

Abstract
Interleukin-10 (IL-10) is an anti-inflammatory cytokine with protective actions on the vasculature. On the other hand, endothelin (ET)-1 has potent vasoconstrictor, mitogenic, and proinflammatory activities, which have been implicated in the pathophysiology of a number of cardiovascular diseases. We hypothesized that, in a condition where ET-1 expression is upregulated, i.e., on infusion of TNF-α, IL-10 confers vascular protection from ET-1-induced injury. Aortic rings and first-order mesenteric arteries from male C57BL/6 (WT) and IL-10-knockout (IL-10−/−) mice were treated with human recombinant TNF-α (220 ng·kg−1·day−1) or vehicle (saline) for 14 days. TNF-α infusion significantly increased blood pressure in IL-10−/−, but not WT, mice. TNF-α augmented vascular ET-1 mRNA expression in arteries from WT and IL-10−/− mice. ET type A (ETA) receptor expression was increased in arteries from IL-10−/− mice, and TNF-α infusion did not change vascular ETA receptor expression in control or IL-10−/− mice. Aorta and mesenteric arteries from TNF-α-infused IL-10−/− mice displayed increased contractile responses to ET-1, but not the ET type B receptor agonist IRL-1620. The ETA receptor antagonist atrasentan completely abolished responses to ET-1 in aorta and mesenteric vessels, whereas the ERK1/2 inhibitor PD-98059 abrogated increased contractions to ET-1 in arteries from TNF-α-infused IL-10−/− mice. Infusion of TNF-α, as well as knockdown of IL-10 (IL-10−/−), induced an increase in total and phosphorylated ERK1/2. These data demonstrate that IL-10 counteracts ETA-mediated vascular responses to ET-1, as well as activation of the ERK1/2 pathway.
Keywords: tumor necrosis factor-α, vascular reactivity, blood pressure
elevated cytokine levels play a role in many cardiovascular diseases and in the associated disturbances of vascular reactivity reported in these pathological conditions. Cytokines seem to influence the balance between vasoconstrictor and vasodilator factors, as well as regional differences in the release and responsiveness to these factors, contributing to the responsiveness within a specific vascular bed (42).
Interleukin-10 (IL-10) is one of the most important anti-inflammatory cytokines involved in the regulatory processes associated with resolution of inflammation. IL-10 inhibits chemokine production, antigen presentation, and antigen-specific T cell proliferation and is a potent inhibitor of monocyte/macrophage activation (1, 14, 17, 20, 39). In polymorphonuclear neutrophils, IL-10 exerts its anti-inflammatory properties by downregulating functions such as NADPH oxidase activation and reactive oxygen species production (6). Many of the anti-inflammatory actions of IL-10 are associated with inhibition of ERK1/2 activity or the MEK/ERK pathway (5, 38, 39).
IL-10 also confers vascular protection against inflammatory stimuli. Exogenous IL-10 prevents proliferative vasculopathy by inhibiting inflammatory cell infiltration, smooth muscle cell proliferation, and chemokine expression (3, 27). This cytokine counteracts the endothelial dysfunction induced by angiotensin II, lipopolysaccharide, and endothelin (ET)-1 (15, 16, 49, 50). To promote changes in vascular reactivity, remodeling, inflammation, and oxidative stress, ET-1 activates MAPK signaling pathways, including ERK1/2 (7, 19, 21, 48). Here we sought to determine whether modulation of ERK1/2 activity is an important point of convergence for the effects of IL-10 and ET-1. We hypothesized that, in a condition where ET-1 expression is upregulated, IL-10 confers vascular protection from ET-1-mediated responses by decreasing ET-1-associated ERK1/2 signaling. To test our hypothesis, IL-10-knockout (IL−/−) and wild-type (WT) mice were infused with tumor necrosis factor-α (TNF-α), and vascular responses to ET-1, as well as activation of the MEK/ERK signaling pathway, were determined. TNF-α was chosen, because it is a proinflammatory cytokine that not only induces ET-1 release but, also, augments contractile responses to ET-1 in different vascular beds (4, 43–47, 51).
METHODS
Animals.
Male B6.129P2-Il10tm1Cgn/J (IL-10−/−) mice and their control C57BL/6J (WT) mice (10 wk old; Jackson Laboratory, Bar Harbor, ME) were used in this study. All procedures were performed in accordance with the National Institutes of Health Guiding Principles in the Care and Use of Animals and approved by the Medical College of Georgia Committee on the Use of Animals in Research and Education. The animals were housed four per cage, exposed to a 12:12-h light-dark cycle, and fed a standard chow diet with water ad libitum.
Animals were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg), and osmotic minipumps (0.25 μl/h; model 1002, Alzet, Cupertino, CA) were implanted subcutaneously in the scapular region. IL-10−/− and WT mice were divided into two groups: one was infused with saline (vehicle) only, and the other was infused with TNF-α (220 ng·kg−1·min−1) for 14 days. This dose of TNF-α results in serum concentrations of ∼0.09 and 0.12 pg/ml in WT and IL-10−/− mice, respectively, at the end of the infusion period. TNF-α concentration in serum of mice infused with saline was ∼0.03 and 0.02 pg/ml in WT and IL-10−/− mice, respectively.
Blood pressure recordings by telemetry and treatment.
