

Abstract
Mitochondria affect cerebrovascular tone by activation of mitochondrial ATP-sensitive K+ (KATP) channels and generation of reactive oxygen species (ROS). Insulin resistance accompanying obesity causes mitochondrial dysfunction, but the consequences on the cerebral circulation have not been fully identified. We evaluated the mitochondrial effects of diazoxide, a putative mitochondrial KATP channel activator, on cerebral arteries of Zucker obese (ZO) rats with insulin resistance and lean (ZL) controls. Diameter measurements showed diminished diazoxide-induced vasodilation in ZO compared with ZL rats. Maximal relaxation was 38 ± 3% in ZL vs. 21 ± 4% in ZO rats (P < 0.05). Iberiotoxin, a Ca2+-activated K+ channel inhibitor, or manganese(III) tetrakis(4-benzoic acid)porphyrin chloride, an SOD mimetic, or endothelial denudation diminished vasodilation to diazoxide, implicating Ca2+-activated K+ channels, ROS, and endothelial factors in vasodilation. Inhibition of nitric oxide synthase (NOS) in ZL rats diminished diazoxide-induced vasodilation in intact arteries, but vasodilation was unaffected in endothelium-denuded arteries. In contrast, NOS inhibition in ZO rats enhanced vasodilation in endothelium-denuded arteries, but intact arteries were unaffected, suggesting that activity of endothelial NOS was abolished, whereas factors derived from nonendothelial NOS promoted vasoconstriction. Fluorescence microscopy showed decreased mitochondrial depolarization, ROS production, and nitric oxide generation in response to diazoxide in ZO arteries. Protein and mRNA measurements revealed increased expression of endothelial NOS and SODs in ZO arteries. Thus, cerebrovascular dilation to mitochondria-derived factors involves integration of endothelial and smooth muscle mechanisms. Furthermore, mitochondria-mediated vasodilation was diminished in ZO rats due to impaired mitochondrial KATP channel activation, diminished mitochondrial ROS generation, increased ROS scavenging, and abnormal NOS activity.
Keywords: diazoxide, depolarization, mitochondrial ATP-sensitive potassium channels, Zucker obese rats
obesity and associated insulin resistance (IR) have been linked to cerebrovascular dysfunction and the increased incidence and severity of stroke in humans (1). Although the mechanisms causing the adverse effects of IR on cerebral arteries are not completely known, there is good evidence that mitochondrial dysfunction may play a major role. Recently, we showed that impaired mitochondrial function in the hearts of Zucker obese (ZO) rats with IR is associated with augmented infarct size and loss of ischemic and pharmacologically induced preconditioning after transient coronary artery occlusion (15). We are unaware of previous studies that have directly examined the cerebrovascular consequences of impaired mitochondrial function in IR.
Recent evidence has shown that mitochondria-derived factors promote relaxation of vascular smooth muscle (VSM) in endothelium-denuded cerebral arteries (4, 27). Diazoxide, a relatively selective activator of putative mitochondrial ATP-sensitive K+ (KATP) channels, has been shown to enhance the generation of reactive oxygen species (ROS) from mitochondria, which sequentially causes activation of ryanodine-sensitive Ca2+ channels on the sarcoplasmic reticulum, generation of Ca2+ transients called “Ca2+ sparks”, and opening of adjacent large-conductance Ca2+-activated K+ (BKCa) channels on the plasma membrane. The resulting K+ efflux leads to hyperpolarization, decreased global intracellular Ca2+, and vasodilation (4, 27). However, the contribution of mitochondrial factors in the endothelium on the integrated response of intact cerebral arteries or the effects of IR on individual and integrated VSM and endothelial contributions to mitochondria-mediated cerebral vascular responses have not been examined.
In this study, we hypothesized that mitochondria-derived factors from the endothelium also promote cerebral artery dilation and that IR leads to impaired mitochondria-dependent vasodilation in ZO rats. We examined the effects of diazoxide on cerebral arteries of young ZO rats and Zucker lean (ZL) control rats. Young ZO rats are normoglycemic and normotensive, but their plasma insulin levels are elevated, indicating IR (7, 8). We also examined the effects of 3-nitropropionic acid (3-NPA), a selective inhibitor of succinate dehydrogenase (SDH), since diazoxide also decreases the activity of this enzyme.
MATERIALS AND METHODS
The animal protocol was approved by the Institutional Animal Care and Use Committee of Wake Forest University Health Sciences. All experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male ZO and ZL rats (n = 30 each) were acquired at 10 wk of age and housed in the animal care facility. The animals received standard rat chow and tap water.
ZO rat model.
The ZO rat, with a leptin receptor mutation (fa/fa, homozygous for the mutation), has been widely used as a model of IR and type 2 diabetes, with ZL rats as genetically appropriate controls (Fa/fa, heterozygous for the mutation) (2). Previous data from our laboratory (7, 8) and others (9, 10, 22, 26) showed that ZO rats develop IR with a metabolic profile very similar to the human condition. As reported previously (7, 8), at 10–12 wk of age, ZO rats had significantly greater body weight and exhibited features typical of metabolic syndrome, including impaired glucose tolerance, higher plasma insulin levels (hyperinsulinemia), hypertriglyceridemia, and hypercholesterolemia. Importantly, young ZO rats used in the present study were IR, but glucose levels and blood pressure were not elevated.
Isolation of cerebral arteries and measurement of arterial diameter.
Videomicroscopy and diameter studies of isolated cerebral arteries were performed as described previously (16). Rats were decapitated under deep anesthesia, and the brains were removed and placed in an ice-cold oxygenated physiological saline solution (PSS). Cerebral arteries were isolated, transferred to a PSS-filled vessel bath (Chueltech Scientific Design, Houston, TX), cannulated with glass pipettes, and secured with 10-0 ophthalmic suture. Oxygenated (20% O2-5% CO2-75% N2) warmed PSS was circulated continuously through the vessel bath. The video camera on the microscope was connected to a video dimension analyzer (Living Systems, Burlington, VT) for measurement of intraluminal diameter. After an equilibration period of ∼20 min, arteries were gradually pressurized (50–60 mmHg) with PSS under no-flow conditions until stable myogenic tone (30–40% of passive diameter) developed. Subsequently, drugs were administered abluminally in the bath solution, and cumulative concentration responses were determined. Viability of arteries was verified by contraction to 60 mM KCl and vasodilation to bradykinin and sodium nitroprusside (SNP). The endothelium was removed by injection of a bolus of 1 ml of air through the arteries, and endothelial denudation was confirmed by the lack of response to bradykinin.
