

Abstract
The Lipid Metabolites and Pathway Strategy (LIPID MAPS) Consortium is a nationwide initiative that has taken on the task of employing lipidomics to advance our understanding of lipid metabolism at the molecular and mechanistic level in living organisms. An important step toward this goal is to craft enabling analytical procedures to comprehensively measure all lipid species, to establish the precise structural identity of the lipid molecules analyzed and to generate accurate quantitative information. The LIPID MAPS Consortium has succeeded in the implementation of a complete infrastructure that now provides the tools for the analysis of the global lipidome in cultured and primary cells. Here we illustrate the advancement of a gas chromatography mass spectrometry (GC/MS) procedure for the analysis of essential fatty acids in RAW 264.7 cells. Our method allows for the specific identification and quantification of over thirty fatty acids present in cells in their free form in a single analytical GC/MS run. Free fatty acids are selectively extracted in the presence of deuterated internal standards that permit subsequent estimation of extraction efficiencies and quantification with high accuracy. Mass spectrometer conditions were optimized for single ion monitoring, which provides an extremely sensitive technology to measure fatty acids from biological samples in trace amounts. These methods will be presented in the context of our broader effort to analyze all fatty acids as well as their metabolites in inflammatory cells.
Introduction
There is an increasing appreciation of the essential roles of lipids in cellular metabolism and signaling that has replaced the notion that lipids only function as an energy reservoir or structural components for cells. As an integral building block of semi-permeable membranes, lipids not only form barriers between cells and extracellular space and between intracellular organelle compartments but they also affect directly the physical and functional properties of membranes [1]. Lipids adjust membrane fluidity, support diverse cellular processes and furnish the physical properties required for rapid membrane fission and fusion during vesicular transport between compartments of the exocytic and endocytic pathways. Specialized lipids serve as pigments, cofactors and detergents. As hormones and cellular messengers, lipids are involved also in many intracellular and intercellular signaling processes. An array of enzymes including phospholipases, kinases and cyclooxygenases can act upon cellular lipids to produce secondary messengers and bioactive metabolites that target the same cell in an autocrine fashion or serve as a matrix for intercellular communication [2-4]. Generally, lipids are not static but change dynamically during normal metabolism or in response to cellular stimuli. Lipids turn over and are biosynthetically remodeled during cell cycle or undergo chemical restructuring to new lipid species with altered properties that serve as bioactive mediators in various signaling pathways. The rapid dynamic adds to the complexity of lipid metabolism. Attempts to produce practical model systems integrating the pathways of all lipids and lipid metabolites into a cellular metabolic network require a series of temporal snapshots of the global lipidome. However, a comprehensive analysis of all cellular lipids has been challenging due to their chemical and physical diversity as well as absence of reliable methods to extract all the lipid species from biological samples. Furthermore, well characterized standards are required to determine the structural identity and to establish quantitatively the amount of each lipid species. The LIPID MAPS Consortium (www.lipidmaps.org) has addressed these issues and has developed the infrastructure to carry out a systematic analysis of all cellular lipids [5]. Systems biology approaches employed by the LIPID MAPS Consortium are designed to quantify temporal and spatial changes in lipids during cell metabolism under normal and pathophysiological conditions of inflammation and to integrate the lipids and metabolites into a dynamic cellular network [6]. In this report, we will focus on the analysis of free fatty acid profiles in macrophages.
Fatty acids represent an elemental class of lipids that are components of all membrane lipids except the sterols, but even they exist also as cholesterol esters. They are integral to membrane structure and represent an important determinant of membrane fluidity and function [7,8]. Acyl chains determine the activities of membrane associated enzymes as well as receptors and influence transport, endocytosis and exocytosis [9]. Fatty acids are also a major source of energy and are efficient forms of energy storage in the mammalian organism. More recently, fatty acids have been implicated in the pathology of various disorders including diabetes [10,11], cystic fibrosis [12,13], asthma [14], allergy [15], cancer [16,17] and atherosclerosis [18]. Fatty acids also play an important role in innate immunity. It is now well established that the endotoxic activity of lipopolysaccharide (LPS) from Gram-negative bacteria resides within the O-acylated saturated fatty acids of the Lipid A moiety [19]. These acylated fatty acids are critical to ligand recognition and activation of toll-like receptors (TLRs) on macrophages. Moreover, previous studies demonstrated that saturated fatty acids in their free form directly activate TLR4 while polyunsaturated fatty acids inhibit the immune response to LPS [20]. Given this wide physiological activity, enormous efforts are dedicated to develop methodologies to reliably assess fatty acid composition and metabolism in biological tissues.
