

Abstract
1. The transport of negatively charged drugs, xenobiotics, and metabolites by epithelial tissues, particularly the kidney, plays critical roles in controlling their distribution, concentration, and retention in the body. Thus, organic anion transporters (OATs) impact both their therapeutic efficacy and potential toxicity.
2. This review summarizes current knowledge of the properties and functional roles of the cloned OATs, the relationships between transporter structure and function, and those factors that determine the efficacy of transport. Such factors include plasma protein binding of substrates, genetic polymorphisms among the transporters, and regulation of transporter expression.
3. Clearly, much progress has been made in the decade since the first OAT was cloned. However, unresolved questions remain. Several of these issues — drug–drug interactions, functional characterization of newly cloned OATs, tissue differences in expression and function, and details of the nature and consequences of transporter regulation at genomic and intracellular sites — are discussed in the concluding Perspectives section.
Keywords: Organic ion transporters, drug transporters, cloning, transport mechanisms, tissue distribution, SLC22A family, expression levels, structure–function
Introduction
It has been recognized for nearly a century and a half that the kidney has the capacity to eliminate certain chemicals very effectively (reviewed by Pritchard and Miller 1993). These early studies were possible because several coloured anionic dyes are highly concentrated in the urine where their presence is easily observed. Subsequently, these observations led to development of the first clinical test for renal function (Rowntree and Geraghty 1909), proof of renal tubular secretion (Marshall and Vickers 1923; Marshall and Grafflin 1928, 1932), and a quantitative measure of renal blood flow (Smith et al. 1938). More recently, research into the nature of organic anion transport has focused on how this strikingly effective transport is achieved.
The earliest in vitro studies using renal slices (Cross and Taggart 1950) demonstrated that the process is dependent upon metabolic energy and on the presence of sodium in the extracellular space, and that a number of metabolic intermediates stimulate transport. Nevertheless, the mechanism of organic anion transport and its coupling to metabolic energy remained uncertain until the mid-1980s and the independent work of Burckhardt (Shimada et al. 1987) and Pritchard (1987, 1988). These authors demonstrated, using isolated basolateral membrane vesicles from rat renal cortex, that organic anion transport is driven by uptake across the basolateral membrane in exchange for intracellular dicarboxylate (physiologically for α-ketoglutarate (α-KG); Pritchard 1990). The required intracellular>extracellular α-KG gradient is maintained by mediated uptake of αKG into the cells across the basolateral membrane by Na+/dicarboxylate co-transport (approximately 60%) and by intracellular production via metabolism (approximately 40%) (Dantzler 2002). Finally, the inwardly directed Na+ gradient driving this process is the product of adenosine triphosphate (ATP) hydrolysis via the Na,K-ATPase at the basolateral membrane (Shimada et al. 1987; Pritchard 1988, 1990; Pritchard and Miller 1993; Schmitt and Burckhardt 1993; Dantzler 2002; Wright and Dantzler 2004). Thus, as depicted in Figure 1, the overall effect of these transporters acting in concert is the net entry of sodium and organic anion into the cell, energized by the hydrolysis of ATP. In contrast, the dicarboxylate counter-ion, αKG, is recycled and undergoes no net change in concentration. The overall process may be termed tertiary active transport, since the transport of the organic anion substrate is separated from ATP hydrolysis by two intervening steps. Despite the complexity of this mechanism, it is very effective, resulting in complete clearance of good substrates like ρ-aminohippurate (PAH) from the renal plasma in a single pass through the kidney (Pritchard and Miller 1993). Subsequent work, discussed below, has led to cloning of the basolateral organic anion transporters (OATs) and demonstrated that the properties of the cloned basolateral organic anion transporters match precisely with the features of the overall process as defined by the membrane vesicle studies.
Figure 1.
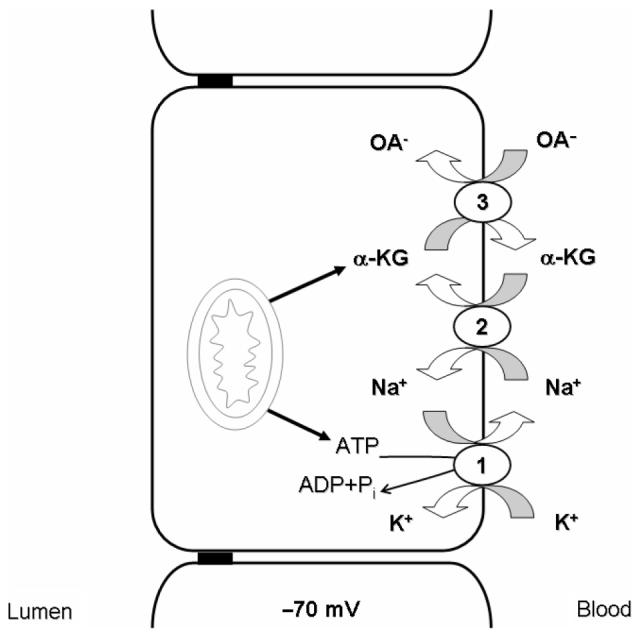
Classical model of basolateral organic anion transport system. Basolateral organic anion uptake is indirectly coupled to the Na+ gradient by a tertiary active transport. (1) Na,K-ATPase derives energy from ATP hydrolysis and pumps Na+ out of the cell. (2) Na+-dicarboxylate co-transporter (NaDC), energetically downhill movement of Na+ into the cell drives dicarboxylate (physiologically αKG) uptake via NaDC, thus, maintaining an outwardly directed αKG gradient. (3) Organic anion transporter (OAT), intracellular αKG is used as a counter-ion by OATs 1 and 3 to drive entry of the organic anions into the cell against the inside negative membrane potential. Cellular metabolism ultimately drives OAT-mediated transport by providing ATP for Na,K-ATPase and the metabolic intermediate, αKG, for OAT-driven exchange.
Given the efficacy of the basolateral uptake process, renal organic anion transport plays a critical role in controlling drug and xenobiotic concentration and retention within the body. The second important feature that is critical for this role is the ability of the organic anion system to transport a diverse array of chemicals, ranging from endogenous metabolites to drugs, xenobiotics and their metabolites. As amply documented by the comprehensive microperfusion studies of Ullrich (e.g., Ullrich and Rumrich 1988; Fritzsch et al. 1989), organic anion system substrates are characterized by a small size (<500 Daltons), a hydrophobic region (optimally 8–10 Å in length), and a negative charge (optimally 6–7 Å from a second such charge). Together, the efficacy of transport and the broad range of compounds transported position the organic anion system to have a major impact on the handling of compounds entering the body as organic anions, as well as on the anionic metabolites of hydrophobic compounds.
The above discussion focuses on the basolateral step in net secretion of organic anions across the renal tubular epithelium. This reflects the fact that for negatively charged compounds, the energetically uphill step in transepithelial transport must be entry from the extracellular fluid into the cell, which is approximately 70 mV negative relative to the extracellular fluid (Wright and Dantzler 2004). Thus, entry of an anion is opposed by electrical and concentration gradients, requiring input of metabolic energy via the tertiary active mechanism summarized above. The apical efflux step for these anions will be energetically downhill. This latter step is less well characterized than the basolateral step, but it too is mediated and transporters playing a role at this cellular face have also been cloned (see below). A number of excellent comprehensive reviews have examined various aspects of organic anion transport including its history, energetics, molecular biology, and specificity (Pritchard and Miller 1993; Burckhardt and Burckhardt 2003; Wright and Dantzler 2004; You 2004; Hediger et al. 2005; Sweet 2005; Anzai et al. 2006; Rizwan and Burckhardt 2007; Zhou and You 2007).
The sections to follow will discuss the properties and functional roles of the cloned organic anion transporters, the relationships between transporter structure and function, and those factors which determine the efficacy of transport, including plasma protein binding of substrates, genetic polymorphisms among the transporters, and regulation of transporter expression.
Characteristics of cloned organic anion transporters (OATs)
The first organic anion transporter, named OAT1, was cloned in 1997 by expression cloning in Xenopus oocytes (Sekine et al. 1997; Sweet et al. 1997) from a rat kidney cDNA library. As noted above and discussed in detail below, its properties match precisely with those characterized in the earlier membrane vesicle studies. Upon sequencing of this transporter, it became apparent that its nucleotide and amino acid sequences were highly homologous to the recently cloned organic cation transporter 1 (OCT1) (Grundemann et al. 1994); reviewed by Ciarimboli et al. in this volume). Thus, both classes of renal transporters were determined to be part of the same superfamily of transporters, the solute carrier 22A family, or SLC22A. All of these transporters have important features in common. First, they are approximately the same size (500–600 amino acids). They have twelve trans-membrane spanning domains (TMD) and both amino and carboxy termini are intracellular. A large extracellular loop is present between TMD 1 and 2 and another large loop is intracellular between TMD 6 and 7. A number of glycosylation sites are located in the extracellular loop and putative phosphorylation sites are present in the intracellular loop. Through a combination of cloning and genomic screening in silico, approximately 25 SLC22A family members have been identified. Of these, six (SLC22A1–5 and 16) transport organic cations and will not be examined here. The remainder handle organic anions (SLC22A6–12, and 20) or are uncharacterized (SLC22A13–15, 17–19, and 21–25) (Sweet 2005; Jacobsson et al. 2007). Several dendrograms depicting the phylogenetic relationships between SLC22A members have been published recently (Eraly et al. 2003; Koepsell and Endou 2004; Wright and Dantzler 2004; Aslamkhan et al. 2006; Jacobsson et al. 2007). In this section, we will discuss the properties and functional roles of the well-characterized transporters — initially, those that are expressed at the basolateral face of the tubule and then those that are apical. Finally, the limited information on several incompletely characterized transporters will be summarized.
Basolateral OATs
OAT1 (SLC22A6)
Cloning
The first member of the OAT family, rOat1, was cloned from a rat kidney cDNA library by two independent groups (Sekine et al. 1997; Sweet et al. 1997). Mouse Oat1 (mOat1) was also cloned at this time, but based on the lack of functional data was called novel kidney transporter (NKT) (Lopez-Nieto et al. 1997) and not functionally determined to be an OAT until 2000 (Pavlova et al. 2000). The winter flounder (Pseudopleuronectes americanus) homologue (fOat1) was also cloned in 1997 (Wolff et al. 1997). Soon thereafter, four isoforms of human OAT1 (hOAT1-1, hOAT1-2, hOAT1-3 and hOAT1-4), as well as the rabbit orthologue (rbOat1) and two isoforms of pig (pOat1 and pOat1A) were cloned and characterized (Reid et al. 1998; Hosoyamada et al. 1999; Lu et al. 1999; Race et al. 1999; Bahn et al. 2000, 2002; Hagos et al. 2002). Finally, Oat1 was also cloned from Caenorhabditis elegans (CeOat1) in 1999 (George et al. 1999) and from monkey (mkOat1) in 2005 (Tahara et al. 2005).
Size and two-dimensional structure
The hOAT1 protein is 550 amino acids in size (Reid et al. 1998; Hosoyamada et al. 1999; Lu et al. 1999; Race et al. 1999) and shares 86–96% sequence identity with other mammalian Oat1 orthologues (rat, mouse, rabbit, pig and monkey). Moreover, as might be expected, it shares only 25 and 47% amino acid identity to the phylogenetically more distant CeOat1 and fOat1, respectively. Secondary structure based on hydropathy analysis indicates that, as noted above, OAT1 contains twelve TMDs, with cytoplasmic amino and carboxyl terminals. The large extracellular loop between TMD 1 and 2 contains three to six potential N-glycosylation sites, depending upon species. Potential phosphorylation sites for protein kinase C (PKC), protein kinase A (PKA), casein kinase II, and tyrosine kinase are clustered in the intracellular loop between TMD 6 and 7 and on the carboxyl terminus (Burckhardt and Wolff 2000; Koepsell and Endou 2004). The molecular weights of hOAT1 gene products vary from 60 to 90 kDa, depending upon glycosylation status (Robertson and Rankin 2006). Molecular weights of Oat1s from other species are in the same range, e.g., rOat1, 57–77 kDa (Robertson and Rankin 2006), fOat1, 62 kDa (Wolff et al. 1997), and mkOat1, 70 kDa (Tahara et al. 2005) (Table I).
Table I.
Properties of organic anion transporters.c
| Name | Species | AA | TMD |
N-linked glycosylation sites |
PKC | PKA | CK II | TK | Predicted MW (kDa) |
Chromosome |
|---|---|---|---|---|---|---|---|---|---|---|
| OAT1 | Rat | 551 | 12 | 39, 56, 92, 97, 113, 184 |
S-129, 271, 278; T-284, 334 |
ND | S-325, 554; T-515 |
ND | 57–77 | 1q43 |
| Mouse | 546 | 11? | 39, 56, 86, 91, 107 | S-265, 270 | ND | S-319, 538; T-515 |
ND | ND | 19 | |
| Human | 550 | 12 | 39, 56, 92, 97, 113 | S-129, 271, 278 521; T-284, 334, 526 |
S-276, 469; T-318, 334 |
S-325, 543; T-122, 515 |
Y-536 | 60–90 | 11q13.1-13.2 | |
| Winter flounder |
562 | 12 | 54, 95, 124 | S-283, 290, 324, 536, 543; T-13 |
S-409; T-351 |
S-237, 543; T-527, 549 |
ND | 62 | ND | |
| OAT2 | Rat | 535 | 12 | 57, 91 | S-279; T-285 | S-529; T-530 |
ND | ND | 52–66 | 9q12 |
| Mouse | 540 | 12 | 57, 91, 356 | S-164, 254, 279 327; T-198, 513 |
ND | ND | ND | 66 | 17C | |
| Human | 546/538 | ND | ND | ND | ND | ND | ND | ND | 6p21.1-21.2 | |
| OAT3 | Rat | 536 | 12 | 54, 81, 86, 102 | S-103, 118, 144, 205, 259, 266, 307; T-88 |
ND | ND | ND | 50–130 | 1 |
| OAT3 | Mouse | 537 | ND | ND | ND | ND | ND | ND | ND | 19 |
| Human | 542 | 12 | 54, 81, 86, 102 | S-266, 511, 528 | ND | S-263, 290; T-2, 310 |
ND | 62–80 | 11q11.7 | |
| URAT1 | Mouse | 535 | 12 | 39, 102, 107 | ND | 405a, 536a | ND | ND | 62/70 | ND |
| Human | 555 | 12 | 39, 56, 102 | ND | 405a, 536a | ND | ND | 40 | 11q13.1 | |
| OAT4 | Human | 550 | 12 | 39, 56, 99, 310, 353 |
S-164, 225, 279 319, 326, 529; T-65, 224, 428 |
ND | ND | ND | 50–83 | 11q13.1 |
| OAT5 | Rat | 551 | 12 | 39, 56, 62, 102 | 216, 272, 279, 313, 536 |
ND | ND | ND | ND | ND |
| Mouse | 551 | 10–12 | 39, 56, 62, 102 | S-46, 60, 68b, T-109b |
ND | ND | ND | 85 | 19 |
Notes:
Consensus sequences are predicted as cAMP-dependent PK phosphorylation sites: 405-RRLC (mUrat1), 405-RRPT (hURAT1), 536-KKVT (mURAT1), and 536-KKAT (hURAT1).
There are an additional eleven sites for serine residue and three additional sites for threonine residue.
For references, see the text in section titled ‘Characteristics of cloned OATs’.
ND, Not determined.
Cell and tissue distribution
Northern blot analysis showed that hOAT1, rOat1, mOat1 transcripts are expressed abundantly in kidney and at lower levels in brain (Lopez-Nieto et al. 1997; Sekine et al. 1997; Sweet et al. 1997; Lu et al. 1999; Race et al. 1999). In situ hybridization demonstrated that rOat1 mRNA is expressed in renal proximal tubules (Lopez-Nieto et al. 1997; Sekine et al. 1997). Immunohistochemistry has shown that hOAT1, rOat1 and mOat1 are expressed at the basolateral membrane of renal proximal tubule cells (Sekine et al. 1997; Sweet et al. 1997; Geng et al. 1999; Hosoyamada et al. 1999). Recently, Hilgendorf et al. (2007) demonstrated by quantitative polymerase chain reaction (PCR) that hOAT1 has the highest expression of 36 drug transporters found in human kidney. Oat1 mRNA expression was also seen in mouse and rat choroid plexus by reverse transcriptase-polymerase chain reaction (RT-PCR) analysis (Sweet et al. 2002). Subsequently, hOAT1 mRNA was also detected in human choroid plexus by Alebouyeh et al. (2003) However, expression in choroid plexus is very low and Oat1 function has not been observed in choroid plexus (Sweet et al. 2002). More recently, mOat1 message was also detected in cerebral cortex and hippocampus (Bahn et al. 2005) and in the olfactory mucosa (Monte et al. 2004). Finally, using radiation hybrid mapping to identify chromosomal location of OAT1 genes, hOAT1 was mapped to human chromosome 11q13.1-2, rOat1 to rat chromosome 1q43, and mOat1 to mouse chromosome 19 (Lopez-Nieto et al. 1997; Hosoyamada et al. 1999) (Tables I and II).
