

Abstract
Humans with central lesions that augment sympathetic nerve activity are predisposed to cardiac arrhythmias, myocardial lesions, and sudden death. Previously, we showed that selectively killing neurons with neurokinin-1 receptors in the nucleus tractus solitarii (NTS) of rats attenuated the baroreflex and, in some animals, led to sudden unexplained death within ∼2 wk. Interruption of arterial baroreflexes is known to increase sympathetic activity. Here we tested the hypothesis that lesions in the NTS lead to fatal cardiac arrhythmias and myocardial lesions. We studied electrocardiograms, echocardiograms, blood pressure, and heart rate in 14 adult male rats after bilateral microinjection into the NTS of stabilized substance P conjugated to the toxin saporin and compared the variables in five sham control rats and in five animals with toxin injected outside the NTS. Only injection of toxin into the NTS led to increased lability of arterial blood pressure, a sign of baroreflex interruption. Two animals treated with toxin died suddenly. All animals engaged in normal activity until, in two, rapid development of asystole and death over 6–8 min. Cardiac function when examined by echocardiography was normal, but pathologic examination of the heart revealed diffuse microscopic areas of acute coagulation necrosis in the myocardium in five animals, focal subacute necrosis in two animals, and both changes in one animal. This study supports the hypothesis that NTS lesions interrupting the baroreflex may induce cardiac arrhythmias and myocardial changes similar to those seen in humans with central lesions and may lead to sudden cardiac death.
Keywords: baroreflex, cardiac arrhythmia, heart injuries, sudden death, sympathetic nervous system
previous studies have suggested that substance P, acting at neurons that express neurokinin-1 (NK1) receptors, may participate in transmission of arterial baroreflexes at the level of the nucleus tractus solitarii (NTS) (17, 23, 26). In an earlier study, we injected into NTS a toxin that selectively targeted neurons with NK1 receptors (24), and we showed that the ensuing lesion attenuated baroreflex responses (24). The altered baroreflex transmission was associated with sudden, unexpected, death in 33% of the experimental animals. Death in these animals was reminiscent of that seen in humans who have sustained central lesions (30). While subarachnoid hemorrhage is a most common cause, other central lesions that lead to enhanced sympathetic nerve activity also predispose an individual to the fatal outcome (30). In fact, sympathetic nerve activity and excessive circulating catecholamines alone may lead to cardiac arrhythmias and death (12, 33). Sudden death in humans with central lesions correlates with cardiac arrhythmias, but the lesions also may lead to cardiac damage (9, 25, 34) even in the absence of arrhythmias. Because NTS lesions interrupt the baroreflex, acutely increase sympathetic nerve activity (28), and lead to lability of arterial pressure (32) we hypothesized that killing neurons that express NK1 receptors in NTS would lead to lability of arterial blood pressure that was accompanied by pathological changes in the heart and in some animals, fatal cardiac arrhythmias. To test that hypothesis we used stabilized substance P conjugated with saporin (SSP-SAP), which selectively kills neurons with NK1 receptors (36).
MATERIALS AND METHODS
Studies were performed in adult male Sprague-Dawley rats, conformed to “Guide for the Care and Use of Laboratory Animals,” and were approved by the institutional animal care and use committees of the University of Iowa and the Iowa City Veterans Affairs Medical Center. In all protocols, every effort was made to minimize or eliminate any pain or distress for the animals. Each rat was anesthetized with halothane (5% induction followed by 1.5 to 2.0% maintenance) delivered in 100% O2 (2 l/min) by a nasal mask. A transmitter (model TL11M2-C50-PXT; Data Sciences, St. Paul, MN) that measured ECG, mean arterial pressure (MAP), heart rate (HR), body temperature, and motor activity was placed in the peritoneal cavity through a midline abdominal incision. A pressure catheter was inserted into the femoral artery and ECG leads were attached to the right thorax (negative lead) and the left hind limb (positive lead). In three animals no further instrumentation or surgery was performed. After closure of the surgical wound, those animals were returned to their home cage for recording of cardiovascular variables. In the remainder of the animals, a venous cannula to administer ketamine for echocardiography was filled with heparin (10,000 U/ml), placed in the femoral vein, plugged at the free end, and externalized through the skin on the back of the neck. The dorsal surface of the brain stem was exposed, as previously described (29), and a glass micropipette filled in 14 animals with SSP-SAP (supplied by Advanced Targeting Systems, San Diego, CA) or in five animals with vehicle (PBS) was stereotactically placed (0.4 mm rostral to the calamus scriptorius, 0.5 mm from the midline, and 0.5 mm below the surface of the brain stem) bilaterally into the dorsolateral and medial portions of the NTS at the level of the area postrema where baroreceptor afferent terminals are most concentrated (35). The dose of SSP-SAP (9.4 ng) was selected from preliminary immunohistological studies that showed its effective reduction in NK1 neurons in NTS. Injections (400 nl) were performed over 15 min, and the pipette was left in place for 15 min to limit efflux of injectate. To avoid effects of volume, individual increments of injection were always 25 nl and were given slowly, not as a bolus. In five additional animals, we injected the same dose of SSP-SAP in 400 nl bilaterally into the brain stem but outside NTS. For those injections, the pipette was placed 0.0 mm rostral to the calamus scriptorius, 1.5 mm from the midline, and 1.0 mm below the surface of the brain stem at that point. In each animal, after withdrawal of the pipette, wounds were closed with silk suture, ampicillin (75 mg in a 250 mg/ml saline solution) was given intramuscularly, 2% lidocaine was delivered to the suture sites, buprenorphine (0.1 mg/kg) was administered subcutaneously for analgesia, and halothane was discontinued. After recovery from anesthesia, the animal was observed at least twice daily until it was killed. The animal's cage was placed on a radio receiver (model RPC-1; DSI, St. Paul, MN), which captured data and transmitted them for storage through a digital data acquisition system. MAP was sampled for 10 s every minute for 10–12 days. This shorter period of recording than was used in our previous study (24) reflected our efforts to reduce the incidence of sudden death. ECG waveforms were sampled for 10-s intervals every 30 s at a rate of 500 Hz. After the recording period, we evaluated the heart with 2-D echocardiography.