Fatty acids show large diversity in structure and physical properties ranging from relatively simple long-chain saturated structures to more complex unsaturated, cyclic or branched configurations. Functional oxygen substituents derived from biochemical oxidation of polyunsaturated fatty acids such as hydroxyl, peroxide, epoxy or keto groups add further to the heterogeneity. Historically, gas chromatography (GC) is the method of choice to separate this composite mixture of fatty acids [21]. Many of the early analytical attempts used flame ionization detector methods. The subsequent coupling of GC with mass spectroscopy (MS) increased sensitivity and additionally provided structural information [22,23]. The combination of GC/MS has become a prominent analytical technique to address structural issues associated with fatty acids such as the number and position of double bonds, branching sites, and the type and position of functional groups. For GC analysis, fatty acids must be derivatized to alkyl derivatives to increase volatility. The derivatization procedure has the potential to introduce unwanted oxidation or isomerization of some fatty acids. To prevent such changes and to streamline sample preparation, liquid chromatography (LC) protocols were developed for the separation of free fatty acids in their non-derivatized form. Initially, these procedures were coupled to UV-detectors. However, a major difficulty arose in the quantitation of non-derivatized fatty acids due to the absence of chromophores that facilitate their detection by UV absorption with reasonable sensitivity. This limitation severely restricts the application and this methodology is not useful for the analysis of trace levels of fatty acids in many biological, biomedical, food and environmental samples. Derivatization with suitable chromophores improved the detection sensitivity but complicated sample preparation [24]. Recent advances in ionization techniques such as electrospray ionization and atmospheric pressure chemical ionization establishes MS as an attractive option to facilitate fatty acid analysis without derivatization. MS detection resolves the issue of detection sensitivity and LC/MS has been employed for the analysis of free fatty acids without pre-column derivatization [25,26]. These LC/MS methodologies provide a cost effective alternative to GC/MS and have been used predominantly for the analysis of selected fatty acids in food samples. Similarly, non-derivatization strategies are also used for structural studies of individual fatty acids but are presently not in use to establish complete fatty acid profiles in biological samples.
As part of the LIPID MAPS Consortium, we developed a GC/MS method for the separation, identification and quantitation of essential fatty acids in cultures of macrophages. The majority of fatty acid in these cells exists as esters or amides in lipids. However, a small but significant portion of the total fatty acids are present in pools of non-esterified or free fatty acids. Free fatty acids, and in particular free arachidonic acid, have received enormous attention due to the important roles as precursors of lipid-derived mediators in disease and inflammation [27]. Recently, we have developed comprehensive procedures to lipidomics analysis of eicosanoids, including their numerous metabolites derived from an array of cyclooxygenases, lipoxygenases, cytochrome P450 and non-enzymatic oxidation [28-30]. We used a similar approach to monitor complete free fatty acid profiles in macrophages. We developed a stable isotope dilution GC/MS method able to detect over thirty saturated and unsaturated fatty acids in the macrophage in a single run. The methodology employs a rapid extraction procedure of free fatty acids from cell cultures followed by a short derivatization step. We found that the extraction was complete and validated the methodology with a detection limit in the fmol to sub-fmol range. We used cultures of macrophages as a model system but the technique is adaptable to other tissues.
Materials and Methods
Cell preparation and fatty acid extraction
RAW 264.7 macrophages were grown to 80% confluency in high glucose DMEM without phenol red (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Hyclone, Thermo Fisher Scientific, Waltham, MA) and 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). The cells were maintained at 37 °C in a humidified 5% CO2/air environment. The cells were harvested by scraping into PBS and counted. For normalization purpose, cellular DNA content was determined using the Quant-iT DNA assay kit according to the manufacturer (Invitrogen). About 0.5×106 cells in a total volume of 250 μl PBS are used for fatty acid analysis. To the cell suspension, 100 μl of a complete set of deuterated internal standards containing each standard at 25 ng/100 μl ethanol was added. All solvents for subsequent extraction and analysis were of highest purity and purchased from EMD Chemicals, Inc. (Gibbstown, NJ) or Fisher Scientific (Pittsburgh, PA). For extraction of free fatty acids, 500 μl of methanol and 25 μl of 1 N HCl were added and a bi-phasic solution was formed by addition of 1.5 ml of isooctane. This solution was vortexed for 30 sec and the phases were separated by centrifugation at 3000 rpm for 2 min. The upper isooctane phase was removed and the extraction was repeated once with an additional 1.5 ml of isooctane. The combined isooctane layers were evaporated to dryness (10 min, room temperature) using a vacuum evaporator.
Fatty acid derivatization
The free fatty acids were analyzed as their pentafluorobenzyl (PFB) bromide derivatives in negative ionization mode [31]. The extracted free fatty acids were taken up in 25 μl of 1% diisopropylethylamine in acetonitrile and derivatized with 25 μl 1% PFB bromide (Sigma-Aldrich, St. Louis, MO) in acetonitrile at room temperature for 20 min in capped glass tubes. The solvent was removed by vacuum evaporator, the residue was dissolved in 50 μl isooctane and 1 μl of the PFB esters was analyzed by GC electron capture MS (GC/EC/MS).
Internal standards and preparation of fatty acid standard curves
Fatty acid quantitation was achieved by the stable isotope dilution method. Two series of standards were prepared from stocks of deuterated and unlabeled fatty acids. First, for each fatty acid a deuterated analog was selected and purchased either from Cayman Chemical Co. (Ann Arbor, MI), from CDN Isotopes Inc. (Pointe-Claire, Canada), or from Cambridge Isotope Laboratories Inc. (Andover, MA). In some cases when a deuterated fatty acid was not commercially available, we employed the deuterated analog with the closest chemical characteristics. Presently, we use 15 deuterated standards to quantitate 31 fatty acids (a list of the deuterated standards is given in Table 1). A stock is made in ethanol that contains each deuterated standard fatty acid at 0.25 ng/μl. All samples and primary standards are spiked with 100 μl of the mixture (25 ng of each deuterated standard fatty acid). Second, standard curves were prepared from dilution of stock solutions of unlabeled primary standards at exact concentrations. The diluted sets of primary standards contained 31 fatty acids in the range of 0.15-500 ng to which 100 μl (25 ng/standard) of the internal standard mixture was added (Table 1). The PFB derivatives were prepared as described above. A standard curve was generated by linear regression analysis of the ratio between primary standard peak area and internal standard peak area plotted versus the amount of primary standard. Figure 2 shows an example of typical standard curves. The fatty acid content in the biological sample was calculated from the standard curve using the ratio between analyte peak area and internal standard peak area.