Surgery was performed on control and experimental mice for implantation of blood pressure radio-telemetry transmitters (model PA-C20, Data Sciences International, Roseville, MN). Briefly, after isoflurane anesthesia, the transmitter was implanted into the carotid artery via a midventral neck incision under aseptic conditions, and the transmitter body was routed to a subcutaneous pocket in the midback region. The incision was infiltrated with bupivacaine and closed with sterile 6.0 Ethicon ophthalmic suture. After recovery (5–7 days), the mice were housed in individual cages in the laboratory animal facilities, and standard laboratory chow and water were provided ad libitum. After 2 days of blood pressure recording, osmotic minipumps were implanted (see above). After 14 days, the mice were euthanized, and aorta and mesentery were isolated for further studies (see below).
RNA extraction, cDNA synthesis, and quantitative real-time RT-PCR.
Total RNA was extracted from aorta using the RNeasy kit (Qiagen). The quantity, purity, and integrity of all RNA samples were determined by NanoDrop spectrophotometry (NanoDrop Technologies, Wilmington, DE). One microgram of total RNA was reverse transcribed in a final volume of 50 μl using the High-Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA), and single-strand cDNA was stored at −20°C. Primers for prepro-ET-1 mRNA were obtained from Applied Biosystems (catalog no. Mn00438656_m1). Real-time (quantitative) RT-PCR was performed using the 7500 Fast Real-Time PCR system (Applied Biosystems) in a total volume of 20 μl of reaction mixture following the manufacturer's protocol and using TaqMan Fast Universal PCR Master Mix (2×; Applied Biosystems) and each primer at 0.1 μM. Negative controls contained water, instead of first-strand cDNA. Each sample was normalized on the basis of its 18S rRNA content. The 18S quantification was performed using a TaqMan rRNA reagent kit (Applied Biosystems) following the manufacturer's protocol. Relative gene expression for prepro-ET-1 mRNA was normalized to a calibrator that was chosen to be the basal condition (untreated sample) for each time point. Results were calculated with the ΔΔCt method and expressed as n-fold differences in prepro-ET-1 gene expression relative to 18S rRNA and to the calibrator and were determined as follows: n-fold = 2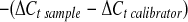 , where Ct (threshold cycle) is defined as the fractional cycle number at which the PCR reporter signal passes a fixed threshold. ΔCt values of the sample and the calibrator were determined by subtraction of the average Ct value of the transcript under investigation from the average Ct value of the 18S rRNA gene for each sample.
, where Ct (threshold cycle) is defined as the fractional cycle number at which the PCR reporter signal passes a fixed threshold. ΔCt values of the sample and the calibrator were determined by subtraction of the average Ct value of the transcript under investigation from the average Ct value of the 18S rRNA gene for each sample.
Vascular functional studies.
After the animals were euthanized, the mesentery and thoracic aorta were rapidly excised and placed in ice-cold physiological saline solution (PSS). First-order branches of mesenteric artery (∼2 mm long, ∼100–200 μm ID) and thoracic aorta (4 mm long) were carefully dissected and mounted as ring preparations on two stainless steel wires. The first-order mesenteric arteries were mounted in an isometric Mulvany-Halpern small vessel myograph (40 μm diameter; model 610 M, Danish MyoTech, Aarhus, Denmark), whereas the aortic rings were mounted in standard organ chambers for isometric tension recording by a PowerLab 8/SP data acquisition system (ADInstruments, Colorado Springs, CO).
One wire was attached to a force transducer and the other to a micrometer. Dissection and mounting of the vessels were carried out in cold (4°C) PSS. The segments were adjusted to maintain a passive force of 3 mN for first-order mesenteric arteries and 5 mN for aortic rings. Vessels were equilibrated for 60 min in PSS at 37°C and continuously bubbled with 5% CO2-95% O2. Arterial integrity was assessed, first, by stimulation of vessels with 120 mM KCl and, after they were washed and restabilized, by contraction of the segments with phenylephrine (PE; 1 μM). The segments were incubated for 30 min with the nitric oxide (NO) synthase inhibitor Nω-nitro-l-arginine methyl ester (l-NAME, 100 μM) + indomethacin (10 μM), an inhibitor of prostanoid synthesis, or with their respective vehicles (control conditions). Endothelium integrity was assessed by contraction of the segments with PE followed by stimulation with ACh (10 μM). Concentration-response curves to ET-1 (0.1 nM–1 μM) were determined in the presence or absence of PD-98059 (10 μM), an ERK1/2 inhibitor in aorta and first-order mesenteric artery. The response to atrasentan (1 μM), a ET-1 type A (ETA) receptor antagonist, was checked in first-order mesenteric artery contracted with ET-1 (1 μM). Concentration-response curves to IRL-1620 (0.1 nM–0.1 μM), an ET type B (ETB) receptor agonist, and to PE (1 nM–30 μM), an α-adrenergic agonist, were also determined in aortas and first-order mesenteric arteries.
Western blot analysis.
Proteins (40 μg) extracted from aorta were separated by electrophoresis on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered saline solution with Tween for 1 h at 24°C. Membranes were then incubated with antibodies (1:1,000 dilution) overnight at 4°C. Antibodies were Thr202/Tyr204-phosphorylated p44/42 MAPK (ERK1/2). Immunoblots for nonphosphorylated p44/42 MAPK (ERK1/2; all antibodies from Cell Signaling Technology) were carried out in the same membranes used to evaluate their phosphorylated forms. ETA receptor expression (1:250 dilution; Alamone Labs) was evaluated. After incubation with secondary antibodies, signals were revealed with chemiluminescence, visualized by autoradiography, and quantified densitometrically. Results were normalized to β-actin protein and are expressed as arbitrary units.