Table II.
Characteristics of the organic anion transporter (OAT) family.
| Gene | Protein | mRNA expression |
Tissue distribution |
Localization | Km (μM) | Substrates/inhibitors |
|---|---|---|---|---|---|---|
| SLC22A6 | rOat1 | Kidney, brain, CPa |
Kidney (cortex> medulla), brain |
Basolateral PT | PAH (11–85.1), ACV (242), adefovir (270), cidofovir (238), AZT (68), OTA (0.57, 2.1), DNP-NAC (2), PMEA (30) |
2,4-D, antiviral agents, PGE2, PGF2α, α-KG, urate, NSAIDs, MTX, β-lactam antibiotics, ACE inhibitors, uraemic toxins, cAMP, cGMP, folate, acetazol- amide, acetylsalicylate, benzylpenecillin (penicillin G), cephaloridine, furosemide, indomethacin, lamivudine, PMEDAP, PMEG, salicylate, stavudine, trifluridine, zalcitabine |
| mOat1 | Kidney, brain, CPa |
Kidney, brain | Basolateral PT | PAH (37, 162) | Ascorbate, captopril, carprofen, cisplatin, diclofenac, enalapril, folate, indomethacin |
|
| hOAT1 | Kidney, brain, CPa |
Kidney, brain, placenta |
Basolateral PT | PAH (3.9–22), ACV (342.3), adefovir (23.8, 30), cidofovir (46, 58), Ganciclovir (896), AZT (45.9), 6- CF (3.93), GA (10.7), MTX (553.8), OTA (0.42), PGE2 (0.97), PGF2α (0.58) |
2,4-D, antiviral agents, α-KG, NSAIDs, uraemic toxins, fluorescein, indole acetate, indoxyl sulfate, penicillin G, BSP, cimetidine, diclofenac, DMPS, furosemide, phenol red, PMEDAP, PMEG, probenecid, tetracycline, doxycycline, bumetanide, betamipron, cefoperazone |
|
| fOat1 | Kidney | Kidney | PAH (21–58) | ES, 2,4-D, antiviral agents (adefovir, cidoforvir), OTA, bumetanide, cimetidine, didanosine, DMPS, ethacrynate, OTA, PMEDAP, PMEG, tienilate |
||
| ceOat1 | PAH (430) | Ascorbate, penicillin G, folate, furosemide |
||||
| rbOat1 | Kidney | Kidney | PAH (15.6) | DMPS, probenecid | ||
| pOat1 | Kidney | PAH (3.7) | Glutarate, probenecid | |||
| mkOat1 | Kidney | PAH (10), 2,4-D (3), hippurate (12.2), indoleacetate (23.6), indoxyl sulfate (32.9), CMPF (85.3) |
Probenecid, furosemide, bumetanide, chlorothiazide, α-KG, penicillin G |
|||
| SLC22A7 | rOat2 | Liver (M:L>K)b, kidney (F:K>L)b, CPa |
Liver, kidney, adipose tissue |
Sinusoidal, apical late PT, TAL and CD |
PGE2 (38.5), salicylate (81/88.9), α-KG (17.8), AZT (26), 2, 3-di- deoxycytidine (3, 080), indomethacin (0.37) |
Glutarate, methotrexate, bumetanide, PAH, PGE2, cAMP, DHEAS, tetracycline, NSAIDs, AZT, MTX, zalcitabine, acetylsalicylate |
| mOat2 | Kidney (F>M), liver, CPa |
Kidney, liver | Apical late PT | PGE2 (0.005), GA (15.8), bumetanide (9) |
PGE2, tetracycline, allopurinol, PAH, α-KG, MTX, OTA, valproate |
|
| hOAT2 | Liver, kidney | Liver, kidney | Sinusoidal, basolateral PT | PGE2 (0.72), PGF2α (0.43), salicylate (3.9), GA (0.43), AZT (26.8), tetracycline (439.9) |
Glutarate, α-KG, MTX, PAH, cAMP | |
| rbOat2 | Kidney, liver, CPa |
|||||
| SLC22A8 | rOat3 | Kidney, liver, CPa |
Liver, kidney, brain |
Basolateral PT | PAH (60–278), ES (2.3-5.3), benzylpenicillin (82.6), 2,4-D (20), cimetidine (40), E2-17-β-G (8.43), indoxyl sulfate (158), OTA (0.74), pravastatin (13.4) |
cAMP, cGMP, glutarate, oestradiol glucuoronide, MTX, MPP |
| mOat3 | Kidney, brain, CPa |
Kidney, brain, eye |
Apical CP | 6-Mercaptopurine (4), 5-fluorouracil (0.054), L-carnitine (0.062) |
ES, PAH, PGE2, PGF2α, allopurinol | |
| hOAT3 | Kidney, brain, skeletal muscle, adrenal tissue |
Kidney, brain, skeletal muscle |
Basolateral PT | PAH (87.2), ES (3.1-9.5), cortisol (2.4), PGE2 (0.35), PGF2a (1.1), AZT (145.1), cimetidine (57.4), MTX (10.9), OTA (0.75), tetracycline (566.2) |
2,4-D, cortisol, cAMP, DHEAS, oestradiol glucuronide, urate, salicylate, GA, salicylate, taurocholate, valacyclovir |
|
| rbOat3 | Kidney | Kidney | ES (4.5) | |||
| pOat3 | Kidney | ES (7.8) | Glutarate, DHEAS | |||
| SLC22A8 | mkOat3 | ES (10.6), penicillin G (49.2), cimetidine (68.5), CMPF (18.6) |
PAH, probenecid, furosemide, bumetanide, chlorothiazide, α-KG, penicillin G |
|||
| SLC22A11 | hOAT4 | Kidney, placenta, adrenal tissues |
Kidney | Apical PT, basal membrane of syncytiotrophoblast |
ES (1, 4.2), 6-CF (108.3), DHEAS (0.6), OTA (23), PGE2 (0.15), PGF2a (692), zonampenal (215), MTX (18) |
tetracycline, steroid sulfates (β-oestradiol-3, 17-disulfate, 17-β-oestradiol 3-disulfate, β-oestradiol 3-disulfate, AZT, cimetidine, urate |
| SLC22A12 | rUrat1 | Kidney, liver, CPa |
||||
| mUrat1 | Kidney | Kidney | Apical PT | Urate (1, 213) | Lactate, nicotinate | |
| hUrat1 | Kidney | Kidney | Apical PT | Urate (199, 371) | PZA, nicotinate | |
| rbUrat1 | Kidney, liver, CPa |
|||||
| SLC22A19 | rOat5 | Kidney | Kidney | Apical PT, outer- and juxta-medullary cortex |
OTA (0.34), ES (18.9), DHEAS (2.3) |
Sulfate conjugates |
| mOat5 | Kidney | Kidney | S2–S3 PT, glomeruli, CCD, MAL |
OTA (2), ES (2.2), DHEAS (3.8) | Sulfate conjugates | |
| rbOat5 | Kidney, liver, CPa |
|||||
| SLC22A20 | mOat6 | Olfactory mucosa |
Olfactory mucosa, vomeronasal organ |
ES (45–110) | Probenecid, 2,4-D, penicillin G, salicylate, propionate, 2 and 3-methylbutyrates, benzoate |
|
| SLC22A9 | hOAT7 | Liver | Liver | Sinusoidal | ES (8.7), DHEAS (2.2) | β-oestradiol sulfate, BSP, SCFAs |
Note:
CP, choroid plexus.
M, male; F, female; L, liver; K, kidney.
Substrate specificity
PAH has been used for many years as the prototypic substrate for renal organic anion transport and is completely cleared from the renal plasma in a single pass (Pritchard and Miller 1993). It is also an excellent substrate for OAT1. The affinity of OAT1 for PAH has been reported in a variety of heterologous expression systems, including Xenopus laevis oocytes and mammalian cell lines. The apparent Km of hOAT1 for PAH is in the 5–9 μM range (Hosoyamada et al. 1999; Lu et al. 1999). Pig (3.7 μM) and monkey (10 μM) Oat1s are similar in affinity (Hagos et al. 2002). Other Oat1s have somewhat lower affinities, with Km's of 14 μM and 70 μM for rOat1 (Sekine et al. 1997; Sweet et al. 1997), 37 μM for mOat1 (Kuze et al. 1999), 15.6 μM for rbOat1 (Bahn et al. 2002), 20 μM fOat (Wolff et al. 1997), and 430 μM for CeOat1 (George et al. 1999) (Table II). Although PAH is also transported by the OAT3 isoform, recent assessment of transport by the Oat1 knockout mouse (Eraly et al. 2006) clearly demonstrates that Oat1 is the major renal transporter for PAH in vivo. Additional studies have shown that probenecid is a potent inhibitor of OAT1 (Sweet et al. 1997), just as it is for organic anion transport in native membranes. The nucleoside phosphate antibiotics, adefovir and cidofovir, are specific substrates for hOAT1 and rOat1 and are not transported by OAT3 isoforms (Cihlar et al. 1999; Aslamkhan et al. 2006). Flounder Oat1 transports adefovir as well as the mammalian OAT1s, but unlike its mammalian counterparts, fOat1 also effectively transports the mammalian OAT3 substrate, oestrone sulfate (ES). Based on these functional characteristics and analysis of its nucleotide sequence, it has been suggested that fOat1 is similar to the ancestral OAT gene that gave rise to mammalian OATs 1 and 3 by gene duplication (Eraly et al. 2003; Aslamkhan et al. 2006). The anionic herbicide, 2,4-dichlorophenoxyacetic acid (2,4-D), is well transported by OAT1 and shows only modest transport by OAT3 (Tahara et al. 2005; Aslamkhan et al. 2006). Rat Oat1 also mediates transport of endogenous compounds including prostaglandin E2 (PGE2), prostaglandin F2α (PGF2α), α-KG, and urate; whereas, hOAT1 transports PGE2, PGF2α, α-KG, but not urate, indicating species differences of organic anion transport among OAT1 isoforms (Sekine et al. 1997; Sweet et al. 1997; Burckhardt et al. 2001; Kimura et al. 2002; Burckhardt and Burckhardt 2003). Finally, inhibition studies also indicate that a variety of organic anions may decrease or block OAT1-mediated transport, including anti-inflammatory drugs, antibiotics, chemotherapeutic drugs, anti-viral drugs, vitamins, anti-hypertensive drugs, uraemic toxins, mycotoxins, heavy metals, and both 2,3-dimercapto-1-propane sulfonic acid (DMPS) and its oxidized form (Sweet et al. 1997; Wolff et al. 1997; Reid et al. 1998; Apiwattanakul et al. 1999; Cihlar et al. 1999; Hosoyamada et al. 1999; Islinger et al. 2001; Jariyawat et al. 1999; Lu et al. 1999; Race et al. 1999; Burckhardt and Burckhardt 2003; Sweet 2005). However, it must be remembered that inhibition studies do not prove that these agents are themselves transported.
Mechanism
The first cloning papers (Sekine et al. 1997; Sweet et al. 1997) demonstrated that OAT1 acts as organic anion/dicarboxylate exchanger. Thus, it is capable of functioning as the basolateral organic anion uptake step in the tertiary active system discussed above (Figure 1), and of mediating active uptake of organic anion substrates against their electrochemical gradients in exchange for an intracellular α-KG. Moreover, the dicarboxylate specificity of OAT1 is also similar to that observed in basolateral membrane vesicles, i.e., dicarboxylates five to ten carbons in length (glutarate, α-KG, adipate, pimelate, suberate, azelate and sebacate) markedly inhibited PAH uptake; whereas, dicarboxylates with three to four carbon atoms (malonate and succinate) did not inhibit (Sweet et al. 1997; Uwai et al. 1998). Glutarate inhibited PAH uptake mediated by hOAT1 wild-type (WT), but not by mutants in the binding pocket of hOAT1–Y230F and F438Y (Perry et al. 2006; and unpublished data), indicating that these tyrosine and phenylalanine residues are important for dicarboxylate recognition.
Physiological role
Given its abundant expression in the kidney (Hilgendorf et al. 2007), its basolateral localization (Sekine et al. 1997; Sweet et al. 1997, 1999; Geng et al. 1999; Hosoyamada et al. 1999), and its ability to mediate organic anion uptake in exchange for α-KG (Sekine et al. 1997; Sweet et al. 1997), OAT1 is a critical component of the overall organic anion secretory process, specifically driving the energy-dependent uphill basolateral step in organic anion secretion. Moreover, knockout mice lacking Oat1 are markedly impaired in their ability to secrete the prototypic organic anion, PAH (Eraly et al. 2006). Thus, OAT1 is positioned to play a key role in controlling the renal elimination of endogenous and exogenous organic anions, and their metabolites from the body (Figure 2).
Figure 2.
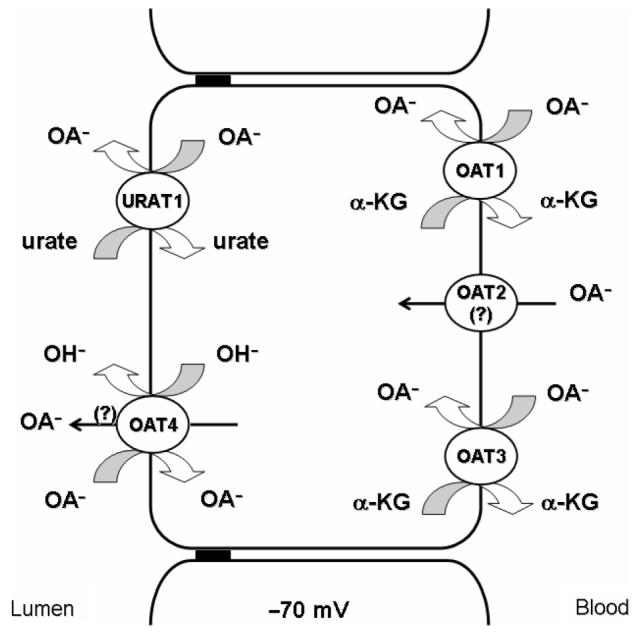
Localization of cloned organic anion transporters (OATs) in human renal proximal tubule cells. Three isoforms of OATs are found at the basolateral membrane of human renal proximal tubular epithelial cells. OAT1 and OAT3 are responsible for transport organic anions across the basolateral membrane via a tertiary active mechanism (Figure 1). The role of the third transporter, OAT2, remains unclear. Two transporters appear to play important roles for organic anion transport at the luminal membrane. URAT1 is capable of reabsorption of urate in exchange for intracellular organic and/or inorganic anions. Human OAT4 may perform a similar role by exchanging organic anions for OH−, but uncertainties remain.
OAT2 (SLC22A7)
Cloning
Rat Oat2 was first cloned in 1994 and called ‘novel liver-specific transporter’ (NLT) (Simonson et al. 1994), because no functional data was obtained for the clone. Somewhat later, Sekine et al. (1998) cloned this transporter from a rat liver cDNA library and showed it to be an organic anion carrier. Two isoforms of the human orthologue were also identified and designated as hOAT2A and hOAT2B (Sun et al. 2001). Finally in 2002, the mouse isoform (mOat2) was cloned from a renal cDNA library (Kobayashi et al. 2002).
Size and two-dimensional structure
OAT2 isoforms range from 535 to 546 amino acids in size (Simonson et al. 1994; Sun et al. 2001; Kobayashi et al. 2002). Rat Oat2 shares 88 and 79% amino acid sequence identity with mOat2 and hOAT2, respectively. Hydropathy analysis indicates that, like OATs 1 and 3, OAT2s have twelve TMDs with potential N-glycosylation sites in the first extracellular loop. A third potential N-glycosylation site is also present at Asn356 in the extracellular loop between TMD 7 and 8 of mOat2 (Kobayashi et al. 2002). There are two potential PKC and PKA phosphorylation sites in rOat2 (Simonson et al. 1994) and six potential PKC phosphorylation sites in mOat2 (Kobayashi et al. 2002). Oat2 proteins from mouse and rat are 52–66 kDa in size (Simonson et al. 1994; Ljubojevic et al. 2007) (Table I). Interestingly, OAT2 homologues share only 37–42 and 34–41% amino acid identity to OAT1 and OAT3 homologues (Table III), implying that functional characteristics of OAT2 may be unique and differ from OAT1 and OAT3.