Table 1.
Experimental conditions and validation data for the GC/MS analysis of fatty acids.
| Precision (RSD)d | ||||||||
|---|---|---|---|---|---|---|---|---|
| Fatty Acid | [M-H]-m/z | tRa (min) | INT STDb | SIM Group | LODc (pg) | Intra-day | Inter-day | R2e |
| Lauric Acid (12:0) | 199 | 3.84 | 12:0-d3 | 1 | 0.1 | 12 | 10 | 0.998 |
| 12:0-d3 | 202 | 3.81 | - | 1 | - | - | - | - |
| Myristic Acid (14:0) | 227 | 5.35 | 14:0-d3 | 2 | 0.1 | 5 | 9 | 0.999 |
| 14:0-d3 | 230 | 5.33 | - | 2 | - | - | - | - |
| Pentadecanoic Acid (15:0) | 241 | 6.11 | 15:0-d3 | 2 | 0.1 | 10 | 7 | 0.999 |
| 15:0-d3 | 244 | 6.09 | 2 | - | - | - | - | |
| Palmitic Acid (16:0) | 255 | 6.87 | 16:0-d3 | 3 | 0.1 | 8 | 8 | 0.992 |
| 16:0-d3 | 258 | 6.84 | - | 3 | - | - | - | - |
| Palmitoleic Acid (16:1) | 253 | 6.67 | 16:0-d3 | 3 | 0.75 | 9 | 15 | 0.994 |
| Margaric Acid (17:0) | 269 | 7.60 | 17:0-d3 | 3 | 0.1 | 9 | 15 | 0.999 |
| 17:0-d3 | 272 | 7.58 | - | 3 | - | - | - | - |
| Heptadecaenoic Acid (17:1) | 267 | 7.42 | 17:0-d3 | 3 | 5 | 12 | 16 | 0.996 |
| Stearic Acid (18:0) | 283 | 8.32 | 18:0-d3 | 4 | 0.1 | 7 | 11 | 0.994 |
| 18:0-d3 | 286 | 8.30 | - | 4 | - | - | - | - |
| Oleic Acid (18:1) | 281 | 8.08 | 18:1-d2 | 4 | 0.1 | 8 | 12 | 0.988 |
| 18:1-d2 | 283 | 8.08 | - | 4 | - | - | - | - |
| Linoleic Acid (18:2) | 279 | 8.02 | 18:2-d4 | 4 | 0.1 | 9 | 12 | 0.983 |
| 18:2-d4 | 283 | 8.01 | - | 4 | - | - | - | - |
| α-linolenic Acid (18:3) | 277 | 7.82 | 18:2-d4 | 4 | 1 | 9 | 10 | 0.998 |
| γ-linolenic Acid (18:3) | 277 | 8.06 | 18:2-d4 | 4 | 1 | 9 | 7 | 0.998 |
| Stearidonic Acid (18:4) | 275 | 7.85 | 18:2-d4 | 4 | 5 | 6 | 8 | 0.997 |
| Arachidic Acid (20:0) | 311 | 9.69 | 20:0-d3 | 5 | 5 | 8 | 8 | 0.999 |
| 20:0-d3 | 314 | 9.68 | - | 5 | - | - | - | - |
| Eicosadienoic Acid (20:2) | 307 | 9.43 | 20:4-d8 | 5 | 5 | 7 | 12 | 0.994 |
| 11,14,17-Eicosatrienoic Acid (20:3) | 305 | 9.08 | 20:4-d8 | 5 | 5 | 6 | 10 | 0.998 |
| Bishomo-γ-linolenic Acid (20:3) | 305 | 9.24 | 20:4-d8 | 5 | 1 | 6 | 13 | 0.997 |
| 5,8,11-Eicosatrienoic Acid (20:3) | 305 | 9.46 | 20:4-d8 | 5 | 1 | 5 | 8 | 0.994 |
| Arachidonic Acid (20:4) | 303 | 9.02 | 20:4-d8 | 5 | 0.1 | 7 | 12 | 0.998 |
| 20:4-d8 | 311 | 8.99 | - | 5 | - | - | - | - |
| Eicosapentaenoic Acid (20:5) | 301 | 9.05 | 20:5-d5 | 5 | 0.5 | 7 | 8 | 0.999 |
| 20:5-d5 | 306 | 9.02 | - | 5 | - | - | - | - |
| Behenic Acid (22:0) | 339 | 10.98 | 22:0-d3 | 6 | 5 | 14 | 15 | 0.999 |
| 22:0-d3 | 342 | 10.97 | - | 6 | - | - | - | - |
| Cis-Erucic Acid (22:1) | 337 | 10.80 | 22:0-d3 | 6 | 0.75 | 9 | 9 | 0.994 |
| Docosadienoic Acid (22:2) | 335 | 10.76 | 22:0-d3 | 6 | 5 | 12 | 15 | 0.997 |
| Docosatrienoic Acid (22:3) | 333 | 10.78 | 22:6-d5 | 6 | 10 | 10 | 14 | 0.989 |
| Docosatetraenoic Acid (22:4) | 331 | 10.39 | 22:6-d5 | 6 | 5 | 8 | 15 | 0.997 |
| Docosapentaenoic Acid (22:5) | 329 | 10.42 | 22:6-d5 | 6 | 0.5 | 4 | 15 | 0.996 |
| Docosahexaenoic Acid (22:6) | 327 | 10.23 | 22:6-d5 | 6 | 0.5 | 7 | 9 | 0.998 |
| 22:6-d5 | 332 | 10.20 | - | 6 | - | - | - | - |
| Tricosanoic Acid (23:0) | 353 | 11.60 | 24:0-d4 | 7 | 0.25 | 13 | 9 | 0.999 |
| Lignoceric Acid (24:0) | 367 | 12.18 | 24:0-d4 | 7 | 1 | 7 | 8 | 0.999 |
| 24:0-d4 | 371 | 12.17 | - | 7 | - | - | - | - |
| Cis-Selacholeic Acid (24:1) | 365 | 12.04 | 24:0-d4 | 7 | 1 | 11 | 17 | 0.982 |
| Cerotic Acid (26:0) | 395 | 12.93 | 26:0-d4 | 8 | 0.5 | 14 | 10 | 0.998 |
| 26:0-d4 | 399 | 12.92 | - | 8 | - | - | - | - |
tR, retention time.