Vascular responses to diazoxide (10, 50, and 100 μmol/l) were determined in the presence and absence of iberiotoxin (a BKCa channel inhibitor, 100 nmol/l), manganese(III) tetrakis(4-benzoic acid)porphyrin chloride [MnTBAP, an SOD mimetic, 100 μmol/l], apocynin (an NADPH oxidase inhibitor, 1 mmol/l), Nω-nitro-l-arginine methyl ester [l-NAME, a nonselective nitric oxide (NO) synthase (NOS) inhibitor, 100 μmol/l], indomethacin [a general cyclooxygenase (COX) inhibitor, 10 μmol/l], and polyethylene glycol (PEG) catalase (an antioxidant enzyme that degrades H2O2, 500 U/ml). The vasodilation response to diazoxide was also determined after pretreatment with 3-NPA at a low concentration (300 μmol/l) known to selectively inhibit SDH. In addition, vasodilation responses solely to 3-NPA (3, 30, and 300 μmol/l) were determined. Pressure-dependent changes in diameter were evaluated in the arteries by increasing the intraluminal pressure in 20-mmHg increments from 20 to 160 mmHg. Pressure-dependent changes in passive diameter were determined in the presence of Ca2+-free PSS, and the myogenic tone for a given pressure was calculated by expression of the difference between active and passive diameters as a percentage of passive diameter. Diameter changes were expressed as percent relaxation after preconstriction or percent constriction with respect to passive diameter.
Measurement of mitochondrial membrane potential and ROS generation in intact arteries.
Fluorescence microscopy was performed as described previously (14). Arteries were treated with the mitochondrial membrane potential-sensitive dye tetramethylrhodamine ethyl ester (TMRE, 100 nmol/l; excitation at 543 nm, emission at >565 nm) or the superoxide indicator hydroethidine (HEt, 5 μmol/l; excitation at 488 nm, emission at >565 nm). For TMRE studies, after 30 min of loading, arteries were washed and allowed to recover myogenic tone before diazoxide administration (100 μmol/l). For HEt studies, arteries were treated with HEt and vehicle (control) or diazoxide (100 μmol/l). Images were acquired using a computer-controlled monochromatic excitation light source (Polychrome II, TILL-photonics, Martinsried, Germany) and a cooled charge-coupled device camera with exposure control. Fluorescence images of TMRE were obtained before and after diazoxide treatment; fluorescence images of HEt were acquired in the presence of vehicle or diazoxide. To minimize photobleaching and photodamage, exposure to excitation light was limited to 30 ms for all fluorescence studies. Image acquisition and offline analysis were performed with TILLvisION software. Under identical settings, the intensity of fluorescence was determined and expressed as relative fluorescence units (RFUs). The change in TMRE fluorescence was expressed as percent decrease in RFU after diazoxide treatment with respect to images before treatment, thus minimizing the effect of variations in TMRE loading. Similarly, the change in fluorescence intensity of HEt after diazoxide treatment was expressed as percent change from the HEt fluorescence measured in the absence of diazoxide.
Measurement of NO generation in intact arteries.
Arteries were prepared as described above, and the vessel bath was placed on an inverted microscope connected to a Zeiss LSM-510 laser scanning confocal system with a Zeiss Achroplan 40×/NA 0.8 water immersion objective. Arteries were treated with the NO-sensitive dye 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate (5 μmol/l; excitation at 488 nm, emission at >505 nm) for 60 min at room temperature. Then the arteries were washed with warm PSS for 15 min and allowed to recover myogenic tone before diazoxide administration (100 μmol/l). Fluorescence images of DAF-FM were acquired before and after diazoxide treatment using Zeiss software, with imaging conditions such as laser power, confocal aperture size, exposure length, and gain level held constant. Image analysis was performed offline with ImageJ software (National Institutes of Health). Under identical settings, the intensity of DAF-FM fluorescence was determined, and the change in fluorescence was expressed as percent increase in fluorescence after diazoxide treatment with respect to images before treatment, thus minimizing the effect of variations in loading.
Western blot analysis.
As previously described (11, 17), Western blot analyses for eNOS, neuronal NOS, Cu/Zn-SOD, Mn-SOD, and cytochrome c oxidase protein were performed using appropriate antibodies. The same amount of extracted protein from whole tissue homogenates of cerebral arteries was loaded for SDS-PAGE/immunoblot analysis. Each immunoband intensity was normalized to the corresponding immunoband intensity of β-actin, which was used as an internal control.
RNA isolation and RT-PCR analysis.
RNA was isolated from arteries using the SV Total Isolation Kit (Promega, Madison, WI) as directed by the manufacturer. The RNA was incubated with RQ1 DNase to eliminate any residual DNA. Real-time RT-PCR experiments were carried out in the ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). From each sample, 50 pg of total RNA were reverse transcribed and amplified using the Quantitect Probe RT-PCR kit (Qiagen, Valencia, CA) and TaqMan gene expression assays. The cycle program was as follows: 30 min at 50°C, 15 min at 95°C, and 40 cycles each at 94°C for 15 s and 60°C for 60 s. All amplifications were conducted with the PreDeveloped TaqMan gene expression assays, with GAPDH serving as an internal control. Assay identification numbers are as follows: Rn00566881_m1 for COX1, Rn00568225_m1 for COX2, Rn00566938_m1 for Cu/Zn-SOD, Rn00566942 for Mn-SOD, and Rn02132634_s1 for eNOS. The results were quantified as cycle threshold (Ct) values, where Ct is defined as the threshold cycle of PCR at which the amplified product is first detected and expressed as the ratio of target to control.
Drugs, chemicals, and solutions.