Table III.
Comparison of OAT2 with OAT1 and OAT3: percent amino acid sequence identity among rat, mouse, and human.
| rOat1 | rOat3 | mOat1 | mOat3 | hOAT1 | hOAT3 | |
|---|---|---|---|---|---|---|
| rOat2 | 42a,b, 38c | 39b, 34c | n.d. | n.d. | 39c | n.d. |
| mOat2 | 41d | 40d | 40d | 41d | 42d | 39d |
| hOAT2 | 37c | 37c | n.d. | n.d. | 39c | 38c |
Notes:
Kumura et al. (1999).
n.d., Not determined.
Cell and tissue distribution
Northern blot analysis indicates that rat and human OAT2 mRNA are expressed predominantly in the liver, with lower levels in kidney and other tissues (Sekine et al. 1998; Sun et al. 2001). In contrast, mOat2 mRNA is strongly expressed in kidney and weakly in liver, indicating that the expression pattern for OAT2 is both species and tissue specific. More recent data using quantitative PCR indicates that hOAT2 has moderate expression in liver and relatively low levels in kidney (Hilgendorf et al. 2007). Oat2 expression is also sex dependent, at least in the mouse, where the livers of female mice have much higher expression than those of males (Kobayashi et al. 2002). Mouse Oat2 was mapped to mouse chromosome 17C using fluorescent in situ hybridization analysis (FISH) (Kobayashi et al. 2002). Immunohistochemistry showed that hOAT2 is expressed at the basolateral membrane of proximal tubules, like OATs 1 and 3 (Enomoto et al. 2002b). In contrast, rOat2 and mOat2 are localized at the apical membrane of late proximal tubules (S3 segments) (Ljubojevic et al. 2007), raising the possibility of species differences in localization of rodent and human OAT2s. This issue remains incompletely resolved, although functional studies may help (see below).
Substrate specificity
Although its substrate specificity has not been extensively examined, initial studies indicate that, like other OATs, OAT2 transports a variety of organic anions (Sekine et al. 1998; Sun et al. 2001; Kimura et al. 2002; Kobayashi et al. 2002, 2005b; Burckhardt and Burckhardt 2003). These include endogenous substrates like PGE2, α-KG, cAMP, and dehydroepiandrosterone sulfate (DHEAS) (Table IV). Additional OAT2 substrates include glutarate, methotrexate (MTX), bumetanide, and PAH. Species differences in substrate specificity are also seen. For example, salicylate is a substrate for hOAT2 and rOat2 (Sekine et al. 1998; Morita et al. 2001; Kobayashi et al. 2005b), but is not transported by mOat2 (Kobayashi et al. 2002). Inhibition studies add potential substrates to this list. For instance, rOat2-mediated salicylate transport is inhibited by organic anions such as bromosulphophthalein (BSP), indocyanine green, erythromycin, rifampicin, benzylpenicillin, and probenecid (Sekine et al. 1998; Morita et al. 2001; Burckhardt and Burckhardt 2003). Cimetidine, oxaloacetate, and enalapril inhibit glutarate transport mediated by mOat2 (Kobayashi et al. 2002; Burckhardt and Burckhardt 2003). Finally, hOAT2-mediated tetracycline transport is also inhibited by diverse structural compounds, including BSP, diclofenac, erythromycin, ibuprofen, bumetanide and furosemide (Burckhardt and Burckhardt 2003; Kobayashi et al. 2005a), indicating overlapping substrate specificity between OAT2 and other OATs.
Table IV.
Substrate specificity (apparent Km) of OAT2 among three different species (μM).
| PGE2 | Salicylate | Glutarate | α-KG | |
|---|---|---|---|---|
| rOat2 | 38.5b, Ta | 81.2b, 88a | n.d. | 17.8a |
| mOat2 | 0.005c | Not transportedc | 15.8c | Tc |
| hOAT2 | 0.71e, Td | Td | Td | Td |
Notes:
Oocyte-expressing rOat2 (Sekine et al. (1998).
LLC-PK-expressing rOat2 (Morita et al. (2001).
Oocyte-expressing mOat2 (Kobayashi et al. (2002).
Oocyte-expressing hOAT2 (Kobayashi et al. (2005b).
S2 cells-expressing hOAT2, S2hOAT2 (Kumura et al. (2002).
n.d., Not determined; T, transported.
Mechanism
The original cloning studies of OAT2 showed that replacement of external sodium by choline or lithium did not change OAT2-mediated transport in Xenopus oocytes, suggesting that transport is sodium-independent (Sekine et al. 1998; Kobayashi et al. 2002, 2005b). Moreover, glutarate did not trans-stimulate organic anion uptake after co-expression of rOat2 and rat sodium dicarboxylate exchanger, rNaDC1, suggesting that rOat2 is not an organic anion/dicarboxylate exchanger (Sekine et al. 1998). More recently, it was also shown that preloading with dicarboxylates, including glutarate, succinate and fumarate does not trans-stimulate OAT2-mediated ES transport (Kobayashi et al. 2005b). These data argue that the mechanism of OAT2 transport is unlike that of OAT1 (discussed above) or OAT3 (see below), but alternative explanations are possible and this aspect of OAT2 function is in need of further study.
Physiological roles
OAT2 is unique in having sex differences in its mRNA expression (Kobayashi et al. 2002) and apparent differences in tissue localization, i.e., basolateral in human kidney and apparently apical in rodents (Enomoto et al. 2002b; Ljubojevic et al. 2007). Moreover, its limited mechanistic understanding further clouds assessment of its potential functional contribution. Hence, the actual physiological role of this transporter in kidney remains unclear. However, hOAT2 has recently been reported to show moderate expression in human liver at the sinusoidal membrane (basolateral membrane) (Hilgendorf et al. 2007), consistent with prior suggestions that it might be responsible for handling both endogenous metabolites and foreign chemicals in the liver and possibly contribute to drug–drug interactions in this tissue (Kimura et al. 2002; Burckhardt and Burckhardt 2003; Kobayashi et al. 2005a).
OAT3 (SLC22A8)
Cloning
Rat Oat3 was first isolated from rat brain in 1999 by RT-PCR based on sequences shared among SLC members cloned at that time (rOat1, rOat2 and rOct1) (Kusuhara et al. 1999). The murine orthologue, mOat3, was initially isolated from an animal model of osteosclerosis (oc), and termed reduced in osteosclerosis transporter, or Roct (Brady et al. 1999). Subsequent studies demonstrated that both rat and mouse Oat3 transport a wide variety of organic anions, as predicted by their similarity to OAT1 in both nucleotide and amino acid sequence (Kusuhara et al. 1999; Ohtsuki et al. 2004). Human OAT3 and mkOat3 have also been cloned and characterized (Race et al. 1999; Cha et al. 2001; Tahara et al. 2005).
Size and two-dimensional structure
Rat Oat3 shares greater amino acid sequence identity with rOat1 (49%) than with either rOat2 (39%) or rOct1 (36%) (Kusuhara et al. 1999). Among the OAT3s, the human OAT3 amino acid sequence is 79 and 78% identical to that of rOat3 and mOat3, respectively. Rat Oat3 protein is 536 amino acids in length, very similar to mOat3 (537 amino acids) and hOAT3 (542 amino acids). The secondary structure of hOAT3, like OATs 1 and 2, has twelve TMDs, with four potential N-glycosylation sites and, depending upon species, three to eight potential phosphorylation sites for PKC (Kusuhara et al. 1999; Burckhardt and Wolff 2000; Cha et al. 2001). The predicted molecular weight of the human OAT3 gene product is 62–80 kDa, depending upon glycosylation status. Rat Oat3 is seen on gels at weights from 50 to 130 kDa, perhaps indicating dimerization or additional protein partners. Monkey Oat3 was 70 kDa (Robertson and Rankin 2006) (Table I).
Cell and tissue distribution
Northern blot analyses have shown that hOAT3 mRNA is predominantly expressed in kidney, with far less expression in brain and skeletal muscle (Cha et al. 2001). Rat Oat3 mRNA is mainly expressed in kidney, liver and brain (Kobayashi et al. 2002); whereas, mOat3 mRNA is abundantly expressed in the kidney, brain, and eye tissues (Kobayashi et al. 2004). Recently, hOAT3 was also shown by RT-PCR to be expressed in adrenal tissue and a human adrenal cell line (NCI-H295R) (Asif et al. 2005). In the kidney, immunohistochemistry demonstrated that both hOAT3 and rOat3 are predominantly expressed at the basolateral membrane of the renal proximal tubule (Cha et al. 2001; Hasegawa et al. 2002). Moreover, quantitative PCR analysis has shown that hOAT3 is highly expressed in kidney — second only to hOAT1 among the 36 drug transporters assessed (Hilgendorf et al. 2007). In choroid plexus (CP), mOat3 is expressed at the apical membrane (Sweet et al. 2002), consistent with its physiological role in transport out of the cerebrospinal fluid (CSF) into the epithelium for efflux into the blood. Mouse Oat3 was mapped to chromosome 19, using single-strand conformation polymorphisms analysis (Brady et al. 1999). Radiation hybrid analysis demonstrated that the hOAT3 gene is on chromosome 11q11.7 (Cha et al. 2001).
Substrate specificity
The substrates for OAT3 are as diverse as those of OAT1. In heterologous expression systems, rOat3 mediated the uptake of PAH, ES, DHEAS, estradiol glucuronide, methotrexate (MTX), ochratoxin A (OTA), PGE2, taurocholate, glutarate, cAMP, urate, and a cationic compound, cimetidine (Kusuhara et al. 1999; Cha et al. 2001; Sugiyama et al. 2001; Kimura et al. 2002; Sweet et al. 2002). The apparent Km of OAT3-mediated PAH transport ranges widely between species. For example, PAH affinity is in the 50–100 μM range for hOAT3 (87.2 μM) (Cha et al. 2001) and rOat3 (60–65 μM) (Kusuhara et al. 1999; Hasegawa et al. 2002) (Table II), but it is not transported at all by rbOat3 (Dantzler and Wright 2003). Inhibition studies suggest that a number of anionic drugs are good inhibitors of mOat3 and may be substrates as well. These include enalapril, tenoxicam, cimetidine, ibuprofen, famotidine, diclofenac, and amantadine (Kobayashi et al. 2004). Several recent studies have demonstrated that ES is a specific substrate for mammalian OAT3 transporters (Sweet et al. 2002; Aslamkhan et al. 2006). Furthermore, the apparent affinities of ES for OAT3 are very high with Km values ranging from 3.1 to 9.5 μM for hOAT3 (Cha et al. 2001; Tahara et al. 2005), from 2.3 to 5.3 μM for rOat3 (Kusuhara et al. 1999; Tahara et al. 2005), 4.5 μM for rbOat3 (Dantzler and Wright 2003), 7.8 μM for pOat3 (Hagos et al. 2005), and 10.6 μM for mkOat3 (Tahara et al. 2005) (Table II). A wide range of drugs inhibit OAT3-mediated ES uptake, including antibiotics, antivirals, H2 antagonists, non-steroidal anti-inflammatory drugs (NSAIDs), diuretics, anti-epileptics, anti-neoplastics, and probenecid (Kusuhara et al. 1999; Race et al. 1999; Berkhin and Humphreys 2001; Dresser et al. 2001; Burckhardt and Burckhardt 2003; Rizwan and Burckhardt 2007; Rizwan et al. 2007). These same studies also indicate that a number of endogenous compounds inhibit ES uptake, including PGE2, PGF2α, and several neurotransmitters (adrenaline, nor-adrenaline and serotonin).
Mechanism
The initial studies to assess the driving forces for rOat3 transport were done using the Xenopus oocyte expression system, and showed no significant effect of sodium replacement on transport and no trans-stimulation of influx or efflux of [3H]-ES by unlabeled ES (Kusuhara et al. 1999; Cha et al. 2001). These results suggested that OAT3 mediated simple facilitated-diffusion of organic anions. However, recent work indicates that the mechanism is more complex (Bakhiya et al. 2003; Sweet et al. 2003). OAT3 was demonstrated to be a dicarboxylate/organic anion exchanger and OAT3-mediated uptake was shown to take place through indirect coupling to metabolic energy via a mechanism identical to that of OAT1. Thus, OAT3 is capable of mediating uphill transport of organic anions across the basolateral membrane of the proximal tubule, the requisite first step in their secretion into the urine.
Physiological roles
Given its similar mechanism, its basolateral localization in the proximal tubule, and its broad specificity, OAT3 is — like OAT1 — uniquely positioned to mediate the energetically uphill uptake step in secretion of its substrates. In particular, OAT3 appears to be important for substrates that are somewhat larger and more lipophilic than those handled by OAT1. Its physiological substrates include ES, DHEAS, cAMP, cGMP, PGE2, and PGF2α (Cha et al. 2001; Burckhardt and Burckhardt 2003). Exogenous substrates include drugs like benzylpenicillin, several NSAIDs, and even cimetidine, a cationic drug. In liver, OAT3 appears to be the primary OAT contributing to sinusoidal uptake of organic anions. In the choroid plexus (blood–CSF barrier), where OAT3 is the only OAT transporter expressed at the apical membrane, it mediates uptake of organic anions from the CSF and thus, plays a crucial role in the regulation of the composition of central spinal fluid CSF (Sweet et al. 2002, 2003). It may also be an important functional component of the blood–brain barrier (BBB) through its uptake of organic anions from the brain into the cerebral capillaries (Kikuchi et al. 2003; Ohtsuki 2004).
Apical OATs
URAT1 (SLC22A12)
Cloning
The first urate transporter was cloned from a mouse kidney cDNA library and identified as a renal-specific transporter (RST) (Mori et al. 1997). However, this study did not provide functional characterization of the transporter. The first functionally characterized urate transporter was cloned from a human kidney cDNA library and identified as URAT1, or hURAT1 (Enomoto et al. 2002a). It appeared to function as an organic anion exchanger (see below) and its loss was shown clinically to lead to reduced urate reabsorption. Subsequently, the mouse urate transporter was cloned again, functionally characterized, and renamed mUrat1 (Hosoyamada et al. 2004). The hURAT1 gene was mapped on human chromosome 11q13.1 (Enomoto et al. 2002a) (Table I).
Size and two-dimensional structure
The hURAT1 cDNA encodes a protein of 555 amino acids (Enomoto et al. 2002a). It shares 42% identity with hOAT4 (discussed below). Mouse Urat1 shares 74% identity to that of hURAT1 (Hosoyamada et al. 2004). Hydropathy analysis predicts that hURAT1 has twelve TMDs, similar to other OAT members. Several potential sites for N-glycosylation are found in a large loop between TMD 1 and 2 of hURAT1. In addition, potential PKC and PKA phosphorylation sites are present in the intracellular loop between TMD 6 and 7 (Mori et al. 1997; Enomoto et al. 2002a). Unlike OATs 1 and 3, hURAT1 contains a PSD-95/Discs Large/ZO-1 (PDZ) domain which plays a role in regulation of this transporter (Anzai et al. 2004). Western blot analysis indicates that hURAT1 has a molecular weight of 40 kDa and mUrat1 ranges between 62 and 70 kDa (Mori et al. 1997; Enomoto et al. 2002a; Hosoyamada et al. 2004).
Cell and tissue distribution
Northern blot analysis revealed a 2.8-kb hURAT1 transcript in both adult and foetal kidney (Enomoto et al. 2002a). Renal expression of mUrat1 mRNA in the male is significantly higher than in the female (Hosoyamada et al. 2004). Immunohistochemistry indicates that both mUrat1 and hURAT1 are most highly expressed at the apical membrane of proximal tubules (Mori et al. 1997; Enomoto et al. 2002a; Hosoyamada et al. 2004). More recently, hURAT1 mRNA and protein were also observed in human vascular smooth muscle cells by RT-PCR and western blot (Price et al. 2006).
Substrate specificity
As suggested by the name, URAT1 protein transports urate with the apparent Km values of 371 and 199 μM for hURAT1 (Enomoto et al. 2002a; Anzai et al. 2004) and 1213 μM for mUrat1 (Hosoyamada et al. 2004). L-lactate, pyrazinecarboxylic acid (PZA), and nicotinate each trans-stimulated urate uptake, indicating that they too were transported by hURAT1 (Enomoto et al. 2002a). These studies also demonstrated that several drugs are able to inhibit hURAT1-mediated transport, including antibiotics, diuretics, and NSAIDs, as well as anti-hypertensive and uricosuric drugs (Enomoto et al. 2002a). However, PAH, ES, and dicarboxylates do not inhibit hURAT1 activity, indicating a marked difference in the specificity of this apical OAT from the basolateral OATs 1 and 3 (see above).