Published methods for echocardiography (7) are summarized as follows. Briefly, 2-D echocardiography was performed under light general anesthesia (ketamine, 25 mg/kg ip), which allowed animals to tolerate confrontation. Ketamine did not diminish HR or respiratory rate or result in loss of consciousness. The anterior chest was shaved, warmed acoustic coupling gel was applied, and the animal was positioned without restraint in the left lateral recumbent position. Parasternal short- and long-axis images were acquired via an 8-MHz sector-array probe, which produced high-quality 2-D images at a rate of ∼100/s. Images were stored off-line for later analysis. In each plane, endo- and epicardial silhouettes were visually identified and electronically measured by planimetry using the leading-edge convention at end diastole and end systole. We used the biplane area-length method (8) to compute left ventricular volumes and mass at both phases of the cardiac cycle. Regions demonstrating akinesis or dyskinesis, if present, would be visually identified, measured by planimetry, and expressed as % of the total left ventricular end-diastolic silhouette. During each imaging session, HR was determined using pulse-wave Doppler interrogation of mitral inflow and/or continuous electrocardiographic monitoring, which was displayed online with the images.
On completion of echocardiography, rats were anesthetized with pentobarbital (50 mg/kg) and perfused through the heart, first with chilled (4°C) PBS for 2 min at a flow rate of 20 ml/min, and then with freshly prepared, chilled 4% paraformaldehyde in PBS (pH 7.4) for 20 min, and also at a flow rate of 20 ml/min. The brain was removed, postfixed in 4% paraformaldehyde for 2 h and then cryoprotected for 2 days in 30% sucrose in PBS at 4°C. Frozen 20-μm coronal slices were cut with a cryostat, collected in 24-well microplates containing chilled 30% sucrose in PBS, and processed as floating sections for immunohistochemistry or fluorescent immunostaining for NK1.
For chromogen-based immunohistochemistry, we followed methods described previously (24). In three animals, SSP-SAP was injected unilaterally to provide comparison between the side with a lesion and the contralateral side that served as an intra-animal control. Tissue sections were treated with 0.3% H2O2, blocked with 10% donkey normal serum in PBS, and then incubated with rabbit anti-NK1 antibody (1:100; Novus Biologicals, Littleton, CO) overnight. Sections were then incubated with biotinylated donkey anti-rabbit antibody (1:500; Jackson ImmunoResearch, West Grove, PA) for 2 h, followed by incubation with streptavidin-conjugated horseradish peroxidase (1:500; Jackson ImmunoResearch) for 1 h. The final visualization of NK1 immunoreactivity (NK1-IR) was achieved by incubating sections with 0.005% 3′-3′-diaminobenzidine tetrahydrochloride in the presence of 0.6% nickel ammonium sulfate and 0.006% H2O2 for 2–4 min. Sections were then transferred to slides, air dried, mounted with Permount (The Science, Denver, CO), and examined with a light microscope. For fluorescent immunostaining of NK1, sections were incubated with rabbit anti-NK1 antibody (1:50) for 24 h, washed, incubated with biotinylated donkey anti-rabbit antibody (1:200; Jackson ImmunoResearch) for 24 h at 4°C, followed by incubation with rhodamine red X-conjugated streptavidin (1:200; Jackson ImmunoResearch) for 24 h at 4°C. Sections were then washed, transferred to slides, air-dried, and mounted with Prolong Gold Antifade Reagents (Molecular Probes, Carlsbad, CA). Fluorescent-labeled sections were analyzed with a Zeiss LSM 510 confocal laser scanning microscope as described in our previous publications (14). Tissue processed without addition of primary antibody served as a negative control for both chromogen-base immunohistochemistry and fluorescent immunostaining. In these control preparations, no NK1-IR was present.