MnTBAP was purchased from Calbiochem (San Diego, CA) and TMRE, HEt, and DAF-FM from Molecular Probes (Eugene, OR); all other chemicals were obtained from Sigma (St. Louis, MO). Stock solutions (10 mM) of diazoxide and apocynin were prepared in DMSO, and the other chemicals were prepared in deionized water. The composition of PSS (mmol/l) was 112 NaCl, 4.8 KCl, 26 NaHCO3, 1.2 KH2PO4·H2O, 1.8 CaCl2, 1.2 MgSO4·7 H2O, and 10 glucose. PSS with 60 mM KCl was prepared by replacement of NaCl with an equimolar quantity of KCl. Ca2+-free PSS was prepared by replacement of CaCl2 with an equimolar quantity of MgSO4·7 H2O with 2 mmol/l EGTA.
Data analysis and statistics.
Values are means ± SE. The number of independent experiments is represented as n, which represents the number of animals used for the experiments. Means were compared by one-way ANOVA. Post hoc analysis was done by Tukey's test, with P < 0.05 considered statistically significant.
RESULTS
Cerebral arteries were of similar passive diameter and, upon pressurization, preconstricted to a similar extent in ZL and ZO rats (Table 1). Although diazoxide induced concentration-dependent dilation of cerebral arteries preconstricted by myogenic tone, the dilation was significantly reduced in ZO compared with ZL rats (Table 1, Fig. 1A). Similar results were observed in cerebral arteries preconstricted by serotonin (Table 1, Fig. 1B), indicating that mechanisms of depolarization are unlikely to affect the diazoxide response.
Table 1.
Changes in arterial diameter after administration of various agents
| ZL | ZO | |
|---|---|---|
| Passive diameter, μm | 188±6 (33) | 189±6 (33) |
| Preconstriction, % | 37.1±2 (18) | 34.1±3 (10) |
| Endothelium intact arteries | ||
| SNP-induced dilation, % | 88.5±8 (7) | 86.5±4 (7) |
| 3-NPA-induced dilation, % | 11±2 (10) | 17±4‡ (9) |
| Diazoxide-induced dilation, % | ||
| Preconstricted by myogenic tone | 38.0±3 (18) | 20.9±4* (10) |
| Preconstricted by serotonin | 29.8±4 (9) | 16.6±2* (8) |
| Pretreated with iberiotoxin | 15.4±2* (10) | 5.2±3†‡ (8) |
| Pretreated with MnTBAP | 13.9±3* (6) | 6.2±3† (7) |
| Pretreated with iberiotoxin + MnTBAP | 17.6±3* (5) | 7.8±1†‡ (5) |
| Pretreated with apocynin | 37.6±2 (6) | 16.9±3‡ (6) |
| Pretreated with l-NAME | 14.8±3* (5) | 14.3±3 (8) |
| Pretreated with indomethacin | 60.1±5* (6) | 13.4±2‡ (6) |
| Pretreated with 3-NPA | 82.8±7* (9) | 75.1±6† (9) |
| Endothelium-denuded arteries | ||
| Diazoxide-induced dilation, % | ||
| Preconstricted by myogenic tone | 28.0±3* (9) | 7.8±2†‡ (10) |
| Pretreated with l-NAME | 24.8±6 (5) | 32.8±5† (8) |
| Pretreated with indomethacin | 26.1±4 (5) | 33.7±6‡ (5) |
Values are means ± SE of number of vessels in the parentheses. Maximal relaxation was calculated as a percent maximal decrease in preconstriction; constriction was calculated by determining the decrease in diameter and expressing it as a percent of passive diameter of the arteries. ZL, Zucker lean; ZO, Zucker obese; 5-HT, serotonin; MnTBAP, manganese (III) tetrakis(4-benzoic acid)porphyrin chloride; l-NAME, Nω-nitro-l-arginine methyl ester; 3-NPA, 3-nitropropionic acid; SNP, sodium nitroprusside.
Significantly different from response to diazoxide alone in ZL arteries (P < 0.05).
Significantly difference from response to diazoxide alone in ZO arteries (P < 0.05).
Significant different from similarly pretreated ZL arteries (P < 0.05).
Fig. 1.

Responses of cerebral arteries from Zucker obese (ZO) and Zucker lean (ZL) rats to diazoxide (10, 50 and 100 μmol/l). A: endothelium-intact and -denuded cerebral arteries preconstricted by myogenic tone. B: cerebral arteries from ZO and ZL rats preconstricted by serotonin. Values are means ± SE of 10–18 experiments. *Significantly different from lean endothelium-intact arteries (P < 0.05); in some cases, 2 points overlap (**). †Significantly different from obese endothelium-intact arteries (P < 0.05).
In ZL arteries, pretreatment with iberiotoxin diminished the diazoxide response, suggesting that BKCa channels mediate the majority of the vasodilation to diazoxide (Fig. 2A). However, a residual iberiotoxin-insensitive vasodilation was observed, suggesting that part of the diazoxide-induced vasodilation was independent of BKCa channels. Similarly, MnTBAP reduced the dilation to diazoxide in ZL arteries, indicating that ROS play a major role in vasodilation (Fig. 2A). A small proportion of the vasodilation was MnTBAP insensitive, suggesting that the diazoxide response was also mediated by ROS-independent mechanisms. Pretreatment with iberiotoxin + MnTBAP resulted in inhibition of the diazoxide response in ZL arteries comparable to that observed when each drug was used alone (Fig. 2A). In contrast, vasodilation to diazoxide in the presence of PEG-catalase failed to decrease the response in ZL arteries [40.4 ± 5%, n = 5, P = not significant (NS)]. This suggests that H2O2 was not the mediator of diazoxide-induced vasodilation in cerebral arteries from ZL rats.
Fig. 2.

A: responses of cerebral arteries from ZL rats to diazoxide (10, 50, and 100 μmol/l) in the presence and absence of iberiotoxin (100 nmol/l), manganese(III) tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP, 100 μmol/l), or iberiotoxin + MnTBAP. B and C: diazoxide-induced vasodilation in endothelium-intact and -denuded arteries in the presence and absence of Nω-nitro-l-arginine methyl ester (l-NAME, 100 μmol/l) and indomethacin (10 μmol/l). Baseline represents diazoxide response in endothelium-intact arteries in the absence of drugs. Values are means ± SE of 5–18 experiments. *Significantly different from lean endothelium-intact arteries (P < 0.05); in some cases, 3 points overlap (***).