Mechanism and physiological role
Both nicotinate and lactate have been shown to drive urate uptake into the oocytes expressing hURAT1 or mUrat1, suggesting that URAT1 acts as urate/organic anion exchanger (Enomoto et al. 2002a; Hosoyamada et al. 2004). Given its expression in the apical membrane of the proximal tubule, exchange of intracellular organic anions, e.g., lactate, for luminal urate would be most consistent with a role for URAT1 in the reabsorption of urate. This interpretation is consistent with the decreased urate reabsorption and low plasma urate levels seen in patients lacking functional URAT1 expression (Enomoto et al. 2002a). However, this same study showed that urate increased Cl− efflux in hURAT1 expressing oocytes, suggesting urate/chloride exchange as well. Moreover, removing external Cl− increased urate uptake in oocytes expressing either hURAT1 or mUrat1 (Enomoto et al. 2002a; Hosoyamada et al. 2004). If exchange for chloride were to prove significant in vivo, the lumen > intracellular Cl− concentration gradient could generate a secretory urate flux. For this reason, the physiological role(s) of URAT1 must, for the moment, remain incompletely resolved (Figure 2).
Nevertheless, it is clear that a more complete understanding of the role of URAT1 in urate homeostasis is very important. On the positive side, urate is a potent scavenger of free radicals (Hediger et al. 2005), helping to protect from oxidative damage. Unfortunately, because of its relatively low aqueous solubility, urate crystals may form — leading to renal stones, gout, and chronic renal failure (Capasso et al. 2005; Hediger et al. 2005). Moreover, in the human, mutational silencing leads to a lack of urate oxidase enzyme activity, resulting in plasma urate concentrations higher than in other species (240–350 μM) (Capasso et al. 2005; Enomoto and Endou 2005; Hediger et al. 2005), increasing potential for stone formation. Finally, there are important species differences in urate handling. Rabbits, pigs and birds have higher fractional urate excretion (FEurate) than filtered load, indicating net secretion. In the human, FEurate is only 10% of filtered load, suggesting net tubular reabsorption of urate (Maesaka and Fishbane 1998; Enomoto and Endou 2005; Anzai et al. 2007). Thus, if URAT1 were the major human transporter responsible for renal urate reabsorption, it should play a key role in determining urate handling and governing its potential to protect from radical damage or to lead to gout and/or renal stone formation. As discussed above, this aspect of the URAT1 function is incompletely understood.
OAT4 (SLC22A11)
Cloning
In 2000, Cha et al. screened a human expressed sequence tag (EST) database for OAT1-like sequences. A novel clone was identified with 30–45% amino acid sequence identity with known SLC22A family members. This clone was designated hOAT4, and is unique in that it is expressed only in higher primates (Sweet 2005; Hagos et al. 2007; Rizwan and Burckhardt 2007). Furthermore, as initially noted by Eraly et al., it occurs as a pair with URAT1 on chromosome 11q13.1 (Eraly et al. 2003).
Size and two-dimensional structure
Human OAT4 cDNA consists of 2210 bp. Its open reading frame encodes a 550-amino acid protein that shares 42% identity to hURAT1 (Cha et al. 2000; Enomoto et al. 2002a). Similar to the other OATs, Kyte–Doolittle hydropathy analysis of hOAT4 predicts twelve TMDs. It has a large, glycosylated extracellular loop between the first two TMDs with five potential N-glycosylation sites, and a large intracellular loop between TMD 6 and 7 containing several possible sites for PKC phosphorylation (Cha et al. 2000). It also has a C-terminal PDZ domain, similar to the other apical OAT, hURAT1 (Anzai et al. 2004).
Cell and tissue distribution
Northern blot and microarray analysis showed that hOAT4 mRNA is expressed in the kidney and placenta (Cha et al. 2000; Bleasby et al. 2006). Human OAT4 message is also shown by RT-PCR to be expressed in adrenal tissue and an adrenal cell line (NCI-H295R) (Asif et al. 2005). Immunohistochemistry demonstrated that hOAT4 is expressed in the apical membrane of renal proximal tubule (Babu et al. 2002a; Ekaratanawong et al. 2004). More recently, quantitative PCR analysis indicated that hOAT4 has the third highest level of expression in human kidney, behind only hOAT1 and hOAT3 (Hilgendorf et al. 2007). In placenta, its localization appears to be basal in the syncytiotrophoblast (Ugele et al. 2003). These authors, like Cha et al. (2000), suggested that its placement at the foetal side of the placenta could mediate the uptake of hormone precursors, especially their sulfate conjugates, like DHEAS.
Substrate specificity
The substrate spectrum of hOAT4 appears to be relatively narrow compared with other OAT isoforms. It mediates transport of ES (Km = 1.0 μM), DHEAS (Km = 0.6 μM), OTA (Km = 23 μM), MTX (Km = 18 μM), and PGE2 (Km = 0.15 μM) with high affinity (Cha et al. 2000; Babu et al. 2002a; Takeda et al. 2002b). Low-affinity substrates (Km ≥ 150 μM) include PGF2α, tetracycline, zonampanel (an AMPA receptor agonist), and the antiviral, zidovudine (or AZT) (Babu et al. 2002b; Kimura et al. 2002; Takeda et al. 2002c; Hashimoto et al. 2004). Inhibition studies suggest that a variety of other drugs may also interact with hOAT4, and thus, are potential substrates. For example, ES transport is effectively inhibited by many NSAIDs (Khamdang et al. 2002). MTX transport is inhibited by penicillin G, probenecid, and several NSAIDs (interestingly, not including salicylate) in mouse S2 cells expressing hOAT4 (Takeda et al. 2002b). Other drugs shown to interact with hOAT4 include diuretics (Hasannejad et al. 2004), cephalosporin antibiotics (Takeda et al. 2002a), ellagic acid (Whitley et al. 2005), as well as the endogenous metabolite, indoxyl sulfate (Enomoto et al. 2003). Human OAT4 also interacts with PAH and dicarboxylates, but these effects are complex, as discussed in detail in the next section.
Mechanism
In 2004, Ekaratanawong et al. demonstrated that preloading mouse S2 cells expressing hOAT4 with PAH or glutarate trans-stimulated uptake of PAH, ES, or glutarate, providing the first evidence that hOAT4 is an organic anion exchanger. Recently, understanding of this aspect of hOAT4 function was further refined by Hagos et al. (2007). Using hOAT4 expressing HEK293 cells, they demonstrated that hOAT4-mediated exchange of PAH or glutarate is asymmetrical. Cell-to-medium flux of PAH and glutarate in exchange for Cl− or organic anions is supported by hOAT4, but the reverse, i.e., medium-to-cell flux, is not seen. These results argue that hOAT4 may contribute to the secretory flux of as least some hOAT4 substrates. However, Hagos et al. (2007) also showed that hOAT4 could mediate urate and ES transport in exchange for hydroxyl ion (OH−). Since the cell interior is more alkaline than luminal fluid, this finding argues that hOAT4 may mediate proximal tubular reabsorption of organic anions. Our own recent work using isolated membrane vesicles from Sf-9 cells (an insect cell line) expressing hOAT4 confirms its ability to transport several substrates including DHEAS, ES, MTX and OTA in exchange for hydroxyl ions (Thompson et al. 2007). Moreover, exchange was shown to be indirectly coupled to the out > in sodium gradient via Na+/H+ exchange, providing a strong driving force for organic anion reabsorption.
Physiological roles
It is clear from the work summarized above that hOAT4 is an apical organic anion transporter. It is less certain what its overall impact on renal organic anion transport may be. One component of its function is clearly pH-driven reabsorption of hOAT4 substrates from the lumen of the proximal tubule. On the other hand, the importance of chloride exchange for intracellular organic anions, e.g., PAH or glutarate, potentially leading to their secretion across the apical membrane must still be resolved (Figure 2).
The physiological role(s) of hOAT4 in placenta remains speculative. However, the work of Ugele et al. (2003) demonstrated that high levels of hOAT4 are expressed at the basal face of the syncytiotrophoblast, i.e., the side that faces the foetal circulation. These authors suggest a role for OAT4 in uptake of DHEAS by these cells for synthesis of placental oestrogens. Moreover, as noted by Hagos et al. (2007), hOAT4 may also play a role in placental homeostasis of urate and thus, through the antioxidant properties of urate, contribute to protection of the foetus from oxidative damage.
Partially characterized
OAT5 (SLC22A19)
Cloning
OAT5 was first isolated from a mouse cDNA library (Youngblood and Sweet 2004). Subsequently, rOat5 was cloned from a rat kidney cDNA library (Anzai et al. 2005). Additionally, a human orthologue, originally named hOAT5, was cloned in 2001 (Sun et al. 2001). However, no functional data has yet been obtained for the hOAT5 clone, and it is now believed that it is not the human orthologue of the rodent Oat5s (Youngblood and Sweet 2004). Thus, the rodent and human forms have been assigned different names within the SLC22A family, with the human form retaining its SLC22A10 systematic name and the rodent Oat5s being designated SLC22A19. Subsequent discussion will be limited to the rodent transporters, since they have functional information associated with them.
Size and two-dimensional structure
Mouse Oat5 cDNA contains 1964 bp, encoding a protein of 551 amino acids, the same size as the rat Oat5 transporter. These two species share 88% amino acid sequence identity, and 37–50% identity to other OATs (Table V). Rat Oat5 is predicted to have twelve TMDs with four glycosylation sites in the first extracellular loop and five PKC-phosphorylation sites in the large extracellular loop between TMD 6 and 7. Mouse Oat5 is similar (Table I). The mouse Oat5 gene is located on chromosome 19 (Youngblood and Sweet 2004). Mouse Oat5 is detected as an 85-kDa protein by Western blot (Kwak et al. 2005b).
Table V.
Percent amino acid sequence identity of rodent OAT5s compared with other OAT isoforms.
Cell and tissue distribution and localization
Rat and mouse Oat5 mRNAs are expressed exclusively in the kidney, with highest expression in S2 and S3 segments of the proximal tubules, moderate expression in glomeruli and cortical collecting duct (CCD), and weak expression in the medullary thick ascending limb (MAL) (Koepsell and Endou 2004; Youngblood and Sweet 2004). Sex-differences in Oat5 expression have been observed in both rodents. Immunohistochemistry demonstrated that rOat5 is expressed at the apical membrane of proximal tubules in the outer medullary and juxtamedullary cortex (Anzai et al. 2005). Mouse Oat5 is only detected at the apical membrane of the late proximal tubule (Kwak et al. 2005b).
Substrate specificity
Oat5 transports OTA with a high affinity in both rat and mouse. The apparent Km of rOat5 for OTA is 0.34 μM (Anzai et al. 2005) and of mOat5 is approximately 2 μM (Youngblood and Sweet 2004; Kwak et al. 2005b). DHEAS (Km = 2.3 μM for rat and 3.8 μM for mouse) and ES (Km = 2.2 μM for mouse and 18.9 μM for rat) are also substrates for Oat5 (Table II). No evidence for potential-driven transport was seen for mOat5-mediated transport of OTA (Youngblood and Sweet 2004). Sulfate conjugates inhibit ES uptake in oocytes expressing mOat5 and rOat5; whereas, glucuronide conjugates do not (Anzai et al. 2005; Kwak et al. 2005b). Curiously, none of dicarboxylates tested (α-KG, glutarate, citric acid, succinate, and malate) inhibit OTA uptake in mOat5-expressing oocytes; whereas, succinate (C4), glutarate (C5), adipate (6), pimetate (C7), suberate (C8) and azelate (C9) all inhibit ES uptake in rOat5 expressing oocytes (Anzai et al. 2005). Moreover, Anzai et al. (2005) showed that rOat5-mediated ES transport was trans-stimulated by succinate, suggesting organic anion/dicarboxylate exchange, at least in this species. The basis for this is disparity is uncertain. It may relate to experimental issues, e.g., the dicarboxylate concentrations tested (250 μM in the mouse studies and 1 mM in the rat), or to species differences in the transporters themselves.
Physiological roles
It appears clear, as discussed above, that the rodent Oat5s are apically expressed, like URAT1 and OAT4. Moreover, they are particularly effective in the transport of sulfate conjugates of both endogenous and foreign chemicals. However, until the issue of mechanism is resolved, any discussion of the physiological role of the rodent Oat5s must remain speculative. If, as proposed based on the rat data (Anzai et al. 2005), OAT5 mediates exchange of organic anions for intracellular dicarboxylates like succinate, OAT5 is positioned to mediate reabsorption of its substrates from the tubular lumen. On the other hand, if Oat5 turns out to be a facilitated diffusion type carrier, which is consistent with the mouse data (Youngblood and Sweet 2004), it could utilize the electrochemical gradient to mediate the efflux of Oat5 substrates from the tubule into the forming urine, i.e., secretion. This issue must be resolved before the physiological significance of the SLC22A19 family members may be understood.
OAT6 (SLC22A20)
Cloning
A novel OAT6 (mOat6) was first identified in the Ensembl mouse genome database by screening for novel SLC22 family genes (Monte et al. 2004). This report included no functional data, but it did establish that mOat6 is unique among the OATs in its complete lack of expression in the kidney. Instead, it is expressed predominantly in the olfactory mucosa.
Size and two-dimensional structure
Mouse OAT6 cDNA consists of 1897 bp, encoding 556 amino acids. In sequence, it is most similar to OATs 1 and 3, with which it shares approximately 40% amino acid identity (Monte et al. 2004). However, unlike OATs 1 and 3 which form a gene pair on both mouse and human chromosomes (Eraly et al. 2003), mOat6 is an un-paired gene, located on chromosome 19. Its structure is similar to other SLC22A family members, having twelve TMDs and large loops between TMD 1 and 2, as well as between TMD 6 and 7 (Monte et al. 2004).
Cell and tissue distribution and localization
Interestingly, as noted above, mOat6 is expressed in the olfactory mucosa. Indeed, it is the only OAT transporter detected there. Moreover, no mOat6 is found in kidney or brain (Monte et al. 2004). Mouse Oat6 is also detected in 7-day-old foetal mouse, indicating a possible involvement of OAT6 in embryogenesis (Monte et al. 2004). Subsequent work indicated that mOat6 expression is found in the main olfactory epithelial and the vomeronasal organ of the nose (Kaler et al. 2006). No signal is detected in neuronal cells of the mucosa, indicating that mOat6 expression is in the non-neuronal cells of olfactory tissue. Mouse Oat6 is also weakly expressed in testis (Monte et al. 2004).
Substrate specificity
Mouse Oat6 transports ES with moderate affinity — apparent Km of 110 μM when expressed in Xenopus oocytes and 45 μM in Chinese hamster ovary (CHO) cells (Schnabolk et al. 2006). Probenecid, 2,4-D, penicillin G, and salicylate effectively inhibit mOat6-mediated ES uptake. Several odorant organic anions including propionate, 2- and 3-methylbutyrates, benzoate, heptanoate and 2-ethylhexanoate also inhibit mOat6-mediated ES transport (Kaler et al. 2006). More recently, an extensive series of anions have been evaluated and significant overlap was seen in the specificity of mOat6 and mOat1 over a wide range of substrates and inhibitors (Kaler et al. 2007).
Mechanism
Schnabolk et al. (2006) also examined the driving forces for mOat6, showing that glutarate was able to trans-stimulate ES transport, indicating mOat6 acts as organic anion exchanger similar to OAT1 and OAT3, its closest relatives among the OATs (Monte et al. 2004).
Physiological roles
As discussed above, mOat6 is mainly expressed in the olfactory mucosa. Kaler et al. (2006, 2007) present the interesting possibility that because OAT6 and OAT1 specificity are so similar, it might be possible for chemicals to be secreted into the urine and subsequently be detected by the olfactory mucosa. Such a system for odorant delivery by renal OAT1 and detection by olfactory OAT6 could play a role in detection of signalling molecules, e.g., pheromones. Finally, it should be noted that because of its localization in the olfactory mucosa, OAT6 may also provide an alternate mode for effective anionic drug administration.
OAT7 (SLC22A9)
Cloning
The last of the reasonably well-characterized OATs to be cloned is now known as OAT7. It was recently cloned and characterized from a human liver cDNA library (Shin et al. 2007). This sequence was previously reported under the name of SLC22A9/hUST3 and hOAT4, but was not functionally characterized (Sun et al. 2001). For consistency with the nomenclature of OATs 1–6 discussed above, this clone was renamed as OAT7 (Shin et al. 2007).
Size and two-dimensional structure
Human OAT7 cDNA consists of 2342 bp, encoding a 554 amino acid protein that shares 35–46% identity to hOAT1, 2, 3 and 4. It is also closely related to the previously cloned, but uncharacterized hOAT5 (SLC22A10), sharing approximately 55% sequence identity (Sun et al. 2001). The hOAT7 gene is located on chromosome 11q12.3 (Jacobsson et al. 2007).