To examine pathological changes in the heart, we removed the heart after the rat had been perfused and stored the heart in 4% paraformaldehyde until it was embedded in paraffin. Transverse sections were then cut, and hematoxylin and eosin-stained slides were prepared at 2-mm intervals throughout the entire heart.
As reported previously (32), we expressed lability of MAP (and HR) as the standard deviation (SD) of the variable and expressed lability in groups of animals as the mean SD (SE). Similarly other physiological variables were expressed as mean (SD). To avoid repeated survival surgeries we did not record any variable in the animals prior to injections into the brain stem. Instead, we accepted data obtained during the first 12 h after instrumentation and injection as basal values and evaluated changes from those values. We acknowledge that those basal values may not reflect normative values for the same animals without any intervention other than instrumentation. For this reason, three animals were instrumented only for recording cardiovascular variables during the first 12 h after recovery from surgery.
As appropriate, we utilized a linear mixed-model analysis for repeated measures with Bonferroni adjustment or two-way ANOVA to compare mean MAP and HR among the experimental, control, and outside NTS groups. Pearson χ2 analysis was applied to associations between lability of arterial pressure and myocardial damage. Comparisons were considered significantly different at P ≤ 0.05.
RESULTS
Effects of SSP-SAP on NTS neurons with NK1 receptors.
Immunofluorescent and immunohistochemical analysis of the brain stem performed 10–12 days after unilateral injections of SSP-SAP into NTS revealed reduction in NK1 neuronal staining in the NTS only on the side of injection as had been seen previously when substance P (SP)-SAP, rather than SSP-SAP had been injected (24). As shown in Fig. 1, there was a marked decrease in NK1-IR in neurons and fibers in the intermediate and medial subnuclei of the NTS of the SSP-SAP-injected rat, compared with that of the control rat. For rats that were studied after bilateral SSP-SAP injection, we also performed immunohistochemical analyses of NK1-IR at the end of the experiments to confirm that there was a reduction in the NK1-IR in the NTS after SSP-SAP injection. We found no change in NK1 staining in the nucleus ambiguus or in the rostral or caudal ventrolateral medulla after injection of SSP-SAP into the NTS. After injection of SSP-SAP bilaterally, but the outside NTS, the injected site showed decreased NK1 staining, but the NTS and other areas outside the injection site showed normal NK1-IR.
Fig. 1.

Representative immunofluorescent labeling of the medial (me) and intermediate (im) subnuclei of the nucleus tractus solitarii (NTS) on the left side where sham treatment was given (A and C) and on the right side where substance P conjugated with saporin (SSP-SAP) had been injected (B and D). Note marked reduction in immunostaining for neurokinin-1 (NK1) receptors in the NTS on the treated side. Scale bar = 20 μm.
Postoperative recovery.
After recovery from anesthesia, the animals engaged in normal activity (eating, grooming, and exploring environment) and appeared to be healthy and in no distress for the entire recording period. Body temperature, which fell slightly in all animals immediately postoperatively, returned to normal (37°C) within the first day of surgery and remained normal. After recovery from surgery, a normal diurnal pattern of motor activity persisted until death (euthanasia or sudden death).
Effects of lesions on arterial pressure.
Rats that received bilateral SSP-SAP injections in the NTS developed significantly (linear mixed model analysis for repeated measures) more labile arterial blood pressures compared with control rats that had received PBS injection only (Fig. 2 and Fig. 3, top). Lability gradually increased over the week after injection of toxin (Fig. 3, top) and remained elevated until animals were euthanized. In contrast, significant lability did not develop in the control group when data during the first 12 h after injection were used as a baseline. The test of fixed effects from the linear mixed-model analysis showed a significant treatment*day interaction effect (P = 0.011). Thus, the effects of treatment became more prominent as time passed after injection of toxin. The plot of the mean (SE) estimates from the fitted linear mixed model for each treatment group over the 10 days is presented in Fig. 3. In the three animals that received no injection into the NTS, the mean SD of MAP was 3.7 mmHg (SE 1.0). In five animals in which injections of toxin were made adjacent to the NTS, significant lability did not develop (Fig. 3, top).
Fig. 2.