Not all deuterated standards are commercially available. Shown is the list of deuterated analogs we are currently employing as internal standards (INT STD). The INT STD are listed in reference to the corresponding fatty acids for which they were used for quantification purpose.
The lower limit of detection (LOD) refers to the actual amount loaded onto the column.
The analytical precision was determined in triplicates for both intra- and inter-day precision and expressed as the relative standard deviation (RSD).
The linearity of the response was determined for calibration curves in the range of 0.003 ng/μl to 10 ng/μl and an GC injection volume of 1 μl. Linear regression analysis was performed and the linear regression coefficient (R2) was computed.
Figure 2.
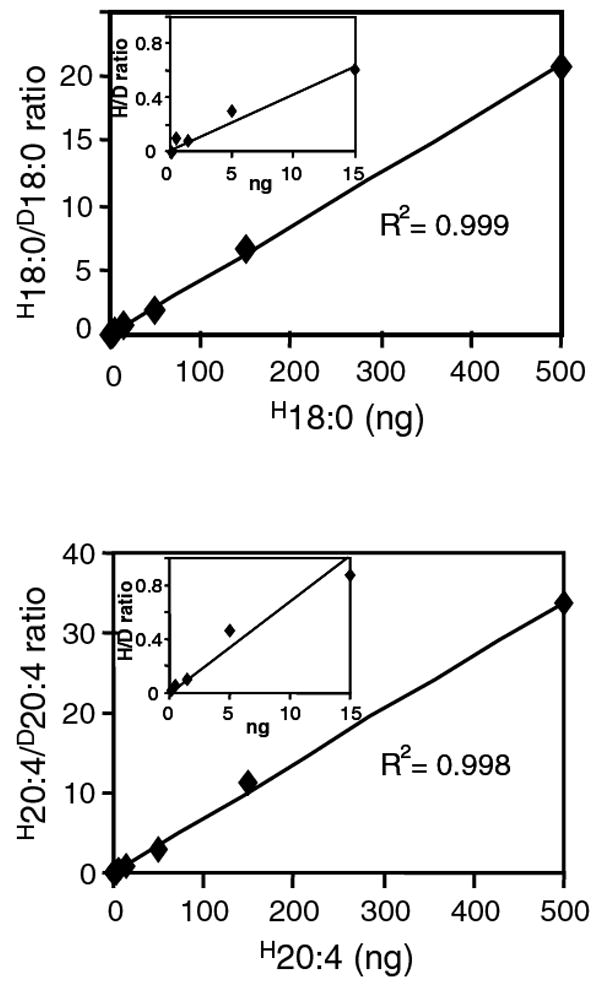
Representative fatty acid calibration curve of saturated and unsaturated fatty acids. Fatty acid standard curves were prepared for each fatty acid by injection of 1 μl of calibration standard mixture containing 0.15 ng to 500 ng of each fatty acid (0.003 ng/μl to 10 ng/μl). For regression analysis, the ratio of unlabeled fatty acid/deuterated fatty acid (H18:0/D18:0, H20:4/D20:4) was calculated as described under “Materials and Methods” and plotted against the absolute amount of unlabeled fatty acid (H18:0, H20:4) in the standard mixture. Inset shows part of the calibration curve at low analyte concentration range. Linear regression parameters were computed and used for quantitative analyses of endogenous fatty acids.
GC/MC analysis
GC/MC analysis was carried out on an Agilent 6890N gas chromatograph equipped with an Agilent 7683 autosampler (Santa Clara CA). The fatty acid esters dissolved in 50 μl isooctance were injected (1 μl) with a pulsed (25 psi) splitless injection mode onto a Zebron ZB-1 column (15 m × 0.25 mm i.d., coated with 50 um 100% dimethylpolysiloxane; Phenomenex, Torrance, CA). Helium (0.9 ml/min was used as a carrier gas. The GC oven temperature was programmed from 150°C to 270°C at 10°C/min, ramped to 240°C at 40°C/min and held at 240°C for 1 min. The injector and transfer line were kept at 250°C and 280°C respectively. Quantitative analysis of fatty acids were performed using an Agilent 5973 mass selective detector. Methane (99.99%) was used as the ionization gas with a source temperature of 150°C. Data were acquired in the selected ion monitoring (SIM) mode, monitoring the [M-H]− anions of the fatty acids. Selected masses were arranged into 8 SIM groups according to elution times. The group assignments and ion masses used for SIM are reported in Table 1.