Cell and tissue distribution and localization
Northern blot and RT-PCR indicate that hOAT7 is expressed exclusively in the liver of both adult and foetus. Immunohistochemistry showed OAT7 to be localized at the sinusoidal membrane of the hepatocyte, and in western blots, it was detected as a 50 kDa protein (Shin et al. 2007).
Substrate specificity
As shown by Shin et al. (2007), hOAT7 has a narrow substrate specificity. ES and DHEAS are both high-affinity substrates for hOAT7, with apparent Km values of 8.7 and 2.2 μM, respectively. Several other sulfate-conjugates such as β-oestradiol sulfate, BSP, and short-chain fatty acids (SCFAs) inhibited ES transport by OAT7. However, PAH, probenecid, glutarate, and glucuronide- and glutathione-conjugates are all unable to inhibit OAT7-mediated ES transport. In addition, SCFAs, particularly butyrate, trans-stimulated ES uptake and butyrate efflux was trans-stimulated by ES in OAT7-expressing oocytes, indicating bi-directional organic anion/butyrate exchange (Shin et al. 2007).
Physiological roles
Because of the unique substrate specificity of OAT7, particularly its ability to transport sulfate conjugates, OAT7 might play a major role in the transport of steroid hormones and/or their conjugates, e.g., ES uptake in exchange with butyrate. The significance of this process is as yet largely uninvestigated and thus, the potential of hOAT7 to modulate normal physiology may be significant, but is as yet uncertain.
Structure–function
Pharmacophore models
Before the cloning of the OATs, the initial work on structure–activity relationships between organic anions and their transporters was carried out through inhibition of radiolabelled PAH uptake in perfused rat kidney tubules (Ullrich and Rumrich 1988; Fritzsch et al. 1989; Ullrich 1997). Results suggested several structural characteristics for basolateral organic anion transporter substrates. Mono-anions must have a hydrophobic moiety of at least 4 Å in length and not more than one electronegative site. Di-anions with a charge separation of 6–7 Å are also preferred, and their hydrophobic region may extend past the distance of charge separation up to 10 Å. Some hydrophobic, non ionizable compounds are also permitted. For these, the ability to form hydrogen bonds is more significant than the degree of ionization (Ullrich 1997).
Additional structure–activity experiments have been conducted with cloned OAT1 isoforms and mimic the earlier findings for the tubular transport of PAH. For example, Sweet et al. (1997) showed that the dicarboxylate specificity of the newly cloned rOat1 mimics that of the basolateral organic anion transport. The dicarboxylate specificity of rOat1 was more systematically assessed by Uwai et al. (1998), using a Xenopus oocyte expression system. They showed that only dicarboxylates with an alkyl chain longer than five carbons (5C) inhibit uptake of PAH (Uwai et al. 1998). Anzai et al. (2005) also examined dicarboxylate inhibition of ES with rOat5, rOat3, and hOAT4 in Xenopus oocytes. Rat Oat3 behaves like rOat1, with transport inhibited by dicarboxylates with 5Cs or greater. The dependency of carbon chain length is not as straightforward for hOAT4 or rOat5. For example, hOAT4 is more effectively inhibited (40% or greater) by succinate, glutarate and azelate (C4, C5, C9, respectively) than by adipate, pimelate or suberate (C6–C8; inhibited only approximately 20%). Rat Oat5 is inhibited by succinate, pimelate, suberate and azelate (C4, C7–C8), while glutarate (C5) and adipate (C6) are not strong inhibitors (Anzai et al. 2005).
Sugawara et al. (2005) have examined uncharged inhibitors structurally similar to xanthine or uric acid. Inhibition of hOAT1-mediated PAH uptake is dependent upon position of the methyl group for xanthine derivatives and on the degree of hydrophobicity for the uric acid derivatives. The presence of an additional methyl group in polycyclic aromatic hydrocarbons appears to be critical for transport. For example, 1-methylpyrenyl mercapturic acid is transported by hOAT1 and hOAT3 and 1,8-dimethylpyrenyl mercapturic acid is not (Bakhiya et al. 2007).
More recently, quantitative structure affinity relationships (QSAR) and 3D pharmacophore modelling have been employed to examine substrate or inhibitor characteristics important for binding and transport by the OATs (Kaler et al. 2007). Inhibition constants for a variety of anions were assessed in mOat1 and mOat6 expressing oocytes. The extent of transport inhibition was compared with the physical properties of compound, e.g., mass, hydrophobicity (logP value), and overall charge, and three-dimensional models were generated based upon a comparative molecular field analysis (CoMFA), which structurally aligns the compounds and correlates biological activity (IC50) with substrate or inhibitor functional groups. This analysis confirmed that hydrophobicity is a major predictor of anion inhibition for both isoforms. However, slight differences in the degree of ionization are important. Di-anions are more effective inhibitors of mOat1; whereas, mOat6 is more effectively inhibited by mono-anions (Kaler et al. 2007).
The same group then applied the QSAR approach to data previously collected in perfused rat kidney tubules (Kaler et al. 2007). Significant correlations were found between the IC50 value and hydrophobicity for monocarboxylate inhibitors of the PAH transporter. In contrast, only a weak correlation with hydrophobicity was present among the dicarboxylate inhibitors. As the number and type of functional groups (aromatic and hydrogen bonding) increased in larger data sets, correlation with hydrophobicity was lost (Kaler et al. 2007). The combined structure–activity work in perfused kidney tubules and in expression systems cloned transporters highlights the large diversity of compounds that are substrates or inhibitors of the OATs. Although commonalities of negative charge, hydrophobicity and the ability to hydrogen bond exist, one must also look at the species and isoforms differences to determine OAT specificity.
Amino acids that contribute to function
To date, over 110 amino acids have been mutated in OATs 1–4 and URAT1 from different species (Feng et al. 2001, 2002; Wolff et al. 2001; Enomoto et al. 2002a; Hong et al. 2004, 2007a; Ichida et al. 2004; Komoda et al. 2004; Tanaka et al. 2004b, c; Zhou et al. 2004a, b, 2005; Bleasby et al. 2005; Erdman et al. 2006; Rizwan et al. 2007). Motivation for the mutations varied, e.g., several studies examined conserved residues most likely directly tied to recognition and transport of organic anions, while other reports examined the role of glycosylation sites (mutating asparagine residues) in protein trafficking (Tanaka et al. 2004c; Zhou et al. 2005) or cysteine residues that may have been linked to disulfide bonding (Tanaka et al. 2004b). In addition, the effects of non-synonymous single nucleotide polymorphisms (SNPs) have also been investigated. Unfortunately, many of the mutant transporters examined (>70%) have reduced surface expression, either from protein instability or trafficking problems. Figure 3 summarizes mutants that have been shown to cause functional increases or decreases in substrate transport regardless of membrane expression levels. This three-dimensional drawing is based on the structures of other members of the major facilitator superfamily (MSF) that have been crystallized, i.e., glycerol 3-phosphate transporter (Huang et al. 2003), oxalate transporter (Hirai et al. 2004) and lactose permease (Abramson et al. 2003b). The model depicts the locations of each residue in the three-dimensional structure. Two groups of amino acids (basic and aromatic) dominate function. These are discussed below, along with examples of other amino acids that have been examined.
Figure 3.
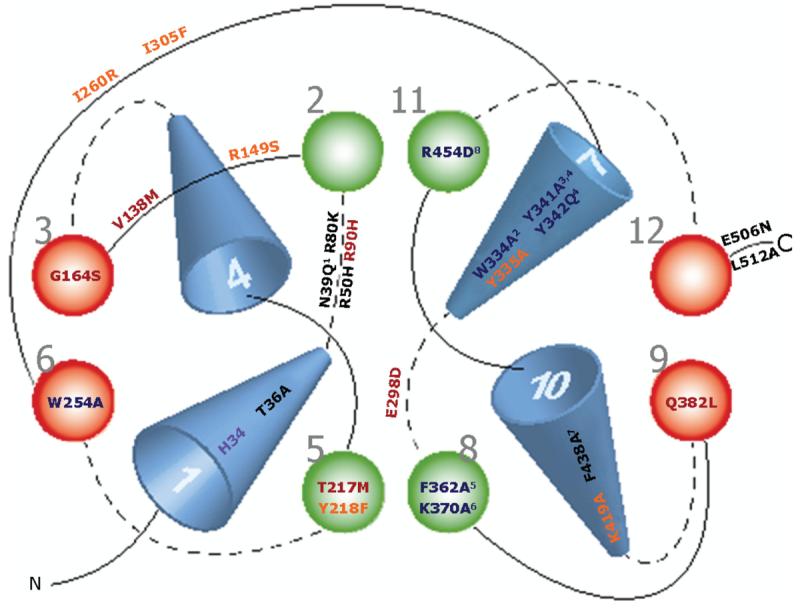
Critical amino acids for transport of SLC22 members. Three-dimensional arrangement of transmembrane domains. Dotted lines depict extracellular loops and solid lines represent cytoplasmic loops. fOat1 mutants are shown in purple, rOat3 mutants in blue, hOAT3 mutants in orange, hOAT1 mutants in black, and URAT1 mutants in red. 1, N39Q of mOat1; 2, residue also mutated to F, which caused functional decreases; 3, residue also mutated to Y, which caused functional decreases; 4, corresponds to Y353A/W/F and Y354A/W/F mutants in hOAT1, respectively; 5, residue also mutated to S, which caused functional decreases; 6, corresponds to K394A mutated in fOat1; 7, corresponds to F426 in hOAT3; 8, corresponds to R478D mutated in fOat1 and R466K mutated in hOAT1.
Basic amino acids
Conservation of the histidine in TMD 1, the lysine in TMD 8, and the arginine in TMD 11 in OAT1, OAT2, and OAT3 isoforms from several species has led investigators to create site specific mutations at those residues. Mutations at H34 reduce PAH uptake by more than 50% in both fOat1 (Wolff et al. 2001) and hOAT1 (Hong et al. 2004). No differences in plasma membrane immunostaining occur for fOat1. Expression patterns have not yet been determined for hOAT1. Kinetic analysis of the H34 mutants in fOat1 indicates that decreases in Vmax lead to reduction of overall PAH uptake. In addition, no change in the handling of dicarboxylates by this mutant is likely, since PAH uptake is significantly cis-inhibited by glutarate in both mutant and wild-type (Wolff et al. 2001). Additional experiments in fOat1 and rOat3 (K394A and K370A, respectively) describe the contributions of the lysine in TMD 8 to OAT function (Feng et al. 2001; Wolff et al. 2001). PAH transport is significantly decreased in both isoforms. Data from fOat1 suggests that PAH affinity is not changed, but that maximal transport rate is decreased in the mutant. Since membrane expression is normal, these findings suggest reduced turnover of the mutant carrier. Finally, the inability of glutarate to cis-inhibit or trans-stimulate PAH uptake, combined with reduced glutarate uptake indicates that K394 is important in dicarboxylate interaction and conformational changes in fOat1 (Feng et al. 2001; Wolff et al. 2001).
Experiments in rOat3 (Feng et al. 2001), fOat1 (Wolff et al. 2001) and more recently hOAT1 (Rizwan et al. 2007) demonstrate a requirement for the arginine in TMD 11 for transport of OAT substrates and dicarboxylates. Mutation effects differ slightly between species. For fOat1, the Km of the R478 mutant for PAH was significantly greater than wild-type, indicating a decrease in PAH affinity. Similar results are seen for rOat3, since PAH only inhibits approximately 15% of wild-type cimetidine uptake, suggesting PAH interacts with R454 in rOat3 (Feng et al. 2001). In contrast, no change in the inhibition profile occurs for OTA and ES. Substrate charge reversal even takes place in the double rOat3 mutant R454D/K370A, resulting in transport of the organic cation, 1-methly-4-phenylpyridinium (MPP+), and the weak base, cimetidine, instead of the anion, PAH (Feng et al. 2001). The hOAT1 R466K mutant also demonstrate decreases in PAH and glutarate uptake. No changes in PAH or glutarate affinity or in protein expression at the membrane are evident, indicating that reduced turnover number is responsible for the reduced uptake. In addition, chloride dependency of wild-type protein is abolished in the R466K mutant, indicating chloride might ion pair to the guanidinium group of arginine (Rizwan et al. 2007).
Similar to the lysine in TMD 8 and arginine in TMD 11, the lysine in TMD 10 also alters PAH transport. Mutating hOAT3 residue K419 yields no change in PAH affinity, little change in transporter membrane expression, and a lower Vmax than wild-type protein (Astorga et al. 2007). Glutarate is not transported by this mutant, and PAH and glutarate uptake are not restored to control levels by the K419R mutation (Perry et al. 2007).
Aromatic amino acids
Tryptophan, tyrosine, and phenylalanine residues in TMD 7 and 8 have been examined to determine their contributions to substrate transport. Rat Oat3 mutants demonstrate the necessity of the hydroxyl group of Y342, the indole ring of W334 and the aromatic rings of Y335, Y341 and F362 for PAH and cimetidine transport (Feng et al. 2002). Interestingly, these residues do not have as great an impact on ES transport. Additional studies of residues Y353, Y354 and W346 in hOAT1 (which correspond to Y341, Y342 and W334 of rOat3) indicated that transport by the Y353A mutant is restored upon mutation to Y353F. However, the Y354F mutant is not similar to wild-type hOAT1 in its transport. This finding implicates potential hydrogen bonding in substrate interaction at Y354 for hOAT1 and rOat3 (Hong et al. 2007b). Our lab recently reported two additional hOAT1 and hOAT3 aromatic residue mutants (Y230A/Y218A, F438A/F426A) in TMD 5 and 10, respectively (Perry et al. 2006, 2007; Astorga et al. 2007). In these studies, kinetic analysis showed a reduced Km for cidofovir transport in the hOAT1 F438 mutant, with no significant change in PAH transport. These results differed from ES transport by the hOAT3 mutants, where no changes in ES uptake were seen for F426Y, but in the Y218F mutant, a significant decrease in the Km values for both ES and PAH were observed.
Additional amino acids
Several analyses suggest that charged amino acids in loops contribute to OAT function (Yang et al. 2002; Bleasby et al. 2005; Erdman et al. 2006; Xu et al. 2006). Mutation of the arginines in the extracellular loop between TMD 1 and 2 (R50H, R80K, R90H) affects uptake transport without changes in Vmax or membrane protein expression for hOAT1 and hURAT1. Likewise, the hOAT3 R149S mutant in intracellular loop between TMD 2 and 3 exhibits reduced transport for both ES and cimetidine (Erdman et al. 2006). One charged hOAT1 mutant also occurs in the C-terminus. That residue (E506) is believed to form a salt bridge with a basic residue, since only substitution with a cationic mutant E506D restored function (Xu et al. 2006).
Amino acids of other types may also contribute to OAT function. For example, several hURAT1 SNPs (V138M, G164S, T217M, E298D, Q382L) are found in TMDs and in extracellular loops (Enomoto et al. 2002b; Ichida et al. 2004; Komoda et al. 2004). Expression of these mutant forms in oocytes leads to changes in substrate transport without changes in membrane protein expression. Likewise, three hOAT3 SNPs occur in the cytoplasmic loops between TMD 2 and 3 and between TMD 6 and 7. These have been shown to result in complete loss of function for R149S and I260R variants and the inability to transport ES for I260R (Erdman et al. 2006). Alanine scanning of TMD1 in hOAT1 identified residue T36 among 22 residues that were not influenced by protein trafficking. The presence of a methyl group on T36 is required for PAH transport (Hong et al. 2004). Finally, residue N39Q in hOAT1 and mOat1 are important for PAH transport. In hOAT1, N39 is glycosylated, unlike that of mOat1. Despite the absence of changes in cell surface expression (Tanaka et al. 2004c), each leads to changes in transport. Additional mutations at each of these residues are needed to determine their individual contributions to OAT function.
Tertiary structure features
The first hypotheses about OAT structure originated from hydropathy models that indicated the presence of twelve TMDs in these carriers, as described in the Introduction. More recently, it has been possible to glean more tertiary structural information for the OATs. First, computational models have been built using consensus secondary structure folding patterns from the hydropathy models and conserved signature sequences present in multiple loops from proteins within the MFS. Subsequently, both hOAT1 and hOAT3 sequences were mapped to the X-ray crystal structure of the glycerol 3-phosphate from E. coli (Huang et al. 2003; Perry et al. 2006, 2007; Astorga et al. 2007). A similar approach has also been taken for the organic cation transporters (Popp et al. 2005; Zhang et al. 2005), whose computational models were generated from MFS X-ray crystal structures of the glycerol 3-phosphate transporter and the lactose permease (Abramson et al. 2003a). Figure 4 shows a computational model of hOAT1 with the putative binding cavity open to the cytoplasm and internal amino- and carboxy-termini.