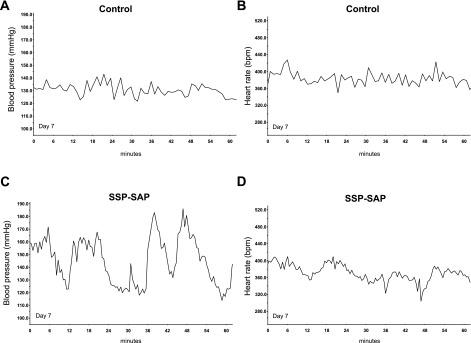
Labile blood pressure [mean arterial pressure (MAP)] seen in a representative treated animal (C) contrasted with more stable blood pressure in a representative control animal (A) on day 7. Lability of heart rate [beats/min (bpm)] did not differ between the control (B) and experimental (D) animal. This treated animal was not one that sustained sudden death, but, at postmortem examination, it did manifest myocardial lesions (see Fig. 5).
Fig. 3.

Lability in control animals (PBS-injected) in those treated with bilateral injection of SSP-SAP into NTS (Exp) and in those treated with SSP-SAP injected adjacent (outside NTS) to NTS (top). Significant (*P = 0.002–0.012) lability developed in rats that had received SSP-SAP in NTS when compared with control animals. Animals treated with SSP-SAP injected adjacent to NTS did not develop significant lability. MAP was significantly (Bonferroni, P = 0.045) higher in treated animals than in controls but was not significantly increased in those animals in which SSP-SAP had been injected adjacent to NTS (bottom).
MAP of the experimental group was significantly greater than in the control group (Fig. 3, bottom). In contrast, HR and lability of HR did not significantly differ between control and experimental animals even when lability of arterial pressure had become maximal. At 2 days, mean HR was 351.1 (SD 35.4) beats/min and SD was 19.3 (SD 4.9) beats/min in controls, whereas in treated animals, mean HR was 365.9 (SD 32) beats/min and SD 20.0 (SD 7.7) beats/min, and in animals with injections outside NTS, mean HR was 331.7 (SD 43.4) and SD 22.9 (SD 4.0). At 7 days, mean HR was 323.2 (SD 21.2) beats/min and SD 30.5 (SD 6.3) in controls, whereas in treated animals, mean HR was 343.2 (SD 28.6) beats/min and SD 24.7 (SD 8.8) beats/min, and in animals with injections outside NTS, mean HR was 330.9 (SD 33.2) and SD 31.9 (SD 6.8). All groups showed a significant effect of time on HR. In other words, whereas HR and its lability did not differ between groups, HR gradually decreased in each group after initial postoperative recordings.
Electrocardiographic changes in animals with NTS lesions.
Two of the 14 rats that had been treated with SSP-SAP suddenly died 9 days after surgery. The rats were engaged in normal daily activity and had a normal ECG until ∼6.5 and 8 min prior to death when their ECG evolved through elevated T waves, ventricular ectopic beats, and finally asystole (Fig. 4). Death occurred in each animal at night (between 9:00 PM and midnight) during a period associated with maximal diurnal activity for rats. In treated animals, blood pressure, motor activity, and temperature were maintained unless a fatal cardiac arrhythmia occurred and led to a precipitous fall in each variable. In those rats that did not experience sudden death and in all control rats, a normal ECG pattern persisted throughout the period of recording.
Fig. 4.
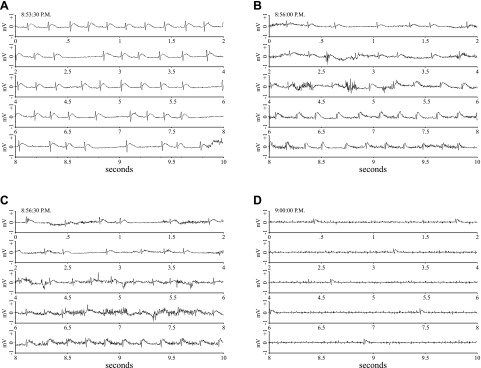
ECG recordings show progression from a normal sinus rhythm at 8:53:30 PM (A) to asystole (D) over the course of 6.5 min. Each segment of this figure depicts a continuous 10-s recording with the time at the beginning of the record posted in the top left hand corner of each.
Echocardiographic analysis.
Echocardiography was performed in 10 animals that had received bilateral injections of SSP-SAP into the NTS in three control animals that had received bilateral injections of PBS into the NTS and in two control animals that had received bilateral injections of SSP-SAP adjacent to the NTS. The echocardiographic images revealed no significant differences between groups. During the echocardiographic analysis, HR for the control group (n = 5) was 419.4 ± 19.3 beats/min compared with the experimental group (n = 10) in which HR was 403.0 ± 33.9 beats/min. The ejection fraction in the control group was 0.8 ± 0.08 ml, whereas it was 0.8 ± 0.04 ml in the experimental group. Despite a normal echocardiogram, pathological changes (see below) in the heart occurred in treated animals.
Pathological changes in the heart.