Removal of the endothelium resulted in reduced dilation in response to diazoxide in ZL arteries (Fig. 1A). In addition, pretreatment of arteries with l-NAME resulted in diminished diazoxide-induced vasodilation in ZL rats (Fig. 2B), suggesting that NO mediates diazoxide-induced vasodilation. In contrast, inhibition of COX with indomethacin resulted in enhanced dilation to diazoxide in ZL arteries (Fig. 2B), indicating that diazoxide activated the generation of vasoactive prostanoids by COX. The dominant effect of these prostanoids, however, appeared to be vasoconstriction. Neither l-NAME nor indomethacin affected vasodilation in endothelium-denuded arteries, suggesting that NOS and COX do not mediate vasodilation in endothelium-denuded arteries (Fig. 2C). Together, these results indicate that vascular actions of diazoxide in ZL arteries were endothelium and VSM dependent.
In ZO arteries, iberiotoxin pretreatment almost abolished the vasodilation. In addition, the maximal vasodilation in response to diazoxide was significantly diminished compared with that in iberiotoxin-pretreated ZL arteries (Fig. 2C). These findings suggest that BKCa channels were proportionally more active in ZO than ZL arteries. However, pretreatment with iberiotoxin reduced the vasodilation to a similar degree in arteries from both groups. Thus the BKCa channel activation by diazoxide was greater in ZO than ZL arteries. In addition, MnTBAP pretreatment diminished vasodilation in response to diazoxide in ZO arteries similar to ZL arteries (Fig. 2C). In contrast to ZL arteries, vasodilation insensitive to iberiotoxin and MnTBAP was small to insignificant in ZO arteries. Pretreatment with iberiotoxin + MnTBAP elicited vasodilation to diazoxide similar to that elicited by iberiotoxin or MnTBAP alone.
Denudation of the endothelium resulted in reduced vasodilation in ZO arteries (Fig. 1A). Notably, the diazoxide response was diminished not only in intact arteries but also in endothelium-denuded arteries compared with the response in corresponding arteries in ZL rats. This suggested that both endothelium- and VSM-dependent vasodilation to diazoxide were impaired in arteries from ZO rats. Moreover, l-NAME pretreatment did not significantly diminish diazoxide-induced vasodilation in intact arteries from ZO rats, indicating that the contribution of NO to vasodilation was minimal in ZO arteries (Fig. 3B). Interestingly, in ZO rats, endothelial denudation diminished vasodilation profoundly compared with l-NAME pretreatment (Fig. 3B). In contrast, l-NAME pretreatment of endothelium-denuded arteries paradoxically enhanced vasodilation to diazoxide (Fig. 3C). This implies that 1) endothelium-denuded arteries in ZO rats exhibit activity by other NOS isoforms and 2) the NOS isoforms in these arteries generate a vasoconstricting factor. Alternatively, it is possible that, in the setting of diminished NO bioavailability, ZO arteries rely on alternative mediators of vasodilation, such as prostacyclin. Incidentally, in contrast to ZL arteries, indomethacin did not enhance the vasodilation response to diazoxide in intact ZO arteries, suggesting that vasoconstrictor COX metabolites did not mediate vascular responses to diazoxide (Fig. 3B). In contrast, indomethacin pretreatment of endothelium-denuded arteries paradoxically enhanced vasodilation to diazoxide (Fig. 3C), suggesting that VSM produces vasoconstrictor prostanoids in ZO arteries.
Fig. 3.

A: responses of cerebral arteries from ZO rats to diazoxide (10, 50, and 100 μmol/l) in the presence and absence of iberiotoxin (100 nmol/l), MnTBAP (100 μmol/l), or iberiotoxin + MnTBAP. B and C: diazoxide-induced vasodilation in endothelium-intact and -denuded arteries in the presence and absence of l-NAME (100 μmol/l) and indomethacin (10 μmol/l). Baseline represents diazoxide response in endothelium-intact arteries in the absence of drugs. Values are means ± SE of 5–18 experiments. *Significantly different from baseline in obese endothelium-intact arteries (P < 0.05); in some cases, 2 points overlap (**). †Significantly different from obese endothelium-denuded arteries (P < 0.05); in some cases, 2 points overlap (††).
Inhibition of SDH with 3-NPA caused modest vasodilation in cerebral arteries. At 0.3 mmol/l 3-NPA induced 1 ± 1% and 1 ± 3% vasodilation in ZL and ZO rats, respectively. In contrast, at 30 mmol/l 3-NPA caused significantly greater vasodilation in ZO than ZL rats (13 ± 4% vs. 5 ± 2%, P < 0.05). However, maximal vasodilation to 3-NPA was statistically not enhanced in ZO compared with ZL arteries (Fig. 4A, Table 1). Additionally, pretreatment with 3-NPA resulted in a profound increase in the diazoxide response in ZO and ZL arteries, with identical maximal relaxation in both groups (Fig. 4B, Table 1).
Fig. 4.

A: responses to 3-nitropropionic acid (3-NPA; 3, 30, and 300 μmol/l) in cerebral arteries from ZL and ZO rats. B: maximal responses in cerebral arteries from ZO and ZL rats to 3-NPA (300 μmol/l), diazoxide (100 μmol/l), and diazoxide + 3-NPA. Values are means ± SE of 9–18 experiments. *Significantly different from lean (P < 0.05).
In contrast, apocynin had no effect on the diazoxide response in ZL or ZO arteries (Fig. 5A), indicating that diazoxide-induced ROS generation was independent of NADPH oxidase. In addition, changes in intraluminal diameter in response to an increase in intraluminal pressure, in the absence of any drugs, were similar in ZL and ZO arteries (Fig. 5B), suggesting that myogenic tone was largely unchanged in ZO arteries.
Fig. 5.