Figure 4.

Three-dimensional structure of hOAT1, looking into the cytoplasmic open face. The putative binding site is surrounding by yellow aliphatic amino acids, orange polar amino acids, red positively charged amino acids (R466 and K370), and green aromatic amino acids (Y230 and F438).
Computational models provide a means to conceptualize experimental data collected to date. For example, they indicate that many of the aromatic and basic amino acids that alter OAT function, as described in the previous section, appear to surround the putative binding site (for hOAT1: R466, K382, Y353, Y354, and F374, as depicted in Figure 4) (Perry et al. 2006). The three-dimensional OCT2 model has also been used to explain the difference in IC50 values in hOAT1 and hOCT2 for inorganic mercury (Zhang et al. 2005; Astorga et al. 2007). Inorganic mercury is believed to interact with accessible cysteines, a number of which appear to be more accessible in hOCT2 than in hOAT1 according to their structural models. This prediction was confirmed experimentally, by showing that a membrane impermeant thiol reactive biotinylation reagent reacted better with hOCT2 than hOAT1. In addition, these models provide an opportunity to select new amino acids that are in, or near, critical sites in the transporter, e.g., the putative substrate binding pocket, for mutation and assessment of their functional consequences. For example, mutation of one such amino acid, F438, significantly increases the Km of cidofovir for hOAT1, although it does not affect PAH affinity (Perry et al. 2006). Likewise, the TMD 5 mutant Y218F increases the Km for both PAH and ES when compared with wild-type values (Astorga et al. 2007; Perry et al. 2007). More recent analysis demonstrates that hOAT3 K419 contributes to dicarboxylate recognition and transport, but has no effect on PAH or ES transport (Perry et al. 2007, and unpublished results).
Experimental confirmation of both computational and hydropathy models of OAT structure appeared in 2007 (Hong et al. 2007a). The amino- and carboxy-termini were confirmed as intracellular through Myc-tagged expression of hOAT1 with immunostaining under permeablized and non-permeablized conditions. In addition, the limited number of lysines present in extracellular loops of hOAT1 has been exploited to determine loop size and orientation with biotin labelling. You and co-workers mutated the native lysines present in extracellular loops to arginines and then selectively mutated amino acids near predicted ends of loops to lysine to determine whether that residue could be detected by cell surface biotinylation (Hong et al. 2007a). Their results indicate that residues in extracellular loops III, V and VI (with mutants V244K, S420I, and L481K) are accessible to biotinylation. This is in agreement with the computational model as both V244 and L481 are in loop locations in the computational model; whereas, S420 is the first residue in TMD 10, a site likely to be accessible to labelling. These studies also revealed that P183 and D359 are not accessible; leading the authors to suggest that those residues may reside in TMDs or in the short loops II and V, thus reducing the extent of labelling. The computational model illustrates that both arguments are possible. D359 is not accessible as it falls within TMD 7; whereas, P183 falls within external loop II in the hOAT1 model (Perry et al. 2006).
New experimental data has also contributed to our knowledge of OAT structural aspects. First, Hong et al. (2005) have suggested that hOAT1 may exist as a homo-oligomer based on chemical cross linking and immunoprecipitation experiments. This organization may affect not only substrate-transporter interactions, but also protein-protein interactions and regulation and signalling pathways. Secondly, truncations of the C-terminus demonstrated that E506 is necessary for transport function and may form a salt bridge with another positively charged residue; whereas, L512 alters substrate Vmax (Xu et al. 2006). These data demonstrate that it will be important to determine the functionally active form of OATs and the role of termini in transport and membrane processing and targeting.
Determinants of transport efficacy
Binding
Plasma protein binding impedes organic anion transport
The use of cloned transporters and expression systems (as reviewed here and in Pritchard and Miller 2005) has increased knowledge about OAT substrate affinity through in vitro uptake assays. Although these in vitro assays are ideal for examining substrate specificity, other results may not mimic in vivo transport. For example, most often these assays do not take into account plasma protein binding of the substrate (primarily to serum albumin), a property that has been historically shown to alter drug pharmacokinetics, bioavailability and half-life (Weiner et al. 1964).
For this reason, Bow et al. (2006) assessed the impact of protein binding on several OAT transporters expressed in vitro. These experiments demonstrated that uptake of two highly albumin bound anions (OTA and ES, Kb >105) is very much less than that seen in the absence of albumin, i.e., that uptake reflects the limited free fraction of substrate in the presence of albumin. For example, the free concentration of OTA (a high-affinity substrate for OATs 1, 3, 4) is only 0.2% of the total concentration and that of ES (an OAT3 substrate) is 3%. Reflecting this fact, uptake of both compounds is significantly reduced at albumin concentrations approximately eightfold lower than that of plasma. In contrast, there is no effect of albumin binding on the uptake of MTX (an OAT1 substrate), which has a free fraction of 47% (Bow et al. 2006). These results are similar to reports that albumin-binding affects hepatic and intestinal organic anion transport by several OATPs and Mrp2 (Berger et al. 2003; Cui and Walter 2003), as well as the importance of plasma protein binding on renal elimination of organic anions (Weiner 1973). Clearly, such factors can profoundly alter the efficacy of drug and xenobiotic transport and need to be remembered as extrapolations are drawn from in vitro experiments with cloned transporters.
Thiol conjugation promotes transport of heavy metals by basolateral OATs
A rather different example of the impact of interactions between reactive chemicals and endogenous chemicals within the body is provided by the formation of metal conjugates with plasma thiols. Several nephrotoxic heavy metals, particularly inorganic mercury, have been shown to be taken up across the basolateral membrane of the proximal tubule, with resulting nephrotoxicity (Zalups 2000). However, pretreatment of rats or mice with an OAT substrate (PAH) or an OAT inhibitor (probenecid), significantly reduced renal uptake of mercury, suggesting a role for the OATs in renal accumulation of mercury (Zalups 2000). Additional experiments have shown that conjugates of mercury with thiols (e.g., homocysteine, cysteine and glutathione) are present in the blood plasma, apparently providing the negative charge and hydrophobicity needed for mercury transport by basolateral OATs. This hypothesis was tested by Aslamkhan et al. (2003) in cells expressing hOAT1. They demonstrated that transport of mercury N-acetylcysteine conjugates is mediated by hOAT1 and that transport and toxicity can be blocked by probenecid. A similar interaction with rOat3 and mercury N-acetylcysteine conjugates is also seen in Xenopus oocytes (Aslamkhan et al. 2003). Subsequently, similar findings were made for conjugates with the sulfhydryl-containing amino acid homocysteine (Zalups and Ahmad 2004). Methylmercury is also conjugated with homocysteine and taken up into MDCK cells expressing hOAT1 (Zalups and Ahmad 2005). Thus, in this example, transport of the conjugate provides a pathway for entry of a toxic agent, and is responsible for increased, rather than decreased, toxicity.
Single nucleotide polymorphisms
DNA sequence variations that occur among individuals are referred to as single nucleotide polymorphisms (SNPs) (Chakravarti 2001). Obviously, SNPs that change the properties of the OAT transporters may well alter the efficacy of transport. Recently, OAT SNPs have been identified in Caucasian, African, Japanese and Pacific Islander populations. The distribution of SNPs has been determined to be one in every 1.2 kb (Iida et al. 2001; Marzolini et al. 2004) and given the limited sample size and ancestry of current data, discovery of more OAT SNPs is likely. Table VI summarizes the non-synonymous SNPs that occur within the coding regions of hOAT1, hOAT2, hOAT3, hOAT4 and URAT1 (see http//www.pharmgkb.org; Ichida et al. 2003; Lee et al. 2003a, b; Nishizato et al. 2003; Iwai et al. 2004; Komoda et al. 2004; Bleasby et al. 2005; Cheong et al. 2005; Fujita et al. 2005; Taniguchi et al. 2005; Wakida et al. 2005; Xu et al. 2005; Bhatnagar et al. 2006; Erdman et al. 2006; Graessler et al. 2006).
Certainly, inter-individual variability in drug disposition and elimination may be dependent upon SNPs occurrence. Clinical studies have investigated the consequences of some non-synonymous variants. Findings indicate that no changes occurred in the in vivo renal secretion clearance of adefovir with the hOAT1 R454Q variant (Fujita et al. 2005). Likewise, no changes in renal tubular secretion of pravastatin, an hOAT3 substrate, are found in the hOAT3 A389V variant (Nishizato et al. 2003). In contrast, non-synonymous variants R90H, W258Stop and R477H in hURAT1 are linked to familial idiopathic hypourecaemia in Japanese populations (Iwai et al. 2004). Clinical analysis showed reduced serum urate levels in these patients, suggesting that these variants are responsible for the increased clearance of urate, the endogenous hURAT1 substrate. The truncated SNP, W258Stop, is believed to be the most predominant hURAT1 mutation responsible for renal hypouricaemia in Japanese and Korean populations (Ichida et al. 2004; Komoda et al. 2004; Cheong et al. 2005).
Functional effects of additional non-synonymous SNPs have also been determined in cell-based systems for hOAT1, hOAT3 and hURAT1. For example, Bleasby et al. (2005) showed that R50H and K525I hOAT1 variants do not affect PAH transport, but the R50H variant shows increased the affinity of cidofovir and adefovir, two lower affinity hOAT1 substrates. No functional changes are observed in PAH, OTA and MTX uptake in six additional hOAT1 SNPs (Fujita et al. 2005). Of the eleven hOAT3 SNPs, 3 variants (R149S, Q239X, I260R) exhibit reduced ES and cimetidine uptake. Variants R277W and I305F do not transport ES, but surprisingly R277W appeared to transport cimetidine more effectively (Erdman et al. 2006). In vitro data on URAT1 SNPs has been well studied by monitoring urate uptake in Xenopus oocyte expression systems. At least eleven variants (R90H, V138M, T217M, G164S, W258Stop, E298D, Dd313-P33 deletion, Q382L, L418R, M430T, and V547fsX602) exhibit reduced urate uptake when compared with wild-type URAT1 (Table VI) (Enomoto et al. 2002a; Ichida et al. 2003; Iwai et al. 2004; Cheong et al. 2005; Wakida et al. 2005). Interestingly, little to no expression of the truncated mutant W258Stop occurred, perhaps explaining some of the loss of function reported in vivo (Enomoto et al. 2002a).
These initial approaches have been helpful, but understanding of SNP impact on OAT function remains limited for many OAT isoforms. For example, the majority of non-synonymous SNPs in hOAT2 and hOAT4 lack data on protein expression, functional effects, and protein structure modification. In addition, larger and more diverse population sets need to be screened for unidentified SNPs that may exist. Furthermore, the roles of synonymous SNPs and non-synonymous SNPs present in non-coding regions (e.g., promoters) remain largely unexplored. Finally, it must be noted that although it may be easier to examine SNP consequences in vitro, cell based systems are still limited by low expression, protein trafficking problems and the inability to examine synergistic transport phenomena present in vivo. Thus, clinical investigations will be needed to determine the phenotypical response to individual variants.
Regulation
It is also possible that differences in transporter function between individuals may be caused, not by differences in genetic makeup, but through up- or down-regulation of OAT transporter expression based on prior exposure to drugs and xenobiotics, or on differences in hormonal status. Although regulation of OATs has not been extensively studied, examples of both longer term control of expression, e.g., via nuclear transcription factors or epigenetic mechanisms, and of short term modulation of transporter activity via signalling pathways have been observed.
Transcriptional regulation
Hepatocyte nuclear factors (HNFs) have been shown to initiate transcriptional regulation of OAT family members. For example, HNF-4α activates hOAT1 promoter activity when HNF-4α and the hOAT1 promoter are co-transfected into opossum kidney cells (Ogasawara et al. 2007). These authors identified a specific binding element for HNF-4α, and suggested that it is important for basal activity of the hOAT1 promoter. Other HNF isoforms, HNF-1α and/or β, can also activate both hOAT1 and mOat1 promoters (Saji et al. 2008). Likewise, HNF-4α activates the hOAT2 promoter in a human hepatoma cell line (Popowski et al. 2005). Interestingly, these data indicate that the farnesoid X receptor (FXR) can reduce HNF-4α transactivation of hOAT2 promoter activity via FXR ligands, e.g., the bile acid chenodeoxycholic acid, suggesting a regulatory network. The hOAT3 promoter is also activated by the HNF-1α homodimer, the HNF-1β homodimer, and the HNF-1α/β heterodimer in HEK-293 cells (Kikuchi et al. 2006). However, the HNF-1β homodimer is less effective in this assay. Kikuchi et al. (2006) also noted that multiple CpG dinucleotides are present in the putative hOAT3 promoter region. Since these sites are a likely target for DNA methylation, they explored the possibility of epigenetic control of hOAT3 expression and function via DNA methylation. As predicted, hOAT3 promoter activity is abolished after methylation in vitro. Conversely, hOAT3 transcription is activated when DNA methylation is inhibited by DNA methyltransferase inhibitor (Kikuchi et al. 2006). Finally, Ogasawara et al. (2006) showed that a cAMP-response element (CRE) affects basal and inducible transcriptional activity of the hOAT3 promoter, and that the hOAT3 promoter is activated by phosphorylation of CRE-binding protein (CRE-B1) and activating transcription factor (ATF1).
Transcription factor control of the apically expressed OAT isoform, URAT1, is very similar to that of the basolateral OATs discussed above (Kikuchi et al. 2007). HNF-1α homodimer, HNF-1β homodimer, and HNF-1α/β heterodimer all increase promoter activity for hURAT1 and mUrat1 in HepG2 cells. Again, HNF-1β homodimer induces a smaller increase in promoter activity with the hURAT1 or mUrat1 promoters than either HNF-1α homodimer or the heterodimer. Tissue expression of mUrat1 is also subject to epigenetic regulation. Hypermethylation is found in both liver and kidney medulla, where mUrat1 is not expressed. However, in kidney cortex, the primary site of mUrat1 expression, its promoter is hypomethylated, supporting the hypothesis that tissue specific expression of mUrat1 is regulated through DNA methylation.
Gender differences have also been seen for several of the OATs. These very likely reflect nuclear transcription factor signalling, but the role of specific transcription factors in control has not yet been assessed and there are substantial isoform and species differences in gender-specific effects. For example, in the female rat, Oat1 mRNA expression is lower than in male (Buist et al. 2002). Consistent with lower mRNA, rOat1 protein expression is also lower in female rats, with a correspondingly smaller PAH clearance as well (Reyes et al. 1998; Cerrutti et al. 2001, 2002a, b). Interestingly, gender differences in rOat1 and rOat3 protein expression are observed in adult, not prepubertal rats (Buist et al. 2002; Ljubojevic et al. 2004; Zhou et al. 2008). Moreover, androgen seems to up-regulate rOat1 and rOat3 expression; whereas, oestrogen reduced expression. However, there are no gender differences in rbOat1 and rbOat3 expression and function (Groves et al. 2006). In contrast to OAT1 and OAT3, female rOat2 mRNA and protein are greater than male rOat2, showing androgen inhibition and oestrogen stimulation (Buist et al. 2002; Ljubojevic et al. 2004). Mouse Oat2 has a similar expression pattern (Ljubojevic et al. 2007). Finally, a gender difference in mUrat1 mRNA expression (male>female) is also seen, but functional data are lacking (Hosoyamada et al. 2004). The data on gender differences in the renal transport was recently reviewed (Sabolic et al. 2007).
Post-translational mechanisms
As discussed above, OAT family members share structural features including a large extracellular loop between TMD 1 and 2 and another large intracellular loop between TMD 6 and 7. Several potential N-glycosylation sites have been predicted in an extracellular loop and a number of potential phosphorylation sites are present in intracellular loops. N-glycosylation appears to play a critical role in membrane targeting/trafficking, protein folding and possibly regulation of OAT function. This has been demonstrated for mOat1 (Kuze et al. 1999), hOAT1 (Tanaka et al. 2004a), and hOAT4 (Zhou et al. 2005), although the molecular basis for the changes in folding and trafficking are not yet fully understood.