Examination of hematoxylin and eosin-stained sections of hearts from 14 rats that had received bilateral SSP-SAP revealed that one had a focus of partially resolving microinfarct and hypereosinophilic myocytes, two demonstrated regions of subacute necrosis, five had scattered hypereosinophilic myocytes (Fig. 5) as their only pathological change, and six had no pathologic changes. Postmortem examination of one of the two animals that had sudden death, demonstrated eosinophilic cardiac myocytes, whereas no pathologic changes were found in the heart of the second animal. None of the rats from either control group displayed any pathologic abnormalities in the heart. Pearson χ2 analysis (see Table 1) supported an association between myocardial damage and lability of arterial pressure in animals treated with bilateral injections of SSP-SAP into the NTS.
Fig. 5.

Eosinophilia of discrete cardiac myocytes (indicated by arrows) was seen diffusely at low (A)- and high (B)-power magnification in sections of left ventricular myocardium stained with hematoxylin and eosin. The box in A is seen at higher magnification in B. Scale bars = 50 μm.
Table 1.
Association between lability of arterial pressure and myocardial damage
| Lability | Myocardial Lesions | No. Myocardial Lesions | Total |
|---|---|---|---|
| SD >12 | 5 | 0 | 5 |
| SD <12 | 1 | 6 | 7 |
| Total | 6 | 6 | 12* |
Two of 14 treated animals were excluded from this analysis because of the failure of their radiotelemetry transmitter. Both animals demonstrated myocardial lesions. The two animals that died are included in this table. With Pearson's χ2: P = 0.0034.
DISCUSSION
This study supports our hypotheses and documents the first animal model of a central lesion leading to cardiac changes like those seen in patients who have sustained subarachnoid hemorrhage or a variety of other lesions within the central nervous system. We show that selectively destroying NK1 receptor neurons in the NTS leads to chronic lability of arterial pressure, a finding consistent with loss of baroreflex responses (4, 24). Although an earlier paper (1) showed a recording of arterial pressure that suggested the presence of lability after damage to NTS neurons with NK1 receptors, our observation is the first to our knowledge to document with data analysis that lability does develop and mimics that seen with less selective NTS lesions (32). Lability of arterial pressure began to appear shortly after injection of toxin into the NTS. It was persistent and gradually increased over the next week. These findings are consistent with previous research (15), which demonstrated gradual evolution of neurotoxic effects after administration of the toxin. Those earlier studies showed that at first neurons failed to efficiently recycle substance P receptors to the plasma membrane; but by day 7, 95% of neurons with the substance P receptor had been ablated. The same studies showed that saporin without the substance P conjugate had no cytotoxic effect, a finding that was also borne out by studies in which saporin was injected as a control into the NTS (1, 22). Thus, those published studies obviated the need for injection of the inactive agent in the current study. Lability of arterial pressure in animals that had been treated with SSP-SAP was consistent with interruption of the baroreflex through lesions in the NTS (32). In the present study, we utilized the standard deviation of MAPs as an index of lability to allow comparison with numerous other studies of lability (2, 3, 10, 19, 27, 32) and found that lability after ablation of NTS neurons with NK1 receptors was similar to that seen with more extensive interruptions of baroreceptor pathways. We found mild, but significant, hypertension in treated animals compared with control animals, a finding that contrasts with a reported lack of neurogenic hypertension in conscious, freely moving animals that are not disturbed during recording after interruption of the baroreflex (4, 32). It is unclear whether the treated animals in our study manifested a significant change in blood pressure with respect to their pretreatment values. However, our ability to fully analyze changes in lability and arterial pressure from true basal values (occurring before any intervention) was limited by our conscious decision to avoid multiple survival surgeries that would have been needed to record variables both in the awake state after implantation of radiotelemetry transducers but before interventions in the brain as well as after the latter interventions. Nonetheless, lability in the three animals studied without injections into the brain stem was fully consistent with that previously reported in intact animals where MAPs of 118 (SD 2.5) mmHg were associated with lability (SD) of 6.0 (SD 0.5) mmHg (32). It would be intriguing indeed if the selective lesion made in this study was itself associated with neurogenic hypertension, but that hypothesis must be tested by further studies. We conjecture that implantation of radiotelemetry monitors in the abdomen may have provided sufficient novel stimulus to our animals to lead to hypertension in the group with attenuated baroreflex control of blood pressure and that relative hypertension was a product of that method of recording, which differed from recording techniques in earlier studies that did not show such hypertension (32).
