731) and SIMAP (IST-2004-027265) projects, the Red Temática de Investigación Cooperativa en Cáncer (RD06/0020/0105), the Fondo de Investigaciones Sanitarias, Carlos III Institute, and the Spanish Ministry of Health.
Footnotes
Published ahead of print on 29 December 2008.
REFERENCES
- 1.Agudo-Ibanez, L., F. Nunez, F. Calvo, I. M. Berenjeno, X. R. Bustelo, and P. Crespo. 2007. Transcriptomal profiling of site-specific Ras signals. Cell. Signal. 192264-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ait-Mamar, B., M. Cailleret, C. Rucker-Martin, A. Bouabdallah, G. Candiani, C. Adamy, P. Duvaldestin, F. Pecker, N. Defer, and C. Pavoine. 2005. The cytosolic phospholipase A2 pathway, a safeguard of beta2-adrenergic cardiac effects in rat. J. Biol. Chem. 28018881-18890. [DOI] [PubMed] [Google Scholar]
- 3.Ajenjo, N., E. Canon, I. Sanchez-Perez, D. Matallanas, J. Leon, R. Perona, and P. Crespo. 2004. Subcellular localization determines the protective effects of activated ERK2 against distinct apoptogenic stimuli in myeloid leukemia cells. J. Biol. Chem. 27932813-32823. [DOI] [PubMed] [Google Scholar]
- 4.Arozarena, I., D. S. Aaronson, D. Matallanas, V. Sanz, N. Ajenjo, S. P. Tenbaum, H. Teramoto, T. Ighishi, J. C. Zabala, J. S. Gutkind, and P. Crespo. 2000. The Rho family GTPase Cdc42 regulates the activation of Ras/MAP kinase by the exchange factor Ras-GRF. J. Biol. Chem. 27526441-26448. [DOI] [PubMed] [Google Scholar]
- 5.Arozarena, I., D. Matallanas, M. T. Berciano, V. Sanz-Moreno, F. Calvo, M. T. Munoz, G. Egea, M. Lafarga, and P. Crespo. 2004. Activation of H-Ras in the endoplasmic reticulum by the RasGRF family guanine nucleotide exchange factors. Mol. Cell. Biol. 241516-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagowski, C. P., M. Stein-Gerlach, A. Choidas, and A. Ullrich. 1999. Cell-type specific phosphorylation of threonines T654 and T669 by PKD defines the signal capacity of the EGF receptor. EMBO J. 185567-5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blagoev, B., I. Kratchmarova, S. E. Ong, M. Nielsen, L. J. Foster, and M. Mann. 2003. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat. Biotechnol. 21315-318. [DOI] [PubMed] [Google Scholar]
- 8.Casar, B., A. Pinto, and P. Crespo. 2008. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaffold complexes. Mol. Cell 31708-721. [DOI] [PubMed] [Google Scholar]
- 9.Chiu, V. K., T. Bivona, A. Hach, J. B. Sajous, J. Silletti, H. Wiener, R. L. Johnson II, A. D. Cox, and M. R. Philips. 2002. Ras signaling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 4343-350. [DOI] [PubMed] [Google Scholar]
- 10.Crespo, P., and J. Leon. 2000. Ras proteins in the control of the cell cycle and cell differentiation. Cell. Mol. Life Sci. 571613-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeFea, K. A., J. Zalevsky, M. S. Thoma, O. Dery, R. D. Mullins, and N. W. Bunnett. 2000. β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 1481267-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhanasekaran, D. N., K. Kashef, C. M. Lee, H. Xu, and E. P. Reddy. 2007. Scaffold proteins of MAP-kinase modules. Oncogene 263185-3202. [DOI] [PubMed] [Google Scholar]
- 13.Frodin, M., and S. Gammeltoft. 1999. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 2565-77. [DOI] [PubMed] [Google Scholar]
- 14.Glading, A., F. Uberall, S. M. Keyse, D. A. Lauffenburger, and A. Wells. 2001. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J. Biol. Chem. 27623341-23348. [DOI] [PubMed] [Google Scholar]
- 15.Harding, A., T. Tian, E. Westbury, E. Frische, and J. F. Hancock. 2005. Subcellular localization determines MAP kinase signal output. Curr. Biol. 15869-873. [DOI] [PubMed] [Google Scholar]
- 16.Ishibe, S., D. Joly, X. Zhu, and L. G. Cantley. 2003. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell 121275-1285. [DOI] [PubMed] [Google Scholar]
- 17.Kolch, W. 2005. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6827-837. [DOI] [PubMed] [Google Scholar]
- 18.Kortum, R. L., H. J. Johnson, D. L. Costanzo, D. J. Volle, G. L. Razidlo, A. M. Fusello, A. S. Shaw, and R. E. Lewis. 2006. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol. Cell. Biol. 262202-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin, L., M. Wartmann, A. Y. Lin, J. L. Knopf, A. Seth, and R. J. Davis. 1993. cPLA2 is phosphorylated and activated by MAP kinase. Cell 72269-278. [DOI] [PubMed] [Google Scholar]