A: responses to diazoxide (10, 50, and 100 μmol/l) in cerebral arteries from ZL and ZO rats in the presence and absence of apocynin (100 μmol/l). Values are means ± SE of 6–18 experiments. B: myogenic tone of isolated cerebral arteries from ZL and ZO arteries in response to increments in intraluminal pressure. Values are means ± SE of 9–12 experiments.
Arteries took ∼20–30 min to attain a stable diameter before and after the administration of pharmacological agents other than diazoxide + 3-NPA. Compared with vehicle-treated arteries (0.85 ± 1% in ZL and 1.3 ± 1% in ZO, n = 10 each), pretreatment with l-NAME [26 ± 5% in ZL (n = 9) and 25 ± 4% in ZO (n = 14), P < 0.05] and iberiotoxin [14 ± 4% in ZL (n = 5) and 15 ± 4% in ZO (n = 4), P < 0.05] caused constriction of all arteries, whereas indomethacin pretreatment induced vasodilation [19 ± 8% in ZL (n = 5) and 13 ± 3% in ZO (n = 8), P < 0.05]. However, pretreatment with MnTBAP (4 ± 2% in ZL and 2 ± 1% in ZO, n = 5 each), apocynin [1 ± 2% in ZL (n = 9) and 2 ± 1% in ZO (n = 7)], and catalase (0.2 ± 0.7% in ZL, n = 5, P = NS) had no significant effect on vascular diameter. The reduction of dilation to diazoxide in arteries from ZO rats is not a generalized response, since arterial responsiveness to SNP was similar in both groups (Table 1).
Fluorescence studies with TMRE showed similar baseline TMRE fluorescence intensity in pressurized arteries from ZL and ZO rats, suggesting that mitochondrial membrane potential was comparable in both groups. TMRE fluorescence (in RFUs) was 200 ± 28 in ZL (n = 6) vs. 190 ± 7 in ZO (n = 5) arteries (P = NS). Application of diazoxide to the cerebral arteries loaded with TMRE decreased TMRE fluorescence intensity, indicating mitochondrial membrane depolarization (Fig. 6, A and B). Control experiments with administration of other vasodilators such as SNP did not reduce TMRE fluorescence, indicating that changes in TMRE fluorescence were not due to diameter changes resulting from vasodilation. However, the decrease in TMRE fluorescence intensity was reduced in ZO compared with ZL arteries, suggesting a diminished mitochondrial depolarization in response to diazoxide (Fig. 6, A and B). In addition, application of diazoxide, along with HEt, to arteries resulted in enhanced HEt fluorescence intensity, indicating increased ROS generation. Diazoxide-induced increases in HEt fluorescence were greater in ZL than ZO arteries, suggesting impaired mitochondrial production of ROS in ZO arteries (Fig. 6, C and D). Measurements of NO generation indicated that vasodilation to diazoxide was accompanied by increased DAF-FM fluorescence. However, the increase in DAF-FM fluorescence following diazoxide administration was markedly reduced in ZO compared with ZL arteries (Fig. 6, E and F), suggesting that diazoxide elicited diminished NO production in ZO arteries.
Fig. 6.
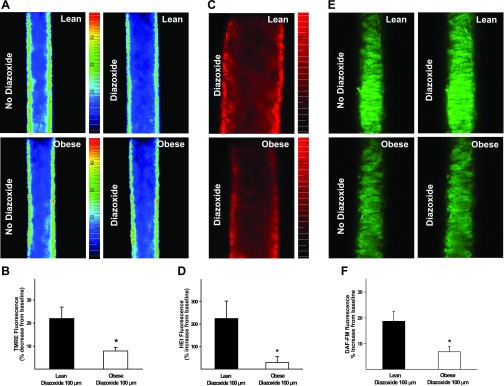
Membrane depolarization, reactive oxygen species (ROS) generation, and nitric oxide (NO) production in isolated pressurized cerebral arteries of ZO and ZL rats before and after diazoxide (100 μmol/l) administration.A: fluorescence images of the mitochondrial membrane potential-sensitive dye tetramethylrhodamine ethyl ester (TMRE) in ZL (top) and ZO (bottom) arteries in the absence and presence of diazoxide. B: TMRE fluorescence response to diazoxide. Reduction of diazoxide-induced decrease in TMRE fluorescence in ZO compared with ZL arteries indicates reduced mitochondrial depolarization. C: fluorescence images of the ROS sensitive dye hydroethidine (HEt) in ZL (top) and ZO (bottom) arteries following diazoxide administration. D: HEt fluorescence response to diazoxide compared with vehicle. E: fluorescence images of the NO-sensitive dye 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM) in ZL (top) and ZO (bottom) arteries in the absence and presence of diazoxide. F: DAF-FM fluorescence response to diazoxide compared with vehicle. Reduction of diazoxide-induced increase in HEt and DAF-FM fluorescence in ZO compared with ZL arteries indicates decreased generation of ROS and NO, respectively. *Significantly different from lean (P < 0.05).
Western blot analysis of artery homogenates showed increased protein levels (expressed in RFUs normalized to β-actin) of eNOS (0.29 ± 0.04 in ZL vs. 0.58 ± 0.05 in ZO, n = 7 each, P < 0.05), Cu/Zn-SOD [0.73 ± 0.06 (n = 6) in ZL vs. 0.98 ± 0.04 in ZO (n = 7), P < 0.05], and Mn-SOD (0.56 ± 0.02 in ZL vs. 0.76 ± 0.05 in ZO, n = 7 each, P < 0.05) in ZO compared with ZL arteries, while neuronal NOS [1.08 ± 0.05 in ZL (n = 9) vs. 1.18 ± 0.09 in ZO (n = 8), P = NS] levels remained unaltered. In addition, cytochrome c oxidase protein levels are similar in both groups (0.93 ± 0.1 in ZL vs. 0.85 ± 0.11 in ZO, n = 7 each, P = NS), indicating that expression of a major enzyme in the electron transport chain of the mitochondria was unchanged in ZO arteries (Fig. 7, A and B).
Fig. 7.