Our findings also support the hypothesis that chronic lesions in NTS will lead to myocardial damage and cardiac arrhythmias similar to that seen in humans with central lesions. Previous studies have demonstrated that patients with subarachnoid hemorrhage can develop focal myocytolysis and discrete areas of coagulative necrosis in the myocardium (16, 25, 37). We found similar cardiac pathological changes in eight of the 14 rats that received SSP-SAP injection bilaterally into the NTS. In humans, those pathological changes are thought to be the result of increased sympathetic nerve activity (30). In this study, the correlation between myocardial damage and lability of arterial pressure, itself dependent on sympathetic nerve activity (2), is consistent with augmented sympathetic activity, which has been shown to occur acutely after lesions of the NTS (6). Although lability of arterial pressure is not correlated with sympathetic nerve activity in chronic fixed lesions of baroreceptor nerves (3), the current study involved a progressively developing NTS lesion that combined both chronic and acute effects of the lesion. Therefore, further study is needed to determine a direct correlation between the cardiac lesions and sympathetic nerve activity.
Pathological changes in the myocardium were diffuse, nonconfluent, and microscopic. In one of the two rats that died suddenly, pathological examination demonstrated hypereosinophilic myocytes. Examination of the myocardium of the other rat did not reveal those changes. Therefore, sudden death due to cardiac arrhythmia may occur independently of pathological changes in the myocardium. The myocardium of another five of the 14 rats did not demonstrate any pathological changes, nor did any of them die from the SSP-SAP injections bilaterally into the NTS. Pathological changes did not follow the territory of coronary arteries but were, instead, scattered and diffuse. The nature of the changes, as well as echocardiographic findings, argue against coronary occlusion or vasospasm as the cause of myocardial damage but are consistent with an effect of sympathetic nerve activation. Although echocardiograms could not be performed in all animals, normal (21) stroke volumes, cardiac output, and left ventricular end-diastolic and end-systolic volumes, as well as normal wall motion, suggested normal myocardial function. There were no segmental wall defects like those that would be expected with coronary occlusion.
In the two rats that died suddenly, the ECG evolved from a normal sinus rhythm through arrhythmia, asystole, and death in 6.5 and 8 min, respectively. It is noteworthy that sudden death occurred in both animals at similar times of day (9:00 PM and 12:00 AM, periods of heightened activity in the nocturnal animal) and similar times after bilateral injections of toxin (9.1 and 9.3 days postoperation). Sudden death occurred at similar times during the night in animals that died in our previous study (24). The progression of ECG changes and death at the same time of day in each animal suggests that a similar etiology could have led to the sudden cardiac death in both animals in this study. In contrast to humans (30) who sustain cardiac arrhythmias and sudden death, the animals in this study did not develop ventricular fibrillation, but developed asystole instead. However, asystole, as seen in the animals of this study, may also be a prominent cause of death in humans with central lesions (20). This study likely failed to demonstrate ventricular tachyarrhythmias as a result of its design in that the animals were not exposed to stress, which is known to accelerate manifestation of ventricular tachyarrhythmias and sudden death in susceptible individuals (11). Instead they were housed individually in a quiet room with controlled temperatures and lighting and ready access to food and water. We anticipate that treated rats will, like humans who are predisposed to cardiac events (18), tend to develop sudden death when exposed to stresses.
We acknowledge that the chemical lesion produced in this study killed neurons that expressed not only NK1 receptors but also other receptors. We have shown colocalization of NK1 receptors with ionotropic glutamate receptors (13) and conjecture that death of neurons in glutamatergic pathways are responsible for loss of baroreflexes and lability of arterial pressure. In addition, GABAergic neurons may also be responsible for these changes, because a decrease in the GABA synthesizing enzyme glutamic acid decarboxylase has been observed after SSP-SAP was injected into the NTS (22). Because saporin did not isolate effects of loss of NK1 function from that of function of other receptors on the same cells, SSP-SAP, while selective for neurons with NK1 receptors, was not selective at the membrane level and somewhat nonspecifically attenuated the baroreflex. Specifically, we do not contend that the cardiac events reported here were directly and specifically related to an effect on the NK1 receptor itself. However, the advantage of using the toxin for that receptor over other means of interrupting the baroreflex at the level of NTS is that baroreflex interruption occurred more slowly with the toxin and allowed the animals to survive without manifesting signs of extreme neurogenic hypertension and pulmonary edema associated with early death as occurs after placement of an acute destructive lesion (5, 31).
This study demonstrates that loss of specific NTS neurons leads to lability of arterial pressure consistent with baroreflex dysfunction and predisposes rats to cardiac arrhythmias, sudden death, and pathological changes in the myocardium.
Perspectives and Significance
The myocardial lesions seen in this animal model of central dysfunction mimic those seen in humans with central nervous system lesions. In humans, those findings are particularly common after subarachnoid hemorrhage. Although there has been considerable speculation about the cause of those cardiac changes in humans, proof of a pathophysiological mechanism and approaches to modify that mechanism have not been possible in the absence of an experimental model of the disease. Therefore, this animal model will allow analysis of mechanisms for cardiac changes and potential discovery of means for more effective treatment to avoid this often fatal complication of human disease. We conjecture that sympathetic input to the heart plays a role in cardiac damage seen here and may play similar roles in cardiac dysfunction associated with stress in the absence of central nervous system disease. Thus, the mechanisms studied in this model may also prove relevant to other causes of sudden death with or without associated intrinsic central or cardiac disease.