Results and Discussion
Method optimization
Our predominant goal was to establish a procedure that maximizes the number of analytes that can be accurately and precisely monitored in a single run. To achieve this, we first optimized the chromatographic procedure and enhanced the resolution. A representative chromatographic separation of long chain saturated and unsaturated fatty acids (<C12) included in our routine reference mixture is shown in Figure 1. The data show that a temperature gradient was developed that achieves high peak resolution and peak capacity. All regio-isomers are well separated, facilitating accurate peak alignment, identification and quantitation of each fatty acid. Due to the large number of analytes (31 unlabeled and 15 deuterated fatty acids) that were monitored in a single run, data acquisition was divided into eight SIM groups based on retention times. In each group, individual ion optics and SIM tuning parameters (10 ms dwell times) were optimized to enhance specificity and sensitivity (Table 1). By fine tuning the chromatographic method, MS parameters and group assignment of ions, we were able to selectively monitor and distinguish all fatty acid species.
Figure 1.
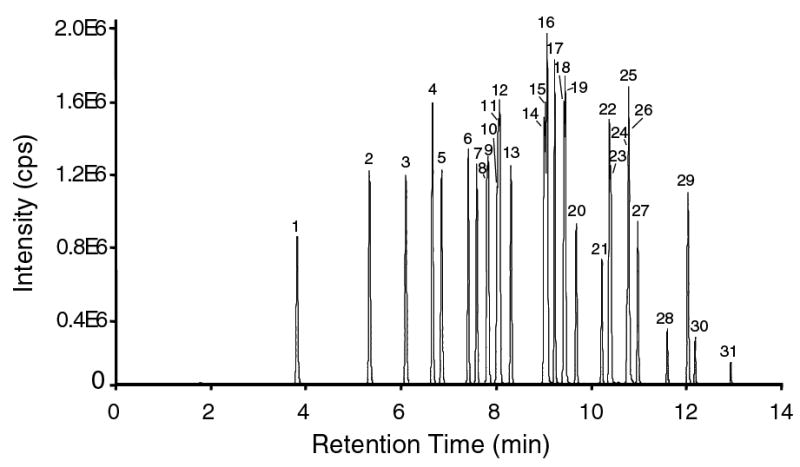
GC/MS chromatogram of a mixture of fatty acid-PBF derivative standards with negative chemical ionization and SIM detection. Shown is a composite chromatogram of all SIM groups. The fatty acids are referenced in the following order: (1), 12:0; (2), 14:0; (3), 15:0; (4), 16:1; (5), 16:0; (6), 17:1; (7), 17:0; (8), α18:3; (9), 18:4; (10), γ18:3; (11), 18:2; (12), 18:1; (13), 18:0; (14), 20:4; (15), 20:5; (16), 11,14,17-20:3; (17), bishomo-20:3; (18), 20:2; (19), 5,8,11-20:3; (20), 20:0; (21), 22:6; (22), 22:4; (23), 22:5; (24), 22:2; (25), 22:3; (26), 22:1; (27), 22:0; (28), 23:0; (29), 24:1; (30), 24:0, (31), 26:0.
Linearity
The linearity of the instrumental response was established with our composite standard solution using the derivatization procedure described above. Calibration curves were established and the slope, intercept and correlation coefficient were calculated (Figure 2). The instrumental response was linear for all fatty acids over a range of 0.003 ng to 10 ng injected (Table 1).
Limit of detection
The limits of detection (LOD) were determined for each of the fatty acids by multiple analysis (n=3) of dilutions of our composite reference mixture of 31 fatty acids and are summarized in Table 1. They were calculated from the chromatograms of dilutions of the standard mixture and defined by the signal to noise ratio S/N>3. The LOD for most fatty acids ranged from 0.1 pg to 1.0 pg. Only docosatrienoic acid showed an LOD that was higher. The variation in LOD was due to differences in the ionization efficiency and sensitivity of the ions of the various fatty acids. In general, a higher sensitivity was observed for shorter chain fatty acids. The LODs achieved with our method is about one order of magnitude higher than was recently reported for trimethylaminoethyl derivatives of fatty acids using LC/MS [32]. Our sensitivity is comparable to the one documented previously for PFB derivatives of fatty acids using GC/MC in the negative ion mode [31]. However, that study optimized instrument settings for sensitivity targeting a single specific fatty acid. Such a setting is technically not practical for the purpose of generating fatty acid profiles and simultaneous quantitation of a large number of fatty acids. We, therefore, balanced our method to achieve high peak capacity while preserving sensitivity.
Analytical precision
The precision of the procedure was determined using designated calibration controls consisting of a mixture of composite unlabeled standards and deuterated internal standards. The intra-day precision was measured by repeating three independent tests on the same day and the inter-day precision was established on three different days. The precision assays were carried out with 5 ng of all 31 fatty acids using routine sample preparation procedures. The ratio of peak areas between specific analyte and corresponding internal standard was used for computing the relative standard deviation (RSD) for each triplicate analysis. As shown in Table 1, the analytical precision for the various fatty acids expressed as the RSD ranged from 4% to 14% for the intra-day and 7% to 17% for the inter-day replicates.