A: immunoblots showing expression of endothelial NO synthase (eNOS), neuronal NO synthase (nNOS), Cu/Zn-SOD, Mn-SOD, and cytochrome c with matching β-actin blots. Protein samples of homogenates from rat cortex were used as positive control (+). L, lean; O, obese. B: cumulative group data. Cyt C, cytochrome c. C: mRNA expression levels of eNOS, cyclooxygenase-1 (COX1), COX2, Cu/Zn-SOD, and Mn-SOD normalized to GAPDH levels. *Significantly different from lean (P < 0.05).
Measurement of mRNA levels in cerebral arteries showed increased expression of eNOS [0.95 ± 0.17 in ZL (n = 8) vs. 2.61 ± 0.66 in ZO (n = 9), P < 0.05] and Cu/Zn-SOD [1.0 ± 0.1 in ZL (n = 9) vs. 1.22 ± 0.1 in ZO (n = 8), P < 0.05] in ZO compared with ZL arteries, while mRNA levels of COX1 (1.04 ± 0.11 in ZL vs. 1.22 ± 0.07 in ZO, n = 8 each, P = NS), COX2 (1.05 ± 0.13 in ZL vs. 0.90 ± 0.11 in ZO, n = 8 each, P = NS), and Mn-SOD (1.02 ± 0.6 in ZL vs. 1.09 ± 0.11 in ZO, n = 10 each, P = NS) were unchanged (Fig. 7C).
DISCUSSION
Our study represents the first report of the integrated vascular responses to mitochondria-derived influences in intact arteries from the cerebral circulation as well as the examination of the detrimental effects of IR. Cerebrovascular dilation to mitochondria-derived factors involves the interaction between VSM and endothelial factors, and IR has a negative impact on vasodilation influences at both levels. The major findings of the present study are as follows. 1) Diazoxide-induced vasodilation was diminished in intact cerebral arteries from ZO compared with ZL rats. BKCa channels and ROS mediate the majority of vasodilation to diazoxide in both groups, although other, yet undefined, mechanisms promote a significant amount of the remaining vasodilation. Diazoxide-induced vasodilation has endothelial and VSM components, both of which were diminished in ZO compared with ZL arteries. 2) Vasodilation to diazoxide was mediated in part by NO in both groups, whereas the contribution of NO was diminished in ZO arteries. Paradoxically, NOS isoforms in ZO arteries promoted vasoconstriction. 3) In contrast to diazoxide, the dilator effects of 3-NPA were not reduced in ZO compared with ZL arteries. Rather, coapplication of 3-NPA restored normal responsiveness to diazoxide in ZO arteries. 4) Diazoxide-induced dilation was associated with impaired depolarization of the mitochondria and an inability to enhance ROS generation in ZO arteries. 5) Levels of eNOS and the antioxidant enzyme Mn-SOD were increased in ZO compared with ZL arteries, thus indicating that IR has a major impact on relevant gene expression.
Mitochondrial depolarization.
In isolated and pressurized cerebral arteries, diazoxide induced mitochondrial depolarization and vasodilation, but both of these responses were diminished in ZO compared with ZL arteries. Diazoxide-induced mitochondrial depolarization was attributed to activation of putative mitochondrial KATP channels. As we previously observed in hearts of ZO rats (15), the reduced mitochondrial membrane depolarization was due to an impaired activation of mitochondrial KATP channels. This is also consistent with our earlier report that plasmalemmal KATP channels of cerebral VSM are impaired in genetic (ZO) (6) and nutritional (fructose-fed) (6) rat models of IR.
The mitochondria-specific membrane potential-sensitive dye TMRE is a cationic, membrane-permeable dye that reversibly accumulates in the negatively charged mitochondrial matrix according to the Nernst equation potential. Thus TMRE fluorescence directly reflects mitochondrial membrane potential. Moreover, TMRE does not bind to the plasma membrane or accumulate in the cytoplasm and, therefore, does not measure plasma membrane potential. However, changes in plasma membrane potential indirectly affect changes in mitochondrial membrane potential in response to various agents by altering the membrane potential gradient between mitochondrial and plasma membrane. Activation of plasmalemmal KATP channels leads to hyperpolarization of the plasma membrane and reduced potential gradient across the mitochondrial membrane, which inhibits mitochondrial depolarization. In contrast, activation of mitochondrial KATP channels leads to mitochondrial depolarization accompanied by decreased TMRE fluorescence. Thus activation of plasmalemmal vs. mitochondrial KATP channels has different effects on TMRE fluorescence (25). Therefore, the reduced mitochondrial membrane depolarization in ZO arteries was unrelated to impaired plasmalemmal KATP channels but indicates specific impairment of mitochondrial KATP channels. Thus, regardless of the site within vascular cells, KATP channels appear to be vulnerable to effects of IR.
Mitochondrial ROS.
Diazoxide-induced ROS generation was also diminished in ZO compared with ZL arteries. Although mitochondrial ROS production is believed to be a result of membrane depolarization, we recently showed that these events are independent of each other. For example, BMS-191095, a selective activator of mitochondrial KATP channels, depolarizes mitochondria without affecting ROS production or ATP synthesis (3, 11). The mechanisms underlying the generation of ROS by mitochondria are complex, and there is no clear consensus concerning the relative role of membrane potential-dependent and -independent mechanisms mediating the phenomenon. Since we have shown in isolated mitochondria that 3-NPA generates ROS but does not cause membrane depolarization, it is also likely that the ROS generation is a result of the direct effect of diazoxide on SDH. Thus, although diazoxide activates mitochondrial KATP channels and inhibits SDH, effects of diazoxide at the doses used were selective for mitochondria. Impairment of ROS production in response to diazoxide was also observed in isolated mitochondria from ZO hearts (15) and from diabetic human myocardium (12). Dilator responses to 3-NPA were modestly enhanced in arteries from ZO rats, although maximal vasodilation was unaffected, indicating that all mitochondria-derived dilator mechanisms were not reduced by IR. Thus the more potent vasodilation caused by diazoxide than by 3-NPA highlights the greater importance of non-SDH mechanisms. In addition, a variable increase in expression of the mitochondrial isoform of SOD, Mn-SOD, as well as the cytosolic isoform, Cu/Zn-SOD, was found in ZO arteries. Although it is likely to be a minor mechanism, enhanced scavenging may contribute to the diminished mitochondrial ROS generation in response to diazoxide in ZO arteries.