GRANTS
This study was funded through National Heart, Lung, and Blood Institute Grants R01-HL-59593 (to W. T. Talman-PI) and R01-HL-088090 (to W. T. Talman and L.-H. Lin-PI) and, in part, through a Veterans Affairs Merit Review Tab 14 (to W. T. Talman, principal investigator).
Acknowledgments
Heart histology was performed in the Core Pathology Research Laboratory, the University of Iowa. The authors gratefully acknowledged the technical assistance provided by Deidre Nitschke Dragon and expert assistance in designing statistical analyses provided by Dr. Miriam B. Zimmerman, Professor of Biostatistics in the College of Public Health at the University of Iowa.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Abdala AP, Schoorlemmer GH, Colombari E. Ablation of NK1 receptor bearing neurons in the nucleus of the solitary tract blunts cardiovascular reflexes in awake rats. Brain Res 1119: 165–173, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Alper RH, Jacob HJ, Brody MJ. Regulation of arterial pressure lability in rats with chronic sinoaortic deafferentation. Am J Physiol Heart Circ Physiol 253: H466–H474, 1987. [DOI] [PubMed] [Google Scholar]
- 3.Barres C, Lewis SJ, Jacob HJ, Brody MJ. Arterial pressure lability and renal sympathetic nerve activity are dissociated in SAD rats. Am J Physiol Regul Integr Comp Physiol 263: R639–R646, 1992. [DOI] [PubMed] [Google Scholar]
- 4.Cowley AW, Liard JF, Guyton AC. Role of the baroreceptor reflex in daily control of arterial blood pressure and other variables in dogs. Circ Res 32: 564–576, 1973. [DOI] [PubMed] [Google Scholar]
- 5.Doba N, Reis DJ. Acute fulminating neurogenic hypertension produced by brainstem lesions in the rat. Circ Res 32: 584–593, 1973. [DOI] [PubMed] [Google Scholar]
- 6.Doba N, Reis DJ. Role of central and peripheral adrenergic mechanisms in neurogenic hypertension produced by brain stem lesions in rat. Circ Res 34: 293–301, 1974. [DOI] [PubMed] [Google Scholar]
- 7.Felder RB, Francis J, Weiss RM, Zhang ZH, Wei SG, Johnson AK. Neurohumoral regulation in ischemia-induced heart failure. Role of the forebrain. Ann NY Acad Sci 940: 444–453, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Francis J, Weiss RM, Wei SG, Johnson AK, Beltz TG, Zimmerman K, Felder RB. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol 281: H2241–H2251, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Karch SB, Billingham ME. Myocardial contraction bands revisited. Hum Pathol 17: 9–13, 1986. [DOI] [PubMed] [Google Scholar]
- 10.LeDoux JE, DelBo A, Tucker LW, Harshfield G, Talman WT, Reis DJ. Hierarchic organization of blood pressure responses during the expression of natural behaviors in rat: mediation by sympathetic nerves. Exp Neurol 78: 121–133, 1982. [DOI] [PubMed] [Google Scholar]
- 11.Leor J, Poole WK, Kloner RA. Sudden cardiac death triggered by an earthquake. N Engl J Med 334: 413–419, 1996. [DOI] [PubMed] [Google Scholar]
- 12.Lepeschkin E, Marchet H, Schroeder G, Wagner R, de Paula S, Raab W. Effect of epinephrine and norepinephrine on the electrocardiogram of 100 normal subjects. Am J Cardiol 5: 594–603, 1960. [DOI] [PubMed] [Google Scholar]
- 13.Lin LH, Taktakishvili OM, Talman WT. Colocalization of neurokinin-1, N-methyl-d-aspartate, and AMPA receptors on neurons of the rat nucleus tractus solitarii. Neuroscience 154: 690–700, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin LH, Talman WT. Vesicular glutamate transporters and neuronal nitric oxide synthase colocalize in aortic depressor afferent neurons. J Chem Neuroanat 32: 54–64, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 278: 275–279, 1997. [DOI] [PubMed] [Google Scholar]
- 16.Mayer SA, Lin J, Homma S, Solomon RA, Lennihan L, Sherman D, Fink ME, Beckford A, Klebanoff LM. Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke 30: 780–786, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Miura M, Takayama K, Okada J. Study of possible transmitters in the solitary tract nucleus of the cat involved in the carotid sinus baro- and chemoreceptor reflex. J Auton Nerv Syst 19: 179–188, 1987. [DOI] [PubMed] [Google Scholar]
- 18.Muller JE, Verrier RL. Triggering of sudden death—lessons from an earthquake. N Engl J Med 334: 460–461, 1996. [DOI] [PubMed] [Google Scholar]