Drugs and solutions.
PSS consisted of (in mM) 130 NaCl, 14.9 NaHCO3, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4·7 H2O, 5.5 glucose, 1.56 CaCl2·2 H2O, and 0.026 EDTA. Indomethacin, l-NAME, phenylephrine hydrochloride (PE), and TNF-α were purchased from Sigma Chemical (St. Louis, MO); ET-1 and IRL-1620 from Tocris (Ellisville, MO); and PD-98059 from Calbiochem (San Diego, CA). Atrasentan was a gift from Abbott Laboratories. All reagents were of analytic grade. Stock solutions were prepared in deionized water, ethanol (indomethacin), DMSO (PD-98059), or saline (TNF-α). Control solutions containing vehicle levels of ethanol and DMSO were also used throughout the experimental protocols.
Data analysis.
Values are means ± SE. Contractions were recorded as changes in displacement (mN) from baseline and are represented as mN for n experiments. Concentration-response curves were fitted using a nonlinear interactive fitting program (Prism 4.0, GraphPad Software, San Diego, CA). Curve-fit parameters were used to fit a sigmoidal curve (see Figs. 2A and 5). Since the curve of Fig. 2B does not fit the standard sigmoidal shape, we used the Gaussian distribution equation to analyze this curve (Gaussian distribution was the model that most closely approximates the shape of this curve). Statistically significant differences were calculated by ANOVA or by Student's t-test where appropriate. P < 0.05 was considered significant.
Fig. 2.

Contractile responses to endothelin (ET)-1 are enhanced in arteries from TNF-α-infused IL-10−/− mice. Cumulative concentration-response curves to ET-1 (0.1 nM–1.0 μM) in 1st-order mesenteric arteries (A) and thoracic aorta (B) were determined in vehicle- and TNF-α-infused WT and vehicle- and TNF-α-infused IL-10−/− mice. Values are means ± SE (n = 5 experiments). *P < 0.05 vs. other groups.
Fig. 5.

ERK1/2 inhibition abrogates augmented contractile responses to ET-1 in arteries from TNF-α-infused IL-10−/− mice. Cumulative concentration-response curves to ET-1 (0.1 nM–1.0 μM) were determined in the presence of PD-98059 (1 μM), an ERK1/2 inhibitor, in 1st-order mesenteric arteries (A) and aorta (B) from vehicle- and TNF-α-infused WT and vehicle- and TNF-α-infused IL-10−/− mice. Values are means ± SE (n = 6 experiments).
RESULTS
Effect of IL-10 and TNF-α on blood pressure and prepro-ET-1 levels.
Mean arterial pressure (MAP) under basal conditions, assessed by telemetry, was lower in IL-10−/− than in WT mice (90.8 ± 3.6 vs. 110.3 ± 2.1 mmHg). After 2–4 days of TNF-α infusion, MAP slightly increased, ∼7–8 mmHg, in both groups. After 14 days of TNF-α infusion, MAP in WT and IL-10−/− mice was 117.0 ± 2.8 and 103.0 ± 5.1 mmHg (n = 5), respectively. Infusion of vehicle did not change MAP in IL-10−/− or WT mice (89.3 ± 3.9 and 111.8 ± 3.7 mmHg, respectively; Fig. 1A).
Fig. 1.

Mean arterial pressure (A) and vascular (aorta) preproendothelin-1 mRNA expression (B) in wild-type (WT) mice infused with vehicle (WT + vehicle) and TNF-α-infused WT mice (WT + TNF-α) and IL-10-knockout (IL-10−/−) mice infused with vehicle (IL-10−/− + vehicle) and TNF-α-infused IL-10−/− mice (IL-10−/− + TNF-α). Values are means ± SE (n = 5 experiments). *P < 0.05 vs. respective control (i.e., vehicle). †P < 0.05, IL-10−/− vs. WT.
Chronic infusion of TNF-α was used as a pharmacological tool to increase ET-1 levels. For evaluation of the tissue levels of ET-1, prepro-ET-1 was measured in aortas using real-time RT-PCR. Indeed, chronic infusion of TNF-α increased levels of prepro-ET-1 in control and IL-10−/− mice (Fig. 1B).
Effect of IL-10 on ET-1-induced vasoconstriction.
In the presence of l-NAME and indomethacin, aorta and first-order mesenteric arteries from TNF-α-infused IL-10−/− mice displayed increased contractile responses to ET-1 compared with arteries from the other groups (Fig. 2). In the absence of l-NAME and indomethacin (i.e., control conditions), endothelium-intact aorta from TNF-α-infused IL-10−/− mice also displayed increased contractile response to ET-1 compared with arteries from the other groups. However, the magnitude of contraction was smaller in these vessels than in vessels treated with l-NAME + indomethacin. Maximum contractions to ET-1 (mN) in aortic rings were as follows: 0.28 ± 0.15 (WT + vehicle), 0.35 ± 0.10 (WT + TNF-α), 0.51 ± 0.30 (IL-10−/− + vehicle), and 8.6 ± 0.20 (IL-10−/− + TNF-α) under control conditions and 1.30 ± 0.87 (WT + vehicle), 0.85 ± 0.88 (WT + TNF-α), 0.74 ± 0.42 (IL-10−/− + vehicle), and 9.91 ± 2.02 (IL-10−/− + TNF-α) in the presence of l-NAME + indomethacin. In small mesenteric arteries, no differences in ET-1-induced contractile responses were observed among the groups under control conditions. Maximum contractions to ET-1 (mN) in mesenteric arteries were as follows: 1.65 ± 0.58 (WT + vehicle), 1.73 ± 0.18 (WT + TNF-α), 1.85 ± 0.07 (IL-10−/− + vehicle), and 3.33 ± 0.39 (IL-10−/− + TNF-α) in control conditions and 1.96 ± 0.27 (WT + vehicle), 2.34 ± 0.3 (WT + TNF-α), 2.19 ± 0.06 (IL-10−/− + vehicle), and 3.65 ± 0.45 (IL-10−/− + TNF-α) in the presence of l-NAME + indomethacin.