Vasodilation.
Diazoxide-induced dose-dependent vasodilation of endothelium-denuded cerebral arteries from ZL and ZO rats was consistent with the characteristics of diazoxide-induced vasodilation reported previously (4, 27). Vasodilation in endothelium-intact arteries was greatly reduced by iberiotoxin, thus confirming a predominant role for BKCa channels. Similarly, diazoxide-induced vasodilation was also reduced by an SOD mimetic, suggesting an essential role for ROS in mediating vasodilation. Importantly, inhibition of BKCa channels and ROS scavenging together or separately elicited similar inhibition of diazoxide-induced vasodilation in all arteries, suggesting that BKCa channels and ROS mediate the vasodilation to diazoxide in sequence.
Diazoxide-induced vasodilation, however, was diminished in cerebral arteries of ZO compared with ZL rats. This was not a result of alterations in the myogenic tone of arteries, because, in contrast to previous reports (24) in adult Zucker rats, the young rats used in our study exhibited identical myogenic tone in all arteries. Moreover, all arteries were preconstricted to a similar degree. Furthermore, the mechanism of vascular depolarization does not appear to affect the diazoxide response in ZO and ZL rats, as preconstriction of arteries with serotonin elicited similar diminished vasodilation to diazoxide in ZO and ZL (Fig. 1). In addition, the diminished vasodilation was specific to diazoxide, since the vasodilation to SNP was preserved in ZO arteries. Despite the reduction in the diazoxide response in ZO arteries, the majority of the dilation in each group appears to be dependent on BKCa channels, ROS, and NO, because inhibition of each of them largely abolished the diazoxide response. However, there was considerable residual vasodilation, which may involve protein kinases (11) and may partly contribute to diazoxide-induced vasodilation. The BKCa channel-independent vasodilation to diazoxide was also reduced in ZO compared with ZL arteries. This finding is consistent with our earlier observation of diminished plasma membrane BKCa channel-mediated vasodilation in ZO rats (6).
Interestingly, apocynin pretreatment had no effect on the diazoxide response in all arteries. Recent observations by Heumuller et al. (13) suggested that apocynin inhibits ROS production in vascular systems by scavenging, rather than inhibiting, NADPH oxidase, as previously reported. Although it is unclear why apocynin and MnTBAP had different effects on the diazoxide response, it is likely that localization of MnTBAP in the vascular cell allowed more effective scavenging of mitochondrial ROS than apocynin. Lack of attenuation of vasodilation in cerebral arteries to diazoxide by catalase suggests that superoxide anion, rather than H2O2, is involved.
Endothelial mechanisms.
Removal of the endothelium diminished the vasodilation to diazoxide, implying that endothelium-derived factors such as NO and prostaglandins contribute to vasodilation. Furthermore, the contribution of NO is supported by the finding that inhibition of NOS greatly diminished the diazoxide-induced vasodilation in ZL arteries. The present findings are consistent with previous reports that K+ channel openers increase endothelial NO production and Ca2+ influx in endothelial cells mediates enhanced NO generation (18–20). However, it is unclear whether diazoxide-induced mitochondrial ROS generation results in generation of Ca2+ transients in endothelial cells similar to the Ca2+ sparks observed in VSM cells that lead to activation of surface KCa channels in endothelial cells. In ZO arteries, diazoxide was relatively ineffective in activating NO production, despite an enhanced expression of eNOS. Fluorescence measurements of NO in arteries confirmed the production of NO in response to diazoxide. However, NO production was reduced in ZO compared with ZL arteries. Although it is unclear why overexpression of eNOS failed to enhance NO generation in response to diazoxide, it is possible that IR may have altered the signaling of eNOS activation (21) or impaired the eNOS activity by uncoupling eNOS (5). Interestingly, in the ZL arteries, NOS inhibition reduced the diazoxide response to a greater degree than endothelial denudation, suggesting that NO modulates vasodilation more than other endothelium-derived factors. Thus it is likely that diazoxide may induce NO release, as well as endothelial factors that counteract the NO, which thereby diminish the effectiveness of NO in vasodilation. Indeed, a major contribution of constrictor prostanoids appears to be involved, since inhibition of COX with indomethacin enhanced the vasodilation to diazoxide in ZL arteries. In ZO arteries, however, COX inhibition reduced dilation, suggesting that counteracting constrictor prostanoids are not involved in modulation of the vascular response to diazoxide. The inhibition of NOS in endothelium-denuded arteries enhanced vasodilation in ZO rats, suggesting the existence of NOS isoforms in endothelium-denuded arteries that release vasoconstrictor factors. It may therefore be speculated that NOS has the ability to generate ROS when uncoupled, and some ROS forms are implicated in vasoconstriction. Further studies are required to fully establish the role of NOS in ZO arteries. Similarly, inhibition of COX also enhanced vasodilation in endothelium-denuded ZO arteries, suggesting that VSM cells generate vasoconstrictor prostanoids. A schematic of the endothelium- and VSM-dependent mechanisms underlying the mitochondrial regulation of cerebrovascular tone, along with the potential pathways where IR causes disruption of normal vasoreactivity, is shown in Fig. 8.
Fig. 8.

Schematic representation of proposed mitochondrial regulation of vascular tone in cerebral arteries. ×, Potential sites of interruption in the pathways leading to impaired mitochondria-dependent vasodilation in ZO arteries; dashed arrows, pathways that have not been resolved. ER, endoplasmic reticulum; SR, sarcoplasmic reticulum; PGs, prostaglandins; KATP channels, ATP-sensitive K+ channels; IR, insulin-resistant; SDH, succinate dehydrogenase; [Ca2+], Ca2+ concentration.
eNOS and SOD expression.
Measurements of protein and mRNA showed a significant increase in expression of eNOS but a very modest increase in expression of Cu/Zn-SOD in ZO arteries. Mn-SOD, however, showed a modest increase in protein but no change in mRNA levels in ZO arteries. Posttranscriptional events, such as stability of mRNA, may possibly explain the variation in Mn-SOD expression. Although it is unclear why eNOS, Mn-SOD, and Cu/Zn-SOD expression was enhanced in arteries from ZO rats, it may be a compensatory response to the increased oxidative stress observed in the vascular wall (7, 10, 15, 16, 24, 26).