A variety of data suggest that phosphorylation of the large intracellular loop and interactions of the OATs with protein partners may markedly alter OAT function. For example, it has been known since the late 1990s that the conventional PKC (cPKC) is an important regulator of OAT1 function (Uwai et al. 1998; Lu et al. 1999; Wolff et al. 2003; You et al. 2000). Activation of cPKC inhibits OAT1-mediated PAH transport in various heterologous expression systems. This effect is seen whether cPKC is activated directly with phorbol 12-myristate 13-acetate (PMA) or by 1,2-dioctanoyl-sn-glycerol (DOG), or more indirectly through activation of receptors and complex signalling pathways ultimately leading to cPKC activation (e.g. via phenylephrin or bradykinin receptors). In addition, inhibition of basolateral fluorescein (FL) uptake is also seen following cPKC activation in an intact renal epithelium (Gekle et al. 1999; Shuprisha et al. 2000). Since FL is a substrate for both OAT1 or OAT3 (Dantzler and Wright 2003; Miller 2003), this result might reflect effects on either transporter. However, rbOat3 does not transport PAH (Dantzler and Wright 2003). Thus, inhibition of basolateral PAH uptake in rabbit proximal tubule must reflect an effect on OAT1 activity. Interestingly, activation of cPKC does not lead to reduced OAT1 transport through phosphorylation of transporter itself (You et al. 2000; Wolff et al. 2003). Instead, it appeared that transport reduction is mediated by retrieval of the transporter from the membrane (Wolff et al. 2003).
Unlike cPKC activation, which reduces OAT-mediated transport, epidermal growth factor (EGF), a mediator for normal tubular growth and regeneration after injury, stimulates basolateral PAH uptake in rabbit tubules via Oat1. This effect is mediated through a complex signalling pathway through activation of mitogen-activated/extracellular-signal regulated kinase (MEK), extracellular signal-regulated kinase isoforms 1 and 2 (ERK1/2), and phospholipase A2 (PLA2), resulting in increased arachidonic acid (AA) release. Subsequently, AA was metabolized to prostaglandin E2 (PGE2) via cyclooxygenase I (COX I). In turn, PGE2 activates adenylate cyclase, leading to cAMP production and activation of PKA and ultimately increased basolateral rbOat1-mediated PAH uptake (Sauvant et al. 2002, 2003, 2006; Dantzler and Wright 2003).
A parallel story has been documented for OAT3. Transport of ES, a specific OAT3 substrate, is decreased by the activation of cPKC through the same regulatory pathway as OAT1 (Takeda et al. 2000). Also like the OAT1 results, EGF stimulates ES uptake in isolated rabbit renal proximal tubules. The signalling process for this effect is identical to that described above for OAT1 up-regulation (Soodvilai et al. 2004). In our laboratory, we have recently shown that both insulin and EGF are able to stimulate ES transport in rat renal cortical slices through the activation of PKCζ, an atypical PKC that is not activated by phorbol esters, calcium, or DOG (Hirai and Chida 2003; Jenny et al. 2005). The signalling pathway is the same as that identified by Soodvilai et al. (2004, 2005) through PKA activation. PKCζ is downstream of PKA, since inhibition of PKCζ blocked PKA induced stimulation of transport. In addition, Barros et al. (unpublished data) showed that microtubule disruption abolished up-regulation of transport, suggesting that like the down-regulation in response to activation of cPKCs (Wolff et al. 2003), trafficking is required for PKCζ-mediated up-regulation to occur. Finally, transport of adefovir, a specific substrate for OAT1, is also increased after insulin or EGF treatment via PKCζ, indicating that PKCζ can up-regulate of OAT1-mediated transport (Barros et al., unpublished data).
At this time, other types of regulation that are known to alter function of transporters have not yet been evaluated for the OATs, but these possibilities need to be explored experimentally. For example, lipid raft microdomains play important roles in membrane trafficking, serve as platforms for cell surface signal transduction and protein targeting as well as regulating cholesterol homeostasis (Simons and Ikonen 1997; Ait Slimane and Hoekstra 2002; Fielding and Fielding 2003). A major component of caveolae, one such lipid raft domain, is the self-associating protein, caveolin. Recently, rOat1 was shown to co-localize with caveolin 2 in the plasma membrane of rat renal proximal tubule cells in primary culture (Kwak et al. 2005a). Furthermore, decreasing expression of either caveolin 1 or 2 leads to the reduction of rOat1 and rOat3 function (Kwak et al. 2005a, c). The full physiological significance of these findings is unknown, but this line of investigation merits further attention.
Down-regulation of hOAT4 occurs following activation of cPKC in BeWo cells, a human placental cell line (Zhou et al. 2007). In addition, progesterone, a steroid hormone produced by human placenta, also reduces hOAT4 function by a mechanism that appears to involve trafficking out of the plasma membrane. Human OAT4 may also interact with two multivalent PDZ proteins, PDZK1 and NHERF1, via its C-terminal portion which contains a PDZ binding motif (Miyazaki et al. 2005). Increased expression of either PDZK1 or NHERF1 (by co-transfection) increases hOAT4 function. More recently, the interaction of hOAT4 with PDZK1 and NHERF1 proteins was investigated in kidney and placental cell lines. Both PDZK1 and NHERF1 increase hOAT4 surface expression and function in a kidney cell line (LLC-PK1) (Zhou et al. 2008). However, similar experiments in a placental cell line expressing hOAT4 do not show enhanced hOAT4 transport activity by either PDZK1 or NHERF1, indicating that regulation is tissue-specific and thus, may involve additional interactions with proteins or membrane lipids (Zhou et al. 2008).
Several lines of investigation indicate that PDZK1 also regulates hURAT1 surface expression and function (Anzai et al. 2004). hURAT1 interacts with PDZK1 in yeast two-hybrid assays and the two proteins are co-localized in the apical membrane of renal proximal tubules. Co-transfection of PDZK1 in hURAT1 expressing cells stimulates hURAT1 surface expression and transport function. Moreover, deletion of the C-terminal portion of URAT1 abolishes this effect. Clearly, protein–protein interactions are an important, and incompletely understood, aspect of OAT function.
Perspectives
A great deal has been learned about the OATs since the first OAT was cloned in 1997. However, as noted in the preceding sections, there are areas where important gaps remain. This section is intended as a brief reminder of some of those issues. For example, because of its very broad acceptance of a variety of drugs and xenobiotics, the OAT family may contribute to drug–drug interactions. This possibility is well appreciated and examples are known, but the extent of such interactions is not yet fully explored. Other important gaps concerning OAT function include: (1) a lack of mechanistic information precludes prediction of the physiological roles of many cloned OATs; (2) an incomplete understanding of the interplay between the OATs themselves, or between OATs and other drug transporters, on the trans-epithelial flux of OAT substrates; (3) an emerging appreciation that the OATs may have unique roles in extra-renal tissues; and (4) the incomplete characterization of genetic and epigenetic mechanisms that may alter expression of OATs and lead to individual differences in drug and xenobiotic transport.
Drug–drug interactions
Since the OATs transport such a wide spectrum of drugs and xenobiotics, the potential for competition between OAT substrates for elimination is substantial. Thus, when multiple OAT substrates are present, profound changes in drug clearance and plasma drug concentrations are often seen. However, there are actually two issues involved here. The first is altered transport of the drug, or of its metabolites, and the second is altered metabolism. In part, this is because metabolism produces substrates for the organic anion system (Pritchard and Miller 1993; Wright and Dantzler 2004). Thus, interactions between drugs, or between drugs and xenobiotics, can occur at the level of transport itself or through altered metabolism leading to a change in the species requiring transport. In part, this is because metabolism and transport are often co-regulated (Klaassen and Slitt 2005; Petrick and Klaassen 2007). Thus, agents may impact drug pharmacodynamics indirectly through these regulatory mechanisms. An excellent example of the complexity of these interactions is provided by Shitara and Sugiyama in their review of the side effects of statin drugs, that emphasizes the close association between metabolism and transport in controlling the fate of these drugs and thus, of their potential to contribute to toxicity or disease (Shitara and Sugiyama 2006).
Research needs
Mechanisms
As discussed in detail above, only for OATs 1 and 3 is there sufficient mechanistic understanding to define their physiological roles. For the other OATs, questions of mechanism and driving forces remain. For example, OAT4 and URAT1 are documented to be anion exchangers and have likely roles in urate reabsorption. However, uncertainties about the contribution of chloride/urate exchange to their function in vivo cloud understanding of their possible roles in organic anion secretion. Finally, for other members of the family, no mechanistic information is available.
Coordinated function
There are multiple OATs expressed at the apical and basolateral membranes of transporting epithelia. Moreover, numerous other drug/xenobiotic transporters are also expressed in tissues like liver, kidney, and barrier epithelia, including the organic anion transporting polypeptides (OATPs; SLCO family), multidrug resistance proteins (MDRs; ABCB family), breast cancer resistance protein (BCRP; ABCG family), and the multidrug resistance-associated proteins (MRPs; ABCC family) (Hagenbuch et al. 2002; Hagenbuch and Meier 2003, 2004; Schinkel and Jonker 2003; Miller et al. 2005; Choudhuri and Klaassen 2006; Shitara et al. 2006). The extent to which their function affects one another, or is coordinated with each other, remains largely uninvestigated. This is particularly true regarding their relative importance for the handling of specific drugs, or how that handling might change in the presence of multiple substrates.
Transporter expression versus tissue function
The distribution of the OATs is largely understood, but, surprisingly, function does not always reflect expression. One example of this perplexing issue is illustrated by data from the Oat3 knockout mouse. In kidney, loss of mOat3 leads to nearly complete loss of the ability to transport Oat3 specific substrates, e.g., taurocholate and ES, and partial loss of the transport of substrates shared with mOat1, e.g., PAH (Sweet et al. 2002). In the choroid plexus of the mOat3 null mice, the handling of some substrates is exactly as predicted. For example, PAH transport is completely lost in the knockout, as would be predicted by the virtual absence of mOat1 expression even in wild-type choroid plexus and the loss of mOat3 in the knockout animals (Sykes et al. 2004). However, transport of the mOat3-specific substrate, ES, was only slightly diminished, not abolished as it was in kidney, despite the lack of both Oats 1 and 3 in choroid plexus. Some of the remaining transport could be attributed to an Oatp, some to a potential-driven transporter, but some did not match the characteristics of any known transporter. Clearly, resolution of such puzzles requires more than the ability simply to list the transporters expressed in a given tissue.
Regulation
As discussed above, individual differences in their ability to transport drugs and xenobiotics may be caused by genomic differences, e.g., SNPs and other mutations, or by their exposure to agents that increase or decrease their ability to transport, e.g., competition driven drug–drug interactions. Alternatively, differences in transport capacity may reflect the consequences of ligand activated nuclear transcription factors or of cellular signalling mechanisms. As discussed in more detail above, many of these signalling events are incompletely understood.
Summary
Thus, although a great deal has been learned about the OATs in the decade since they were first cloned, much remains unresolved regarding their mechanisms, physiological roles, structure–function relationships, and regulation. Fortunately, many of the necessary tools are in place. Thus, the next ten years promise to be equally exciting and illuminating.
Table VI.
Non-synonymous SNPs in OATs and URAT1 coding regions.
| Isoform | Amino acid change |
Functional effect | References |
|---|---|---|---|
| hOAT1 | L7P | n.d. | Xu et al. (2005) |
| R50H |
Km change in adefovir cidofovir and tenofovir |
Bleasby et al. (2005), Fujita et al. (2005), Xu et al. (2005) |
|
| P104L | No significant change in PAH, OTA or MTX uptake |
Fujita et al. (2005), Lee et al. (2003b) | |
| A190T | n.d. | Lee et al. (2003b) | |
| I226T | No significant change in PAH, OTA or MTX uptake |
Fujita et al. (2005) | |
| A256V | No significant change in PAH, OTA or MTX uptake |
Fujita et al. (2005) | |
| R293W | No significant change in PAH, OTA or MTX uptake |
Fujita et al. (2005) | |
| R454Q | No significant change in PAH, OTA or MTX uptake |
Fujita et al. (2005) | |
| Q454R | No significant change in PAH, OTA or MTX uptake |
Fujita et al. (2005) | |
| K525I | No Km change for PAH or adefovir |
Bleasby et al. (2005) | |
| hOAT2 | T110I | n.d. | Xu et al. (2005) |
| V192I | n.d. | Xu et al. (2005) | |
| G507D | n.d. | Xu et al. (2005) | |
| hOAT3 | F129L | No significant change in ES or cimetidine uptake |
Erdman et al. (2006) |
| R149S | Significantly reduced uptake of ES and cimetidine |
Erdman et al. (2006) | |
| I175V | n.d. | Xu et al. (2005) | |
| Q239X | Significantly reduced uptake of ES and cimetidine |
Erdman et al. (2006) | |
| I260R | Significantly reduced uptake of ES and cimetidine |
Erdman et al. (2006) | |
| R277W | No significant change in ES or cimetidine uptake |
Erdman et al. (2006) | |
| V281A | No significant change in ES or cimetidine uptake |
Erdman et al. (2006) | |
| I305F | Decreased ES uptake, increased cimetidine uptake |
Erdman et al. (2006) | |
| A310V | No significant change in ES or cimetidine uptake |
Erdman et al. (2006) | |
| A399S | No significant change in ES or cimetidine uptake |
Erdman et al. (2006) | |
| V448I | No significant change in ES or cimetidine uptake |
Erdman et al. (2006) | |
| hOAT4 | V13M | n.d. | Xu et al. (2005) |
| R48Stop | n.d. | Xu et al. (2005) | |
| T62R | n.d. | Xu et al. (2005) | |
| V155M | n.d. | Xu et al. (2005) | |
| A244V | n.d. | Xu et al. (2005) | |
| E278K | n.d. | Xu et al. (2005) | |
| V339M | n.d. | Xu et al. (2005) | |
| T392I | n.d. | Xu et al. (2005) | |
| URAT1 | R90H | Significantly reduced uptake of urate |
Cheong et al. (2005), Ichida et al. (2004), Iwai et al. (2004) |
| R92C | n.d. | Graessler et al. (2006) | |
| V138M | Significantly reduced uptake of urate |
Ichida et al. (2004) | |
| G164S | Significantly reduced uptake of urate |
Ichida et al. (2004) | |
| T217M | Significantly reduced uptake of urate |
Enomoto et al. (2002a) | |
| A226V | n.d. | Iwai et al. (2004) | |
| R228E | No significant change in urate uptake |
Iwai et al. (2004) | |
| W258Stop | Significantly reduced uptake of urate |
Cheong et al. (2005), Enomoto et al. (2002a), Ichida et al. (2004), Iwai et al. (2004), Komoda et al. (2004), Taniguchi et al. (2005), Wakida et al. (2005) |
|
| E298D | Significantly reduced uptake of urate |
Enomoto et al. (2002a) | |
| Q297Stop | n.d. | Takahashi 2005 | |
| Q312L | n.d. | Iwai et al. (2004) | |
| D313-P333 deletion | Significantly reduced uptake of urate |
Iwai et al. (2004) | |
| Q382L | Significantly reduced uptake of urate |
Ichida et al. (2004), Wakida et al. (2005) | |
| L418R | Significantly reduced uptake of urate |
Wakida et al. (2005) | |
| M430T | Significantly reduced uptake of urate |
Ichida et al. (2004) | |
| R477H | No significant change in urate uptake |
Iwai et al. (2004) | |
| V547fsX602 | Significantly reduced uptake of urate |
Ichida et al. (2004), Wakida et al. (2005) |
n.d., Not determined.
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. The authors also thank Drs David Miller, Deborah Thompson and Ramsey Walden for their helpful discussion of this manuscript.