We were surprised to find contractile responses to ET-1 in thoracic aorta from TNF-α-infused IL-10−/− mice, since evidence from the literature shows that mouse thoracic aorta does not develop force in response to ET-1 (52).
Addition of atrasentan (1 μM), a selective ETA antagonist, almost completely abolished ET-1 contraction in first-order mesenteric arteries and aorta (Fig. 3).
Fig. 3.

Contractile responses to ET-1 are mediated by ET type A (ETA) receptor activation. Responses of small mesenteric arteries (A) and aortas (B) to ET-1 were determined in the absence or presence of atrasentan (1 μM), a selective ETA receptor antagonist. Values are means ± SE (n = 6 experiments). *P < 0.05 vs. ET-1.
Since activation of ETB receptors can also cause vasoconstriction, concentration-response curves to IRL-1620 (0.1 nM–0.1 μM), an ETB receptor agonist, were determined in first-order mesenteric arteries and aorta (data not shown). IRL-1620 did not produce a significant contractile response in these vascular segments.
In addition, concentration-response curves to PE were determined in all groups. In aortas, TNF-α infusion slightly increased PE-induced contractions in WT and IL-10−/− mice. However, contractions to PE were slightly greater in aortas from TNF-α-infused WT than IL-10−/− mice. No differences in PE responses in small mesenteric arteries were observed among the groups in the absence or presence of l-NAME + indomethacin (data not shown). These data indicate that the increased responses to ET-1 displayed by TNF-α-infused IL-10−/− mice are selective to ET-1.
ETA receptor expression.
To determine whether increased responses to ET-1 in arteries from TNF-α-infused IL-10−/− mice are associated with changes in ETA receptor expression, protein levels of ETA receptor were evaluated by Western blot analysis. As shown in Fig. 4, ETA receptor expression was increased in arteries from IL-10−/− mice. Infusion of TNF-α, in WT or IL-10−/− mice, did not change vascular ETA receptor expression. These data support the idea that ETA is the main receptor recruited in ET-1-induced contractile response but that augmented expression of ETA receptors per se is not responsible for the increased response to ET-1 in TNF-α-infused IL-10−/− mice.
Fig. 4.

ETA receptor expression is augmented in vessels from IL-10−/− mice. Top: representative immunoblots for ETA receptor and β-actin expression in murine aorta. Bottom: corresponding bar graphs demonstrating regulatory role of IL-10 in ETA receptor expression. Densitometric analysis was performed in samples from vehicle- and TNF-α-infused WT and vehicle- and TNF-α-infused IL-10−/− mice. Values, expressed in arbitrary units, are means ± SE (n = 5 experiments) and were normalized to β-actin protein expression. *P < 0.05 vs. respective control (i.e., WT).
ERK1/2 activation and ET-1 contractile responses.
We then determined whether increased responses to ET-1 in TNF-α-infused IL-10−/− mice are related to upregulation of intracellular signaling pathways. Because the ERK1/2 MAPK represents a point of convergence in signaling activated by IL-10 and ET-1, we evaluated the effects of an ERK1/2 inhibitor, PD-98059, on ET-1-induced contraction, as well as ERK1/2 expression and activity in vessels from WT and IL-10−/− animals.
Incubation with 1 μM PD-98059 drastically reduced responses to ET-1 in aorta and first-order mesenteric arteries from TNF-α-infused IL-10−/− mice (Fig. 5). In the presence of PD-98059, no contractile responses were observed in aortas from TNF-α-infused IL-10−/− mice, and contractile responses to ET-1 were similar among mesenteric arteries from all groups.
As shown at Fig. 6, arteries from IL-10−/− mice express increased levels of p44/42 compared with arteries from their respective WT controls. TNF-α infusion did not significantly alter ERK1/2 expression in WT mice (Fig. 6A). Arteries from IL-10−/− mice also exhibited increased levels of phosphorylated ERK1/2. Infusion of TNF-α further augmented ERK1/2 phosphorylation in vessels from IL-10−/−, but not WT, mice (Fig. 6B). The ratio of phosphorylated ERK1/2 to total ERK1/2 protein levels shows that ERK1/2 activity is increased only in arteries from TNF-α-infused IL-10−/− mice (Fig. 6C). These results clearly indicate that arteries from TNF-α-infused IL-10−/− mice display increased activity of the ERK1/2 pathway.
Fig. 6.