Application of the method to biological samples
The developed method was applied to establish a complete free fatty acid profile of RAW 264.7 cells, an established murine macrophage cell line. Shown in Figure 3 is a representative chromatogram of free fatty acids detected as their [M-H]- ions. Even though not all fatty acids were completely baseline resolved on the capillary column, they were distinguished by MS based on the difference in m/z. The SIM group assignment and SIM parameters were optimized to assure complete discrimination from other fatty acid ions that co-elute or have similar retention times (Table 1). This procedure yields not only complete fatty acid profiles but it also produces reliable quantitative data of all major and trace free fatty acids. To assure accurate peak identity and precise quantitative evaluation, a set of standards was always run before and after the unknown sample. We analyzed three independent cultures of RAW macrophages and identified thirty one fatty acids present in their free form (Table 2). The major free fatty acids were palmitic acid, stearic acid and oleic acid. Together they comprise over 83% of all fatty acids. Free myristic acid, linoleic acid and arachidonic acid were present at 3%, 2% and 2% respectively. Other free fatty acids were detectable in trace amounts.
Figure 3.
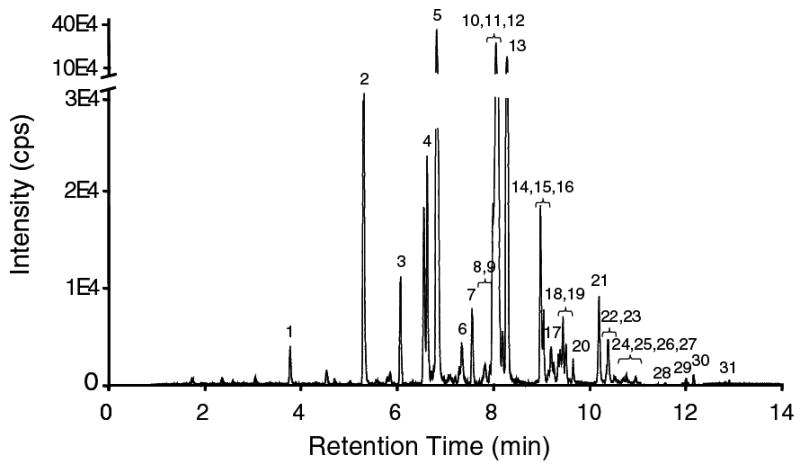
GC/MS chromatogram of cellular free fatty acids extracted from RAW macrophages. The fatty acids were extracted, derivatized and analyzed as described under “Materials and Methods”. No internal standard was added to this representative sample to clearly illustrate the profile of endogenous free fatty acids. For quantification, a mixture of deuterated internal standards was added to the sample prior to the extraction step. The free fatty acid content in the biological sample was calculated from the standard curve using the ratio between analyte peak area and corresponding internal standard peak area. The fatty acids in the chromatogram are referenced as described under Figure 1.
Table 2. Free fatty acid profile of RAW macrophages.
Free fatty acids were extracted from three independent cultures of RAW cells and analyzed separately. All values are normalized to cellular DNA and are expressed as ng fatty acid/μg DNA.
| Fatty Acid | Biological Replicates (ng/μg DNA) | Mean | sDev. | ||
|---|---|---|---|---|---|
| 1 | 2 | 3 | ng/μg | ng/μg | |
| Lauric Acid (12:0) | 0.873 | 0.563 | 0.848 | 0.761 | 0.172 |
| Myristic Acid (14:0) | 3.792 | 3.849 | 3.273 | 3.638 | 0.318 |
| Pentadecanoic Acid (15:0) | 0.806 | 0.636 | 0.391 | 0.611 | 0.209 |
| Palmitic Acid (16:0) | 28.796 | 27.342 | 22.416 | 26.185 | 3.344 |
| Palmitoleic Acid (16:1) | 1.765 | 1.747 | 1.880 | 1.797 | 0.072 |
| Heptadecanoic Acid (17:0) | 0.901 | 0.939 | 0.419 | 0.753 | 0.290 |
| Heptadecaenoic Acid (17:1) | 0.147 | 0.130 | 0.155 | 0.144 | 0.012 |
| Stearic Acid (18:0) | 17.587 | 27.112 | 16.418 | 20.372 | 5.866 |
| Oleic Acid (18:1) | 49.667 | 48.904 | 45.623 | 48.065 | 2.149 |
| Linoleic Acid (18:2) | 2.355 | 2.479 | 1.931 | 2.255 | 0.287 |
| α-linolenic Acid (18:3) | 0.089 | 0.116 | 0.110 | 0.105 | 0.014 |
| γ-linolenic Acid (18:3) | 0.062 | 0.043 | 0.049 | 0.051 | 0.010 |
| Stearidonic Acid (18:4) | 0.004 | 0.005 | 0.003 | 0.004 | 0.001 |
| Arachidic Acid (20:0) | 0.303 | 0.271 | 0.334 | 0.303 | 0.031 |
| Eicosadienoic Acid (20:2) | 0.058 | 0.088 | 0.049 | 0.065 | 0.021 |
| 11,14,17-Eicosatrienoic Acid (20:3 | 1.359 | 1.406 | 1.074 | 1.279 | 0.180 |
| Bishomo-γ-linolenic Acid (20:3) | 0.441 | 0.514 | 0.321 | 0.425 | 0.097 |
| 5,8,11-Eicosatrienoic Acid (20:3 | 0.003 | 0.004 | 0.002 | 0.003 | 0.001 |
| Arachidonic Acid (20:4) | 2.331 | 3.431 | 2.269 | 2.677 | 0.654 |
| Eicosapentaenoic Acid (20:5) | 0.293 | 0.282 | 0.250 | 0.275 | 0.022 |
| Behenic Acid (22:0) | 0.039 | 0.169 | 0.055 | 0.087 | 0.071 |
| Cis-Erucic Acid (22:1) | 0.005 | 0.006 | 0.007 | 0.006 | 0.001 |
| Docosadienoic Acid (22:2) | 0.003 | 0.007 | 0.002 | 0.004 | 0.003 |
| Docosatrienoic Acid (22:3) | 0.002 | 0.001 | 0.002 | 0.002 | 0.001 |
| Docosatetraenoic Acid (22:4) | 0.425 | 0.529 | 0.386 | 0.447 | 0.074 |
| Docosapentaenoic Acid (22:5) | 0.950 | 1.101 | 0.791 | 0.947 | 0.155 |
| Docosahexaenoic Acid (22:6) | 1.490 | 1.768 | 1.069 | 1.442 | 0.352 |
| Tricosanoic Acid (23:0) | 0.019 | 0.027 | 0.022 | 0.023 | 0.004 |