Limitations.
Several experimental limitations should be considered. 1) Mitochondria-active pharmacological agents are limited in number, have cytotoxic effects beyond a narrow dose range, and/or have unproven specificity. While diazoxide can have other effects, administration of this drug at the doses used in this study appear to be specific for selective activation of mitochondria-centered mechanisms, which lead to vasodilation. Although the involvement of physiological and pathological stimuli in activation of mitochondria-derived dilation mechanisms has not been specifically studied, it seems likely that factors such as increased shear stress, ischemia-reperfusion, and clinical drugs could play a role (15, 16, 23, 28). Thus the present study attempts to highlight the impaired mitochondrial mechanisms of regulation of vascular tone in ZO rats. 2) ROS measurements in our studies determined the total cellular ROS but not specifically mitochondria-derived ROS. Under the conditions of our studies, HEt elicited reliable responses compared with mitochondria-specific probes of ROS. However, ROS generation in response to diazoxide has been reported to be mitochondrial in origin. To our knowledge, diazoxide has not been shown to enhance ROS generation from nonmitochondrial sources in the cells. Thus it is reasonable to assume that an increase in HEt fluorescence in response to diazoxide represents mitochondrial ROS generation.
Perspectives and Significance
Diazoxide-induced vasodilation involves an interaction between VSM and endothelial mechanisms and was diminished in cerebral arteries from ZO rats. This appears to be due to impairment of mitochondrial depolarization and ROS generation in response to diazoxide. In addition, decreased NO production, despite increased eNOS expression, appears to contribute to diminished vasodilation. Our study is the first to indicate that mitochondria-derived factors in the endothelium are able to affect the total vasoactive responses in cerebral arteries. Furthermore, we present new data that indicate that derangements of mitochondrial function have substantial, negative effects on cerebral arterial responsiveness. Therefore, the present study establishes a new mechanism contributing to the cerebral vascular dysfunction in IR.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-077731, HL-030260, and HL-065380 and Hungarian Scientific Research Fund Grant OTKA K68976.
Acknowledgments
We thank Nancy Busija for help with editing the manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Arenillas JF, Moro MA, Davalos A. The metabolic syndrome and stroke: potential treatment approaches. Stroke 38: 2196–2203, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Bray GA The Zucker-fatty rat: a review. Fed Proc 36: 148–153, 1977. [PubMed] [Google Scholar]
- 3.Busija DW, Katakam P, Rajapakse NC, Kis B, Grover G, Domoki F, Bari F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res Bull 66: 85–90, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol 556: 755–771, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest 116: 1071–1080, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erdos B, Simandle SA, Snipes JA, Miller AW, Busija DW. Potassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke 35: 964–969, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Erdos B, Snipes JA, Miller AW, Busija DW. Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes 53: 1352–1359, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Erdos B, Snipes JA, Tulbert CD, Katakam P, Miller AW, Busija DW. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol Heart Circ Physiol 290: H1264–H1270, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Frisbee JC Hypertension-independent microvascular rarefaction in the obese Zucker rat model of the metabolic syndrome. Microcirculation 12: 383–392, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Frisbee JC, Stepp DW. Impaired NO-dependent dilation of skeletal muscle arterioles in hypertensive diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol 281: H1304–H1311, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Gaspar T, Snipes JA, Busija AR, Kis B, Domoki F, Bari F, Busija DW. ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095. J Cereb Blood Flow Metab 28: 1090–1103, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Hassouna A, Loubani M, Matata BM, Fowler A, Standen NB, Galinanes M. Mitochondrial dysfunction as the cause of the failure to precondition the diabetic human myocardium. Cardiovasc Res 69: 450–458, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Jarajapu YP, Oomen C, Uteshev VV, Knot HJ. Histamine decreases myogenic tone in rat cerebral arteries by H2-receptor-mediated KV channel activation, independent of endothelium and cyclic AMP. Eur J Pharmacol 547: 116–124, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Katakam PV, Jordan JE, Snipes JA, Tulbert CD, Miller AW, Busija DW. Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol 292: R920–R926, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Katakam PV, Tulbert CD, Snipes JA, Erdos B, Miller AW, Busija DW. Impaired insulin-induced vasodilation in small coronary arteries of Zucker obese rats is mediated by reactive oxygen species. Am J Physiol Heart Circ Physiol 288: H854–H860, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Kis B, Snipes JA, Simandle SA, Busija DW. Acetaminophen-sensitive prostaglandin production in rat cerebral endothelial cells. Am J Physiol Regul Integr Comp Physiol 288: R897–R902, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Langheinrich U, Mederos y Schnitzler M, Daut J. Ca2+-transients induced by K+ channel openers in isolated coronary capillaries. Pflügers Arch 435: 435–438, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Luckhoff A, Busse R. Activators of potassium channels enhance calcium influx into endothelial cells as a consequence of potassium currents. Naunyn Schmiedebergs Arch Pharmacol 342: 94–99, 1990. [DOI] [PubMed] [Google Scholar]
- 20.Luckhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflügers Arch 416: 305–311, 1990. [DOI] [PubMed] [Google Scholar]
- 21.Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase C-β inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes 55: 691–698, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Oltman CL, Richou LL, Davidson EP, Coppey LJ, Lund DD, Yorek MA. Progression of coronary and mesenteric vascular dysfunction in Zucker obese and Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 291: H1780–H1787, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol 292: H93–H100, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Phillips SA, Sylvester FA, Frisbee JC. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R522–R530, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Poppe M, Reimertz C, Dussmann H, Krohn AJ, Luetjens CM, Bockelmann D, Nieminen AL, Kogel D, Prehn JH. Dissipation of potassium and proton gradients inhibits mitochondrial hyperpolarization and cytochrome c release during neural apoptosis. J Neurosci 21: 4551–4563, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stepp DW Impact of obesity and insulin resistance on vasomotor tone: nitric oxide and beyond. Clin Exp Pharmacol Physiol 33: 407–414, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res 97: 354–362, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292: H2023–H2031, 2007. [DOI] [PubMed] [Google Scholar]