ERK1/2 activity is augmented in vessels from TNF-α-infused IL-10−/− mice. A and B: representative immunoblots for total ERK1/2, phosphorylated ERK1/2, and β-actin expression in murine aorta (top) and corresponding bar graphs (bottom) demonstrating regulatory role of IL-10 on total and phosphorylated ERK1/2. C: ratio of phosphorylated to total ERK1/2 vascular proteins. Densitometric analysis was performed in samples from vehicle- and TNF-α-infused WT and vehicle- and TNF-α-infused IL-10−/− mice. Values, expressed in arbitrary units, are means ± SE (n = 5 experiments) and were normalized to β-actin protein expression. *P < 0.05 vs. respective control (i.e., WT). †P < 0.05 vs. respective vehicle-infused group.
DISCUSSION
The main findings of the present study are as follows: 1) chronic infusion of TNF-α increases MAP, as well as prepro-ET-1 levels; 2) vascular reactivity to ET-1 is increased in arteries from TNF-α-infused IL-10−/− mice, via ETA receptor and ERK1/2-dependent mechanisms; 3) the ERK1/2 pathway is upregulated in IL-10−/− mice, and this increased activity is further augmented after TNF-α infusion.
TNF-α infusion caused a temporary hypertensive effect in the WT mice (from day 5 to 11), and after day 12 the changes in blood pressure were restored to normal, in agreement with studies performed in female rats (8, 23). TNF-α infusion significantly increased MAP in IL-10−/− mice (from day 7 to 14), but MAP remained below the levels in vehicle-infused WT mice. Recently, Nonaka-Sarukawa and colleagues (32) showed that the anti-inflammatory cytokine IL-10 has a significant antihypertensive effect. Similarly, serum IL-10 levels are decreased in congestive heart failure patients (38), and exogenous IL-10 administration retards progression of vascular injury in many cardiovascular disease models (20). Interestingly, resting MAP was lower in IL-10−/− mice, which may be associated with changes in blood volume or nonspecific effects of IL-10 gene ablation. Accordingly, IL-10-deficient animals display a decrease in the number of blood erythrocytes and decreased hemoglobin concentration. Since blood volume depends on the amount of plasma and red blood cells, this may be a factor contributing to lower blood pressure in IL-10−/− mice. In addition, these animals are prone to infection, and the reduction in MAP may be due to nonspecific effects. Therefore, more detailed studies are needed to better characterize whether IL-10 has antihypertensive properties in this experimental model.
TNF-α was chronically infused to induce increased ET-1 expression, as previously described (2, 10, 18, 29, 47, 51). We used prepro-ET-1 mRNA as a biomarker for ET-1 levels, since increased ET-1 plasma values may not reflect local production (12, 30).
In TNF-α-infused IL-10−/− mice, increased vascular contractile responses to ET-1 were observed in small mesenteric arteries and thoracic aorta. We were surprised to find contractile responses only in thoracic aortas from TNF-α-infused IL-10−/− mice. The lack of response to ET-1 in murine aorta in the present study is consistent with a report from Zhou and colleagues (52) that ET-1 induces responses in abdominal, but not thoracic, aorta from mice. Moreover, Russell and Watts (36) showed a weak response to ET-1 by thoracic aorta from mice. Although in control conditions, increased contractile responses to ET-1 were observed in aortas, but not small mesenteric arteries, from TNF-α-infused IL-10−/− mice in the presence of l-NAME and indomethacin, aortas and small mesenteric arteries exhibited augmented contractions to ET-1. These results show that responses to ET-1 are altered in a different pattern in conductance and resistance arteries and suggest that, when NO bioavailability is decreased, the effects of ET-1 will be further augmented. Accordingly, counterregulatory interactions between the NO and ET systems are well established (26, 34).
Consistent with previous studies (3, 15, 16, 42), we have found that IL-10 plays a protective role in the alterations of vascular reactivity. Regional differences in cytokine-induced release and responsiveness to mediators appear to contribute to the dilator or constrictor responses observed in a specific vascular bed (42). Vascular cells may also respond to anti-inflammatory counterregulatory mechanisms, including anti-inflammatory cytokines, which maintain vascular wall integrity and homeostasis (40, 42). IL-10, one of the main anti-inflammatory cytokines, displays vasculoprotective properties (42), and we can speculate that IL-10 is playing an important role, i.e., preventing ET-1-induced vascular injuries. TNF-α and ET-1 are described to augment IL-10 levels. Accordingly, TNF-α stimulates IL-10 production by macrophages (28), and treatment of mice with ETA/ETB antagonist significantly reduces IL-10 levels (41). Additionally, TNF-α and ET-1 activate MAPKs, which can regulate IL-10 promoter activity and increase IL-10 production (11, 24).
One reservation in the present study is that knockout animals are likely to develop compensatory mechanisms after ablation of a given gene. Rescue experiments, where the gene product (IL-10 in the present study) is made available to the animal for a limited/specific period of time or, alternatively, chronic pharmacological treatment with antibodies against IL-10 is used to mimic the phenotype or the functional changes displayed by the knockout animal, may partially overcome this concern. We have characterized the mechanisms involved in this unusual response to ET-1. Several reports have shown that IL-10 can negatively regulate proinflammatory signaling pathways via activation of Janus kinases/suppressor of cytokine signaling-3. However, the NF-κB and MAPK pathways also seem important for the anti-inflammatory actions of IL-10. IL-10 blocks activation of NF-κB induced by lipopolysaccharide, TNF-α, and reactive oxygen species (9, 37). IL-10 also suppresses cyclooxgenase-2 expression by downregulation of p38 MAPK and ERK2 (31). Moreover, several reports have shown that IL-10 inhibits ERK1/2 activity and that this is important, for example, for the downregulation of polymorphonuclear neutrophil functions and CD40-mediated induction of IL-1β and TNF-α synthesis by monocytes (6, 33, 39).