- 20.Matallanas, D., I. Arozarena, M. T. Berciano, D. S. Aaronson, A. Pellicer, M. Lafarga, and P. Crespo. 2003. Differences in the inhibitory specificities of H-Ras, K-Ras and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J. Biol. Chem. 2784572-4581. [DOI] [PubMed] [Google Scholar]
- 21.Matallanas, D., V. Sanz-Moreno, I. Arozarena, F. Calvo, L. Agudo-Ibanez, E. Santos, M. T. Berciano, and P. Crespo. 2006. Distinct utilization of effectors and biological outcomes resulting from site-specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol. Cell. Biol. 26100-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matheny, S. A., C. Chen, R. L. Kortum, G. L. Razidlo, R. E. Lewis, and M. A. White. 2004. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 427256-260. [DOI] [PubMed] [Google Scholar]
- 23.Morrison, D. K., and R. J. Davis. 2003. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 1991-118. [DOI] [PubMed] [Google Scholar]
- 24.Northwood, I. C., F. A. Gonzalez, M. Wartmann, D. L. Raden, and R. J. Davis. 1991. Isolation and characterization of two growth factor-stimulated protein kinases that phosphorylate the epidermal growth factor receptor at threonine 669. J. Biol. Chem. 26615266-15276. [PubMed] [Google Scholar]
- 25.Pavoine, C., and N. Defer. 2005. The cardiac beta2-adrenergic signalling a new role for the cPLA2. Cell. Signal. 17141-152. [DOI] [PubMed] [Google Scholar]
- 26.Perez de Castro, I., T. G. Bivona, M. R. Philips, and A. Pellicer. 2004. Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi apparatus. Mol. Cell. Biol. 243485-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prior, I. A., A. Harding, J. Yan, J. Sluimer, R. G. Parton, and J. F. Hancock. 2001. GTP-dependent segregation of H-Ras from lipid rafts is required for biological activity. Nat. Cell Biol. 3368-375. [DOI] [PubMed] [Google Scholar]
- 28.Prior, I. A., C. Muncke, R. G. Parton, and J. F. Hancock. 2003. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 160165-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson, M. J., and M. H. Cobb. 1997. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 9180-186. [DOI] [PubMed] [Google Scholar]
- 30.Robinson, M. J., S. A. Stippec, E. Goldsmith, M. A. White, and M. H. Cobb. 1998. A constitutively active and nuclear form of MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr. Biol. 81141-1150. [DOI] [PubMed] [Google Scholar]
- 31.Rocks, O., A. Peyker, M. Kahms, P. J. Verveer, C. Koerner, M. Lumbierres, J. Kuhlmann, H. Waldmann, A. Wittinghofer, and P. I. Bastiaens. 2005. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 3071746-1752. [DOI] [PubMed] [Google Scholar]
- 32.Roy, S., B. Wyse, and J. F. Hancock. 2002. H-Ras signaling and K-Ras signaling are differentially dependent on endocytosis. Mol. Cell. Biol. 225128-5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanz-Moreno, V., B. Casar, and P. Crespo. 2003. p38alpha isoform Mxi2 binds to extracellular signal-regulated kinase 1 and 2 mitogen-activated protein kinase and regulates its nuclear activity by sustaining its phosphorylation levels. Mol. Cell. Biol. 233079-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto, T., S. Stewart, M. Han, and K. L. Guan. 1998. The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. EMBO J. 171717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teis, D., W. Wunderlich, and L. A. Huber. 2002. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 3803-814. [DOI] [PubMed] [Google Scholar]
- 36.Tohgo, A., K. L. Pierce, E. W. Choy, R. J. Lefkowitz, and L. M. Luttrell. 2002. β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 2779429-9436. [DOI] [PubMed] [Google Scholar]
- 37.Torii, S., M. Kusakabe, T. Yamamoto, M. Maekawa, and E. Nishida. 2004. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev. Cell 733-44. [DOI] [PubMed] [Google Scholar]
- 38.Xiong, S., Q. Zhao, Z. Rong, G. Huang, Y. Huang, P. Chen, S. Zhang, L. Liu, and Z. Chang. 2003. hSef inhibits PC-12 cell differentiation by interfering with Ras-mitogen-activated protein kinase MAPK signaling. J. Biol. Chem. 27850273-50282. [DOI] [PubMed] [Google Scholar]
- 39.Yamabhai, M., and R. G. Anderson. 2002. Second cysteine-rich region of epidermal growth factor receptor contains targeting information for caveolae/rafts. J. Biol. Chem. 27724843-24846. [DOI] [PubMed] [Google Scholar]
- 40.Yoon, S., and R. Seger. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 2421-44. [DOI] [PubMed] [Google Scholar]