| Lignoceric Acid (24:0) | 0.403 | 0.551 | 0.498 | 0.484 | 0.075 |
| Cis-Selacholeic Acid (24:1) | 0.014 | 0.040 | 0.018 | 0.024 | 0.014 |
| Cerotic Acid (26:0) | 0.146 | 0.099 | 0.177 | 0.140 | 0.039 |
Conclusions
Macrophages play an essential role in many facets of immunity. They are integral to innate immunity but they also represent an important cellular switch to activate adaptive immune responses [33]. Ligation of surface receptors including TLRs induces a signaling program that culminates in the transcription and activation of immune response genes [34]. Saturated fatty acids have been shown to be potent activator of TLR4, while polyunsaturated fatty acid neutralize the endotoxic effects of bacterial lipopolysaccharides [20]. The important role of fatty acids at the interface between lipid metabolism and inflammation prompted us to establish a comprehensive GC/MS procedure that is suitable for complete fatty acid profiling in macrophages. Analysis of fatty acids with GC/MS has become routine and a number of procedures are available. Our procedure takes into account the requirement of establishing complete fatty acid profiles and providing quantitative data of all fatty acids. We have improved the resolution to such an extent that we can detect over thirty fatty acids (31 endogenous fatty acids and 15 internal controls) in a single run. We have extended the dynamic linear range so we can accurately measure all major and minor fatty acids at physiological concentrations. Our sensitivity allows the detection of trace amounts of fatty acids. This makes this protocol ultimately suitable not only for profiling but also for precise quantification of fatty acids. The relative short GC running time and streamlined extraction procedure make this protocol also applicable for high throughput screening. We initially developed the method for the analysis of free fatty acids in macrophages, however, it is readily adaptable to other cells and tissues. With the appropriate modifications of the extraction procedure and with an additional saponification step, the method can be expanded from free fatty acids to the analysis of total cellular fatty acids as well. This versatility makes this protocol widely applicable.
Acknowledgments
This work was supported by the LIPID MAPS Large Scale Collaborative Grant GM069338.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 3.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 4.Rocca B, FitzGerald GA. Cyclooxygenases and prostaglandins: shaping up the immune response. Int Immunopharmacol. 2002;2:603–630. doi: 10.1016/s1567-5769(01)00204-1. [DOI] [PubMed] [Google Scholar]
- 5.Schmelzer K, Fahy E, Subramaniam S, Dennis EA. The lipid maps initiative in lipidomics. Methods Enzymol. 2007;432:171–183. doi: 10.1016/S0076-6879(07)32007-7. [DOI] [PubMed] [Google Scholar]
- 6.Fahy E, Cotter D, Byrnes R, Sud M, Maer A, Li J, Nadeau D, Zhau Y, Subramaniam S. Bioinformatics for lipidomics. Methods Enzymol. 2007;432:247–273. doi: 10.1016/S0076-6879(07)32011-9. [DOI] [PubMed] [Google Scholar]
- 7.Huang CH. Mixed-chain phospholipids: structures and chain-melting behavior. Lipids. 2001;36:1077–1097. doi: 10.1007/s11745-001-0818-1. [DOI] [PubMed] [Google Scholar]
- 8.Dowhan W, Mileykovskaya E, Bogdanov M. Diversity and versatility of lipid-protein interactions revealed by molecular genetic approaches. Biochim Biophys Acta. 2004;1666:19–39. doi: 10.1016/j.bbamem.2004.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stubbs CD, Smith AD. Essential fatty acids in membrane: physical properties and function. Biochem Soc Trans. 1990;18:779–781. doi: 10.1042/bst0180779. [DOI] [PubMed] [Google Scholar]
- 10.Phillips C, Owens D, Collins P, Tomkin GH. Low density lipoprotein non-esterified fatty acids and lipoprotein lipase in diabetes. Atherosclerosis. 2005;181:109–114. doi: 10.1016/j.atherosclerosis.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 11.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Turkmani MR, Freedman SD, Laposata M. Fatty acid alterations and n-3 fatty acid supplementation in cystic fibrosis. Prostaglandins Leukot Essent Fatty Acids. 2007;77:309–318. doi: 10.1016/j.plefa.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Innis SM, Davidson AG. Cystic Fibrosis and Nutrition: Linking Phospholipids and Essential Fatty acids with Thiol Metabolism. Annu Rev Nutr. 2008 doi: 10.1146/annurev.nutr.27.061406.093625. [DOI] [PubMed] [Google Scholar]