Because ET-1 activates ERK1/2 to promote changes in vascular reactivity, remodeling, inflammation, and oxidative stress (7, 19, 21, 48), we hypothesized that the protective actions of IL-10 on ET-1-induced changes in vascular reactivity occur via downregulation of ERK1/2 activity. The immunoblot data showed that the lack of IL-10 leads to increased vascular expression of total and phosphorylated ERK1/2 and that infusion of TNF-α further increased vascular ERK1/2 activity in IL-10−/− mice. In these mice, contractile responses to ET-1 were augmented in two different vessels, mesenteric resistance arteries and thoracic aorta. Furthermore, PD-98059, an ERK1/2 inhibitor, completely abolished the increased responses to ET-1 in both vessels, demonstrating that ERK1/2 is recruited in this abnormal response and suggesting that increased ERK1/2 activity results in augmented contractile responses to ET-1. In this sense, it is known that ERK1/2 phosphorylates caldesmon and calponin, blocking their ability to inhibit the actin-myosin interaction, leading to augmented contractions (22, 25, 35). In addition, inhibition of ERK attenuates ET-1-induced force development in porcine carotid artery by lowering myosin light chain phosphorylation (5).
The following considerations might suggest that TNF-α may be modifying other mechanisms that enable exacerbated contractions to ET-1: 1) TNF-α infusion similarly upregulates vascular prepro-ET-1 expression in WT and IL-10−/− mice; 2) ETA receptor expression, as well as ERK1/2 activation, is increased in arteries from IL-10−/−, but not WT, mice; and 3) ET-1-induced responses are increased only in TNF-α-infused IL-10−/− mice. Possible mechanisms include TNF-α-induced changes in NO bioavailability, as previously discussed. In addition, if we consider that MAPK signaling is regulated through phosphorylation and dephosphorylation by kinases and phosphatases, respectively, it is possible that TNF-α, along with IL-10 ablation, increases ERK1/2 phosphorylation by increasing MEK1 activity or by attenuating MAPK phosphatase-1 activity.
Since ETA and ETB receptors, by different mechanisms, can lead to an activation of ERK1/2 (13), we determined which receptor subtype was involved in ET-1-induced vascular contractions in these preparations. In accordance with a previous report (52), our data suggest that the ETA receptor is involved in the increased response to ET-1 in vessels from TNF-α-infused IL-10−/− mice, since ETA blockade with atrasentan was able to completely abolish ET-1 responses and no contraction was observed on stimulation with the ETB agonist IRL-1620.
Collectively, these findings demonstrate that IL-10 attenuates ETA receptor-mediated abnormal vascular responses to ET-1, induced by TNF-α infusion, by a negative modulation of ERK1/2 activity. This is a novel mechanism for the vasculoprotective effects of a classic anti-inflammatory cytokine.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL-71138 and HL-74167 and Fundacao de Amparo a Pesquisa do Estado de Sao Paulo Grant 2006/01773-0.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy—review of a new approach. Pharmacol Rev 55: 241–269, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Boldrini L, Gisfredi S, Ursino S, Lucchi M, Melfi F, Mussi A, Basolo F, Fontanini G. Tumour necrosis factor-α: prognostic role and relationship with interleukin-8 and endothelin-1 in non-small cell lung cancer. Int J Mol Med 17: 887–892, 2006. [PubMed] [Google Scholar]
- 3.Chen S, Kapturczak MH, Wasserfall C, Glushakova OY, Campbell-Thompson M, Deshane JS, Joseph R, Cruz PE, Hauswirth WW, Madsen KM, Croker BP, Berns KI, Atkinson MA, Flotte TR, Tisher CC, Agarwal A. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc Natl Acad Sci USA 102: 7251–7256, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corder R, Carrier M, Khan N, Klemm P, Vane JR. Cytokine regulation of endothelin-1 release from bovine aortic endothelial cells. J Cardiovasc Pharmacol 26 Suppl 3: S56–S58, 1995. [PubMed] [Google Scholar]
- 5.D'Angelo G, Adam LP. Inhibition of ERK attenuates force development by lowering myosin light chain phosphorylation. Am J Physiol Heart Circ Physiol 282: H602–H610, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Dang PM, Elbim C, Marie JC, Chiandotto M, Gougerot-Pocidalo MA, El-Benna J. Anti-inflammatory effect of interleukin-10 on human neutrophil respiratory burst involves inhibition of GM-CSF-induced p47PHOX phosphorylation through a decrease in ERK1/2 activity. FASEB J 20: 1504–1506, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Daou GB, Srivastava AK. Reactive oxygen species mediate endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 signaling, as well as protein synthesis, in vascular smooth muscle cells. Free Radic Biol Med 37: 208–215, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Davis JR, Giardina JB, Green GM, Alexander BT, Granger JP, Khalil RA. Reduced endothelial NO-cGMP vascular relaxation pathway during TNF-α-induced hypertension in pregnant rats. Am J Physiol Regul Integr Comp Physiol 282: R390–R399, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Dokka S, Shi X, Leonard S, Wang L, Castranova V, Rojanasakul