- 19.Nathan MA, Severini WH, Tucker LW, Reis DJ. Enhancement of conditioned arterial pressure responses in cats after brainstem lesions. Science 201: 71–73, 1978. [DOI] [PubMed] [Google Scholar]
- 20.Noritomi DT, de Cleva R, Beer I, Dalbem AG, Liborio AB, Frota NA, Gama-Rodrigues JJ. Doctors awareness of spontaneous subarachnoid haemorrhage as a cause of cardiopulmonary arrest. Resuscitation 71: 123–124, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Pawlush DG, Moore RL, Musch TI, Davidson WR Jr. Echocardiographic evaluation of size, function, and mass of normal and hypertrophied rat ventricles. J Appl Physiol 74: 2598–2605, 1993. [DOI] [PubMed] [Google Scholar]
- 22.Potts JT, Fong AY, Anguelov PI, Lee S, McGovern D, Grias I. Targeted deletion of neurokinin-1 receptor expressing nucleus tractus solitarii neurons precludes somatosensory depression of arterial baroreceptor-heart rate reflex. Neuroscience 145: 1168–1181, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potts JT, Fuchs IE. Naturalistic activation of barosensitive afferents release substance P in the nucleus tractus solitarius of the cat. Brain Res 893: 155–164, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Riley J, Lin LH, Chianca DA Jr, Talman WT. Ablation of NK1 receptors in rat nucleus tractus solitarii blocks baroreflexes. Hypertension 40: 823–826, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Samuels MA The brain-heart connection. Circulation 116: 77–84, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Seagard JL, Dean C, Hopp FA. Modulation of the carotid baroreceptor reflex by substance P in the nucleus tractus solitarius. J Auton Nerv Syst 78: 77–85, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Snyder DW, Nathan MA, Reis DJ. Chronic lability of arterial pressure produced by selective destruction of the catecholamine innervation of the nucleus tractus solitarii in the rat. Circ Res 43: 662–671, 1978. [DOI] [PubMed] [Google Scholar]
- 28.Sved AF Peripheral pressor systems in hypertension caused by nucleus tractus solitarius lesions. Hypertension 8: 742–747, 1986. [DOI] [PubMed] [Google Scholar]
- 29.Talman WT Kynurenic acid microinjected into the nucleus tractus solitarius of rat blocks the arterial baroreflex but not responses to glutamate. Neurosci Lett 102: 247–252, 1989. [DOI] [PubMed] [Google Scholar]
- 30.Talman WT Cardiovascular presentations in primary central neurological disease. In: Systemic Diseases, edited by Goetz CG, Tanner CM, and Aminoff MJ. Amsterdam: Elsevier, 1993, part 1, p. 229–247.
- 31.Talman WT, Perrone MH, Reis DJ. Acute hypertension after the local injection of kainic acid into the nucleus tractus solitarii of rats. Circ Res 48: 292–298, 1981. [DOI] [PubMed] [Google Scholar]
- 32.Talman WT, Snyder DW, Reis DJ. Chronic lability of arterial pressure produced by destruction of A2 catecholaminergic neurons in rat brainstem. Circ Res 46: 842–853, 1980. [DOI] [PubMed] [Google Scholar]
- 33.Verrier RL, Thompson P, Lown B. Ventricular vulnerability during sympathetic stimulation: role of heart rate and blood pressure. Cardiovasc Res 8: 602–610, 1974. [DOI] [PubMed] [Google Scholar]
- 34.Weidler DJ Myocardial damage and cardiac arrhythmias after intracranial hemorrhage. A critical review. Stroke 5: 759–764, 1974. [DOI] [PubMed] [Google Scholar]
- 35.Weston M, Wang H, Stornetta RL, Sevigny CP, Guyenet PG. Fos expression by glutamatergic neurons of the solitary tract nucleus after phenylephrine-induced hypertension in rats. J Comp Neurol 460: 525–541, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Wiley RG, Lappi DA. Targeting neurokinin-1 receptor-expressing neurons with [Sar9,Met(O2)11] substance P-saporin. Neurosci Lett 277: 1–4, 1999. [DOI] [PubMed] [Google Scholar]
- 37.Wu DJ, Fujiwara H, Matsuda M, Ishida M, Kawamura A, Takemura G, Kida M, Uegaito T, Fujiwara T, Kawai C. Clinicopathological study of myocardial infarction with normal or nearly normal extracardiac coronary arteries. Quantitative analysis of contraction band necrosis, coagulation necrosis, hemorrhage, and infarct size. Heart Vessels 6: 55–62, 1990. [DOI] [PubMed] [Google Scholar]