- 14.Kompauer I, Demmelmair H, Koletzko B, Bolte G, Linseisen J, Heinrich J. Association of fatty acids in serum phospholipids with lung function and bronchial hyperresponsiveness in adults. Eur J Epidemiol. 2008;23:175–190. doi: 10.1007/s10654-007-9218-y. [DOI] [PubMed] [Google Scholar]
- 15.Chilton FH, Rudel LL, Parks JS, Arm JP, Seeds MC. Mechanisms by which botanical lipids affect inflammatory disorders. Am J Clin Nutr. 2008;87:498S–503S. doi: 10.1093/ajcn/87.2.498S. [DOI] [PubMed] [Google Scholar]
- 16.Lupu R, Menendez JA. Pharmacological inhibitors of Fatty Acid Synthase (FASN)--catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Curr Pharm Biotechnol. 2006;7:483–493. doi: 10.2174/138920106779116928. [DOI] [PubMed] [Google Scholar]
- 17.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 18.Thijssen MA, Mensink RP. Fatty acids and atherosclerotic risk. Handb Exp Pharmacol. 2005:165–194. doi: 10.1007/3-540-27661-0_5. [DOI] [PubMed] [Google Scholar]
- 19.Raetz CR, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Jr, Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, Dennis EA. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, Hwang DH. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Schrade W, Boehle E, Biegler R, Meder V, Teicke R. Gas chromatographic studies on the serum fatty acids in man. Part I. On the fatty acid composition of serum fats in healthy subjects, arteriosclerotics and diabetics. Klin Wochenschr. 1960;38:126–134. doi: 10.1007/BF02189078. [DOI] [PubMed] [Google Scholar]
- 22.Murphy RC, Fiedler J, Hevko J. Analysis of nonvolatile lipids by mass spectrometry. Chem Rev. 2001;101:479–526. doi: 10.1021/cr9900883. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths WJ. Tandem mass spectrometry in the study of fatty acids, bile acids, and steroids. Mass Spectrom Rev. 2003;22:81–152. doi: 10.1002/mas.10046. [DOI] [PubMed] [Google Scholar]
- 24.Miwa H. High-performance liquid chromatographic determination of mono-, poly- and hydroxycarboxylic acids in foods and beverages as their 2-nitrophenylhydrazides. J Chromatogr A. 2000;881:365–385. doi: 10.1016/s0021-9673(00)00284-3. [DOI] [PubMed] [Google Scholar]
- 25.Perret D, Gentili A, Marchese S, Sergi M, Caporossi L. Determination of free fatty acids in chocolate by liquid chromatography with tandem mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:1989–1994. doi: 10.1002/rcm.1582. [DOI] [PubMed] [Google Scholar]
- 26.Lacaze JP, Stobo LA, Turrell EA, Quilliam MA. Solid-phase extraction and liquid chromatography--mass spectrometry for the determination of free fatty acids in shellfish. J Chromatogr A. 2007;1145:51–57. doi: 10.1016/j.chroma.2007.01.053. [DOI] [PubMed] [Google Scholar]
- 27.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 28.Deems R, Buczynski MW, Bowers-Gentry R, Harkewicz R, Dennis EA. Detection and quantitation of eicosanoids via high performance liquid chromatography-electrospray ionization-mass spectrometry. Methods Enzymol. 2007;432:59–82. doi: 10.1016/S0076-6879(07)32003-X. [DOI] [PubMed] [Google Scholar]
- 29.Buczynski MW, Stephens DL, Bowers-Gentry RC, Grkovich A, Deems RA, Dennis EA. TLR-4 and sustained calcium agonists synergistically produce eicosanoids independent of protein synthesis in RAW264.7 cells. J Biol Chem. 2007;282:22834–22847. doi: 10.1074/jbc.M701831200. [DOI] [PubMed] [Google Scholar]
- 30.Harkewicz R, Fahy E, Andreyev A, Dennis EA. Arachidonate-derived dihomoprostaglandin production observed in endotoxin-stimulated macrophage-like cells. J Biol Chem. 2007;282:2899–2910. doi: 10.1074/jbc.M610067200. [DOI] [PubMed] [Google Scholar]
- 31.Pawlosky RJ, Sprecher HW, Salem N., Jr High sensitivity negative ion GC-MS method for detection of desaturated and chain-elongated products of deuterated linoleic and linolenic acids. J Lipid Res. 1992;33:1711–1717. [PubMed] [Google Scholar]
- 32.Pettinella C, Lee SH, Cipollone F, Blair IA. Targeted quantitative analysis of fatty acids in atherosclerotic plaques by high sensitivity liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;850:168–176. doi: 10.1016/j.jchromb.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 33.Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111:927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 34.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]