Y. Interleukin-10-mediated inhibition of free radical generation in macrophages. Am J Physiol Lung Cell Mol Physiol 280: L1196–L1202, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Finsnes F, Skjonsberg OH, Lyberg T, Christensen G. Endothelin-1 production is associated with eosinophilic rather than neutrophilic airway inflammation. Eur Respir J 15: 743–750, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Gee K, Angel JB, Ma W, Mishra S, Gajanayaka N, Parato K, Kumar A. Intracellular HIV-Tat expression induces IL-10 synthesis by the CREB-1 transcription factor through Ser133 phosphorylation and its regulation by the ERK1/2 MAPK in human monocytic cells. J Biol Chem 281: 31647–31658, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Giachini FR, Callera GE, Carneiro FS, Tostes RC, Webb RC. Therapeutic targets in hypertension: is there a place for antagonists of the most potent vasoconstrictors? Expert Opin Ther Targets 12: 327–339, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Grantcharova E, Reusch HP, Beyermann M, Rosenthal W, Oksche A. Endothelin A and endothelin B receptors differ in their ability to stimulate ERK1/2 activation. Exp Biol Med (Maywood) 231: 757–760, 2006. [PubMed] [Google Scholar]
- 14.Groux H, Cottrez F, Rouleau M, Mauze S, Antonenko S, Hurst S, McNeil T, Bigler M, Roncarolo MG, Coffman RL. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J Immunol 162: 1723–1729, 1999. [PubMed] [Google Scholar]
- 15.Gunnett CA, Heistad DD, Berg DJ, Faraci FM. IL-10 deficiency increases superoxide and endothelial dysfunction during inflammation. Am J Physiol Heart Circ Physiol 279: H1555–H1562, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Gunnett CA, Heistad DD, Faraci FM. Interleukin-10 protects nitric oxide-dependent relaxation during diabetes: role of superoxide. Diabetes 51: 1931–1937, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Hagenbaugh A, Sharma S, Dubinett SM, Wei SH, Aranda R, Cheroutre H, Fowell DJ, Binder S, Tsao B, Locksley RM, Moore KW, Kronenberg M. Altered immune responses in interleukin 10 transgenic mice. J Exp Med 185: 2101–2110, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hohlfeld T, Klemm P, Thiemermann C, Warner TD, Schror K, Vane JR. The contribution of tumour necrosis factor-α and endothelin-1 to the increase of coronary resistance in hearts from rats treated with endotoxin. Br J Pharmacol 116: 3309–3315, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishihata A, Tasaki K, Katano Y. Involvement of p44/42 mitogen-activated protein kinases in regulating angiotensin II- and endothelin-1-induced contraction of rat thoracic aorta. Eur J Pharmacol 445: 247–256, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Ikeda U. Inflammatory cytokines and cardiovascular disease. Curr Drug Targets Inflamm Allergy 2: 257–265, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Endothelin-mediated vascular growth requires p42/p44 mitogen-activated protein kinase and p70 S6 kinase cascades via transactivation of epidermal growth factor receptor. Endocrinology 140: 4659–4668, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Je HD, Gangopadhyay SS, Ashworth TD, Morgan KG. Calponin is required for agonist-induced signal transduction—evidence from an antisense approach in ferret smooth muscle. J Physiol 537: 567–577, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension 46: 82–86, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Leghmari K, Contreras X, Moureau C, Bahraoui E. HIV-1 Tat protein induces TNF-α and IL-10 production by human macrophages: differential implication of PKC-βII and -δ isozymes and MAP kinases ERK1/2 and p38. Cell Immunol 254: 46–55, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Leinweber BD, Leavis PC, Grabarek Z, Wang CL, Morgan KG. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem J 344: 117–123, 1999. [PMC free article] [PubMed] [Google Scholar]
- 26.Luscher TF, Yang Z, Tschudi M, von Segesser L, Stulz P, Boulanger C, Siebenmann R, Turina M, Buhler FR. Interaction between endothelin-1 and endothelium-derived relaxing factor in human arteries and veins. Circ Res 66: 1088–1094, 1990. [DOI] [PubMed] [Google Scholar]
- 27.Mazighi M, Pelle A, Gonzalez W, Mtairag el M, Philippe M, Henin D, Michel JB, Feldman LJ. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am J Physiol Heart Circ Physiol 287: H866–H871, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Meisel C, Vogt K, Platzer C, Randow F, Liebenthal C, Volk HD. Differential regulation of monocytic tumor necrosis factor-α and interleukin-10 expression. Eur J Immunol 26: 1580–1586, 1996. [DOI] [PubMed] [Google Scholar]
- 29.Molet S, Furukawa K, Maghazechi A, Hamid Q, Giaid A. Chemokine- and cytokine-induced expression of endothelin 1 and endothelin-converting enzyme 1 in endothelial cells. J Allergy Clin Immunol 105: 333–338, 2000. [DOI] [PubMed] [Google Scholar]
- 30.Naya M, Tsukamoto T, Morita K, Katoh C, Furumoto T, Fujii S, Tamaki N, Tsutsui H. Plasma interleukin-6 and tumor necrosis factor-α can predict coronary endothelial dysfunction in hypertensive patients. Hypertens Res 30: 541–548, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Niiro H, Otsuka T, Ogami E, Yamaoka K, Nagano S, Akahoshi M, Nakashima H, Arinobu Y, Izuhara K, Niho Y. MAP kinase pathways as a route for regulatory mechanisms of IL-10 and IL-4 which inhibit COX-2 expression in human monocytes. Biochem Biophys Res Commun 250: 200–205, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Nonaka-Sarukawa M, Okada T, Ito T, Yamamoto K, Yoshioka T, Nomoto T, Hojo Y, Shimpo M, Urabe M, Mizukami H, Kume A, Ikeda U, Shimada K, Ozawa K. Adeno-associated virus vector-mediated systemic interleukin-10 expression ameliorates hypertensive organ damage in Dahl salt-sensitive rats. J Gene Med 10: 368–374, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Park PH, McMullen MR, Huang H, Thakur V, Nagy LE. Short-term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor-α (TNF-α) expression via ERK1/2 activation and Egr-1 expression: role of TNF-α in adiponectin-stimulated interleukin-10 production. J Biol Chem 282: 21695–21703, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quaschning T, Kocak S, Bauer C, Neumayer HH, Galle J, Hocher B. Increase in nitric oxide bioavailability improves endothelial function in endothelin-1 transgenic mice. Nephrol Dial Transplant 18: 479–483, 2003. [DOI] [PubMed] [Google Scholar]
- 35.Rokolya A, Singer HA. Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am J Physiol Cell Physiol 278: C537–C545, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Russell A, Watts S. Vascular reactivity of isolated thoracic aorta of the C57BL/6J mouse. J Pharmacol Exp Ther 294: 598–604, 2000. [PubMed] [Google Scholar]
- 37.Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS Jr. Interleukin-10 signaling blocks inhibitor of κB kinase activity and nuclear factor κB DNA binding. J Biol Chem 274: 31868–31874, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Stumpf C, Lehner C, Yilmaz A, Daniel WG, Garlichs CD. Decrease of serum levels of the anti-inflammatory cytokine interleukin-10 in patients with advanced chronic heart failure. Clin Sci (Lond) 105: 45–50, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Suttles J, Milhorn DM, Miller RW, Poe JC, Wahl LM, Stout RD. CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (IL)-4 and IL-10 anti-inflammatory action. J Biol Chem 274: 5835–5842, 1999. [DOI] [PubMed] [Google Scholar]
- 40.Tedgui A, Mallat Z. Anti-inflammatory mechanisms in the vascular wall. Circ Res 88: 877–887, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Todd KE, Lewis MP, Gloor B, Lane JS, Ashley SW, Reber HA. An ETa/ETb endothelin antagonist ameliorates systemic inflammation in a murine model of acute hemorrhagic pancreatitis. Surgery 122: 443–450, 1997. [DOI] [PubMed] [Google Scholar]
- 42.Vila E, Salaices M. Cytokines and vascular reactivity in resistance arteries. Am J Physiol Heart Circ Physiol 288: H1016–H1021, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Wagner EM TNF-α-induced bronchial vasoconstriction. Am J Physiol Heart Circ Physiol 279: H946–H951, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Wen Y, Wang JY, Liu P. [Tumor necrosis factor-α enhances the effect of endothelin on renal vasoconstriction in isolated perfused rat kidney]. Zhonghua Gan Zang Bing Za Zhi 11: 583–585, 2003. [PubMed] [Google Scholar]
- 45.Woods M, Mitchell JA, Wood EG, Barker S, Walcot NR, Rees GM, Warner TD. Endothelin-1 is induced by cytokines in human vascular smooth muscle cells: evidence for intracellular endothelin-converting enzyme. Mol Pharmacol 55: 902–909, 1999. [PubMed] [Google Scholar]
- 46.Woods M, Wood EG, Mitchell JA, Warner TD. Signal transduction pathways involved in cytokine stimulation of endothelin-1 release from human vascular smooth muscle cells. J Cardiovasc Pharmacol 36: S407–S409, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Xu J, Zhong NS. The interaction of tumour necrosis factor α and endothelin-1 in pathogenetic models of asthma. Clin Exp Allergy 27: 568–573, 1997. [PubMed] [Google Scholar]
- 48.Yogi A, Callera GE, Montezano AC, Aranha AB, Tostes RC, Schiffrin EL, Touyz RM. Endothelin-1, but not ANG II, activates MAP kinases through c-Src independent Ras-Raf dependent pathways in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27: 1960–1967, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Zemse SM, Hilgers RH, Simkins GB, Rudic RD, Webb RC. Restoration of endothelin-1-induced impairment in endothelium-dependent relaxation by interleukin-10 in murine aortic rings. Can J Physiol Pharmacol 86: 557–565, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zemse SM, Hilgers RH, Webb RC. Interleukin-10 counteracts impaired endothelium-dependent relaxation induced by ANG II in murine aortic rings. Am J Physiol Heart Circ Physiol 292: H3103–H3108, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Zhao RZ, Chen X, Yao Q, Chen C. TNF-α induces interleukin-8 and endothelin-1 expression in human endothelial cells with different redox pathways. Biochem Biophys Res Commun 327: 985–992, 2005. [DOI] [PubMed] [Google Scholar]
- 52.Zhou Y, Dirksen WP, Zweier JL, Periasamy M. Endothelin-1-induced responses in isolated mouse vessels: the expression and function of receptor types. Am J Physiol Heart Circ Physiol 287: H573–H578, 2004. [DOI] [PubMed] [Google Scholar]