Appendix: Abbreviations
| Abbreviation | Definition |
|---|---|
| α-KG | α-Ketoglutarate |
| ACE | Angiotensin-converting enzyme |
| ACV | Acyclovir |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| AZT | Zidovudin |
| BSP | Bromosulphophthalein |
| CCD | Cortical collecting duct |
| CHO | Chinese hamster ovary cell line |
| CK II | Casein kinase II |
| CMPF | 3-Carboxy-4-methyl-5-propyl-2-furan-propionate |
| COX I | Cyclooxygenase I |
| CP | Choroid plexus |
| CpG | 2′-Deoxyribo-cytidine-phosphate-guanosine |
| DHEAS | Dehydrodrosterone sulfate |
| DMPS | 2,3-Dimercapto-1-propane sulfonate |
| DNP-NAC | N-acetyl-S-2,4-dinitrophenyl-L-cysteine |
| DOG | 1,2-Dioctanoyl-sn-glycerol |
| ERK1/2 | Extracellular signal-regulated kinase isoforms 1 and 2 |
| ES | Oestrone sulfate |
| HEK | Human embryonic kidney cell line |
| MAL | Medullary thick ascending limb |
| MEK | Mitogen-activated/extracellular-signal regulated kinase |
| MFS | Major facilitator superfamily |
| MPP+ | 1-Methyl-4-phenylpyridinium |
| MTX | Methotrexate |
| NSAIDs | Nonsteroidal inflammatory drugs |
| OTA | Ochratoxin |
| PAH | p-Aminohippurate |
| PDZ | PSD-95/Discs Large/ZO-1 |
| PGE2 | Prostaglandin E2 |
| PGF2α | Prostaglandin F2α |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLA2 | Phospholipase A2 |
| PMA | Phorbol 12-myristate 13-acetate |
| PMEA | 9-(2-Phosphonylmethoxy-oxyethyl) adenine |
| PMEDAP | 9-(2-Phosphonylmethoxyethyl diaminopurine) |
| PMEG | 9-(2-Phosphonylmethoxy-oxyethyl) guanidine |
| PT | Proximal tubule |
| PZA | Pyrazinecarboxylic acid |
| SCFAs | Short-chain fatty acids |
| SNPs | Single nucleotide polymorpholisms |
| TAL | Thick ascending limb |
| TK | Tyrosine kinase |
| TMD | Transmembrane domain |
Footnotes
Publisher's Disclaimer: The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
References
- Abramson J, Smirnova I, Kasho V, Verner G, Iwata S, Kaback HR. The lactose permease of Escherichia coli: Overall structure, the sugar-binding site and the alternating access model for transport. Federation of European Biochemical Societies Letters. 2003a;555:96–101. doi: 10.1016/s0014-5793(03)01087-1. [DOI] [PubMed] [Google Scholar]
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003b;301(5633):610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Ait Slimane T, Hoekstra D. Sphingolipid trafficking and protein sorting in epithelial cells. Federation of European Biochemical Societies Letters. 2002;529:54–59. doi: 10.1016/s0014-5793(02)03183-6. [DOI] [PubMed] [Google Scholar]
- Anzai N, Jutabha P, Enomoto A, Yokoyama H, Nonoguchi H, Hirata T, Shiraya K, He X, Cha SH, Takeda M, et al. Functional characterization of rat organic anion transporter 5 (Slc22a19) at the apical membrane of renal proximal tubules. Journal of Pharmacology and Experimental Therapeutics. 2005;315:534–544. doi: 10.1124/jpet.105.088583. [DOI] [PubMed] [Google Scholar]
- Anzai N, Kanai Y, Endou H. Organic anion transporter family: Current knowledge. Journal of Pharmacological Sciences. 2006;100:411–426. doi: 10.1254/jphs.crj06006x. [DOI] [PubMed] [Google Scholar]
- Anzai N, Kanai Y, Endou H. New insights into renal transport of urate. Current Opinion in Rheumatology. 2007;19:151–157. doi: 10.1097/BOR.0b013e328032781a. [DOI] [PubMed] [Google Scholar]
- Anzai N, Miyazaki H, Noshiro R, Khamdang S, Chairoungdua A, Shin HJ, Enomoto A, Sakamoto S, Hirata T, Tomita K, et al. The multivalent PDZ domain-containing protein PDZK1 regulates transport activity of renal urate-anion exchanger URAT1 via its C terminus. Journal of Biological Chemistry. 2004;279:45942–45950. doi: 10.1074/jbc.M406724200. [DOI] [PubMed] [Google Scholar]
- Apiwattanakul N, Sekine T, Chairoungdua A, Kanai Y, Nakajima N, Sophasan S, Endou H. Transport properties of nonsteroidal anti-inflammatory drugs by organic anion transporter 1 expressed in Xenopus laevis oocytes. Molecular Pharmacology. 1999;55:847–854. [PubMed] [Google Scholar]
- Asif AR, Steffgen J, Metten M, Grunewald RW, Muller GA, Bahn A, Burckhardt G, Hagos Y. Presence of organic anion transporters 3 (OAT3) and 4 (OAT4) in human adrenocortical cells. Pflügers Archiv European Journal of Physiology. 2005;450:88–95. doi: 10.1007/s00424-004-1373-3. [DOI] [PubMed] [Google Scholar]
- Aslamkhan AG, Han YH, Yang XP, Zalups RK, Pritchard JB. Human renal organic anion transporter 1-dependent uptake and toxicity of mercuric-thiol conjugates in Madin–Darby canine kidney cells. Molecular Pharmacology. 2003;63:590–596. doi: 10.1124/mol.63.3.590. [DOI] [PubMed] [Google Scholar]
- Aslamkhan AG, Thompson DM, Perry JL, Bleasby K, Wolff NA, Barros S, Miller DS, Pritchard JB. The flounder organic anion transporter fOat has sequence, function, and substrate specificity similarity to both mammalian Oat1 and Oat3. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2006;291:R1773–R1780. doi: 10.1152/ajpregu.00326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astorga BO, Pelis RM, Wunz TM, Perry JA, Pritchard JR, Wright SH. Molecular basis for the differential sensitivity of OCT2 and OAT1 toward Hg2+ Journal of the Federation of American Societies for Experimental Biology. 2007;21(6):A908. [Google Scholar]
- Babu E, Takeda M, Narikawa S, Kobayashi Y, Enomoto A, Tojo A, Cha SH, Sekine T, Sakthisekaran D, Endou H. Role of human organic anion transporter 4 in the transport of ochratoxin A. Biochimica et Biophysica Acta: Molecular Cell Research. 2002a;1590:64–75. doi: 10.1016/s0167-4889(02)00187-8. [DOI] [PubMed] [Google Scholar]
- Babu E, Takeda M, Narikawa S, Kobayashi Y, Yamamoto T, Cha SH, Sekine T, Sakthisekaran D, Endou H. Human organic anion transporters mediate the transport of tetracycline. Japanese Journal of Pharmacology. 2002b;88:69–76. doi: 10.1254/jjp.88.69. [DOI] [PubMed] [Google Scholar]
- Bahn A, Knabe M, Hagos Y, Rodiger M, Godehardt S, Graber-Neufeld DS, Evans KK, Burckhardt G, Wright SH. Interaction of the metal chelator 2,3-dimercapto-1-propanesulfonate with the rabbit multispecific organic anion transporter 1 (rbOAT1) Molecular Pharmacology. 2002;62:1128–1136. doi: 10.1124/mol.62.5.1128. [DOI] [PubMed] [Google Scholar]
- Bahn A, Ljubojevic M, Lorenz H, Schultz C, Ghebremedhin E, Ugele B, Sabolic I, Burckhardt G, Hagos Y. Murine renal organic anion transporters mOAT1 and mOAT3 facilitate the transport of neuroactive tryptophan metabolites. American Journal of Physiology: Cell Physiology. 2005;289:C1075–C1084. doi: 10.1152/ajpcell.00619.2004. [DOI] [PubMed] [Google Scholar]
- Bahn A, Prawitt D, Buttler D, Reid G, Enklaar T, Wolff NA, Ebbinghaus C, Hillemann A, Schulten HJ, Gunawan B, et al. Genomic structure and in vivo expression of the human organic anion transporter 1 (hOAT1) gene. Biochemical and Biophysical Research Communications. 2000;275:623–630. doi: 10.1006/bbrc.2000.3230. [DOI] [PubMed] [Google Scholar]
- Bakhiya A, Bahn A, Burckhardt G, Wolff N. Human organic anion transporter 3 (hOAT3) can operate as an exchanger and mediate secretory urate flux. Cellular Physiology and Biochemistry. 2003;13:249–256. doi: 10.1159/000074539. [DOI] [PubMed] [Google Scholar]
- Bakhiya N, Batke M, Laake J, Monien BH, Frank H, Seidel A, Engst W, Glatt H. Directing role of organic anion transporters in the excretion of mercapturic acids of alkylated polycyclic aromatic hydrocarbons. Drug Metabolism and Disposition. 2007;35:1824–1831. doi: 10.1124/dmd.107.016964. [DOI] [PubMed] [Google Scholar]
- Berger V, Gabriel AF, Sergent T, Trouet A, Larondelle Y, Schneider YJ. Interaction of ochratoxin A with human intestinal Caco-2 cells: Possible implication of a multidrug resistance-associated protein (MRP2) Toxicology Letters. 2003;140-141:465–476. doi: 10.1016/s0378-4274(03)00043-2. [DOI] [PubMed] [Google Scholar]
- Berkhin EB, Humphreys MH. Regulation of renal tubular secretion of organic compounds. Kidney International. 2001;59:17–30. doi: 10.1046/j.1523-1755.2001.00461.x. [DOI] [PubMed] [Google Scholar]
- Bhatnagar V, Xu G, Hamilton BA, Truong DM, Eraly SA, Wu W, Nigam SK. Analyses of 5′ regulatory region polymorphisms in human SLC22A6 (OAT1) and SLC22A8 (OAT3) Journal of Human Genetics. 2006;51:575–580. doi: 10.1007/s10038-006-0398-1. [DOI] [PubMed] [Google Scholar]
- Bleasby K, Castle JC, Roberts CJ, Cheng C, Bailey WJ, Sina JF, Kulkarni AV, Hafey MJ, Evers R, Johnson JM, et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: A resource for investigations into drug disposition. Xenobiotica. 2006;36:963–988. doi: 10.1080/00498250600861751. [DOI] [PubMed] [Google Scholar]
- Bleasby K, Hall LA, Perry JL, Mohrenweiser HW, Pritchard JB. Functional consequences of single nucleotide polymorphisms in the human organic anion transporter hOAT1 (SLC22A6) Journal of Pharmacology and Experimental Therapeutics. 2005;314:923–931. doi: 10.1124/jpet.105.084301. [DOI] [PubMed] [Google Scholar]
- Bow DA, Perry JL, Simon JD, Pritchard JB. The impact of plasma protein binding on the renal transport of organic anions. Journal of Pharmacology and Experimental Therapeutics. 2006;316:349–355. doi: 10.1124/jpet.105.093070. [DOI] [PubMed] [Google Scholar]
- Brady KP, Dushkin H, Fornzler D, Koike T, Magner F, Her H, Gullans S, Segre GV, Green RM, Beier DR. A novel putative transporter maps to the osteosclerosis (oc) mutation and is not expressed in the oc mutant mouse. Genomics. 1999;56:254–261. doi: 10.1006/geno.1998.5722. [DOI] [PubMed] [Google Scholar]
- Buist SC, Cherrington NJ, Choudhuri S, Hartley DP, Klaassen CD. Gender-specific and developmental influences on the expression of rat organic anion transporters. Journal of Pharmacology and Experimental Therapeutics. 2002;301:145–151. doi: 10.1124/jpet.301.1.145. [DOI] [PubMed] [Google Scholar]
- Burckhardt BC, Burckhardt G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Reviews of Physiology, Biochemistry and Pharmacology. 2003;146:95–158. doi: 10.1007/s10254-002-0003-8. [DOI] [PubMed] [Google Scholar]
- Burckhardt G, Bahn A, Wolff NA. Molecular physiology of renal p-aminohippurate secretion. News in Physiological Sciences. 2001;16:114–118. doi: 10.1152/physiologyonline.2001.16.3.114. [DOI] [PubMed] [Google Scholar]
- Burckhardt G, Wolff NA. Structure of renal organic anion and cation transporters. American Journal of Physiology: Renal Physiology. 2000;278:F853–F866. doi: 10.1152/ajprenal.2000.278.6.F853. [DOI] [PubMed] [Google Scholar]
- Capasso G, Jaeger P, Robertson WG, Unwin RJ. Uric acid and the kidney: Urate transport, stone disease and progressive renal failure. Current Pharmaceutical Design. 2005;11:4153–4159. doi: 10.2174/138161205774913219. [DOI] [PubMed] [Google Scholar]
- Cerrutti JA, Brandoni A, Quaglia NB, Torres AM. Sex differences in p-aminohippuric acid transport in rat kidney: Role of membrane fluidity and expression of OAT1. Molecular and Cellular Biochemistry. 2002a;233:175–179. doi: 10.1023/a:1015563021602. [DOI] [PubMed] [Google Scholar]
- Cerrutti JA, Quaglia NB, Brandoni A, Torres AM. Effects of gender on the pharmacokinetics of drugs secreted by the renal organic anions transport systems in the rat. Pharmacological Research. 2002b;45:107–112. doi: 10.1006/phrs.2001.0912. [DOI] [PubMed] [Google Scholar]
- Cerrutti JA, Quaglia NB, Torres AM. Characterization of the mechanisms involved in the gender differences in p-aminohippurate renal elimination in rats. Canadian Journal of Physiology and Pharmacology. 2001;79:805–813. [PubMed] [Google Scholar]
- Cha SH, Sekine T, Fukushima JI, Kanai Y, Kobayashi Y, Goya T, Endou H. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Molecular Pharmacology. 2001;59:1277–1286. doi: 10.1124/mol.59.5.1277. [DOI] [PubMed] [Google Scholar]
- Cha SH, Sekine T, Kusuhara H, Yu E, Kim JY, Kim DK, Sugiyama Y, Kanai Y, Endou H. Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. Journal of Biological Chemistry. 2000;275:4507–4512. doi: 10.1074/jbc.275.6.4507. [DOI] [PubMed] [Google Scholar]
- Chakravarti A. To a future of genetic medicine. Nature. 2001;409(6822):822–823. doi: 10.1038/35057281. [DOI] [PubMed] [Google Scholar]
- Cheong HI, Kang JH, Lee JH, Ha IS, Kim S, Komoda F, Sekine T, Igarashi T, Choi Y. Mutational analysis of idiopathic renal hypouricemia in Korea. Pediatric Nephrology. 2005;20:886–890. doi: 10.1007/s00467-005-1863-3. [DOI] [PubMed] [Google Scholar]
- Choudhuri S, Klaassen CD. Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters. International Journal of Toxicology. 2006;25:231–259. doi: 10.1080/10915810600746023. [DOI] [PubMed] [Google Scholar]
- Cihlar T, Lin DC, Pritchard JB, Fuller MD, Mendel DB, Sweet DH. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Molecular Pharmacology. 1999;56:570–580. doi: 10.1124/mol.56.3.570. [DOI] [PubMed] [Google Scholar]
- Cross RJ, Taggart JV. Renal tubular transport: Accumulation of p-aminohippurate by rabbit kidney slices. American Journal of Physiology. 1950;161:181–190. doi: 10.1152/ajplegacy.1950.161.1.181. [DOI] [PubMed] [Google Scholar]
- Cui Y, Walter B. Influence of albumin binding on the substrate transport mediated by human hepatocyte transporters OATP2 and OATP8. Journal of Gastroenterology. 2003;38:60–68. doi: 10.1007/s005350300007. [DOI] [PubMed] [Google Scholar]
- Dantzler WH. Renal organic anion transport: A comparative and cellular perspective. Biochimica et Biophysica Acta: Biomembranes. 2002;1566:169–181. doi: 10.1016/s0005-2736(02)00599-0. [DOI] [PubMed] [Google Scholar]
- Dantzler WH, Wright SH. The molecular and cellular physiology of basolateral organic anion transport in mammalian renal tubules. Biochimica et Biophysica Acta: Biomembranes. 2003;1618:185–193. doi: 10.1016/j.bbamem.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Dresser MJ, Leabman MK, Giacomini KM. Transporters involved in the elimination of drugs in the kidney: Organic anion transporters and organic cation transporters. Journal of Pharmaceutical Sciences. 2001;90:397–421. doi: 10.1002/1520-6017(200104)90:4<397::aid-jps1000>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Ekaratanawong S, Anzai N, Jutabha P, Miyazaki H, Noshiro R, Takeda M, Kanai Y, Sophasan S, Endou H. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. Journal of Pharmaceutical Sciences. 2004;94:297–304. doi: 10.1254/jphs.94.297. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Endou H. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease. Clinical and Experimental Nephrology. 2005;9:195–205. doi: 10.1007/s10157-005-0368-5. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002a;417(6887):447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Takeda M, Shimoda M, Narikawa S, Kobayashi Y, Kobayashi Y, Yamamoto T, Sekine T, Cha SH, Niwa T, et al. Interaction of human organic anion transporters 2 and 4 with organic anion transport inhibitors. Journal of Pharmacology and Experimental Therapeutics. 2002b;301:797–802. doi: 10.1124/jpet.301.3.797. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Takeda M, Taki K, Takayama F, Noshiro R, Niwa T, Endou H. Interactions of human organic anion as well as cation transporters with indoxyl sulfate. European Journal of Pharmacology. 2003;466:13–20. doi: 10.1016/s0014-2999(03)01530-9. [DOI] [PubMed] [Google Scholar]
- Eraly SA, Hamilton BA, Nigam SK. Organic anion and cation transporters occur in pairs of similar and similarly expressed genes. Biochemical and Biophysical Research Communications. 2003;300:333–342. doi: 10.1016/s0006-291x(02)02853-x. [DOI] [PubMed] [Google Scholar]
- Eraly SA, Vallon V, Vaughn DA, Gangoiti JA, Richter K, Nagle M, Monte JC, Rieg T, Truong DM, Long JM, et al. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. Journal of Biological Chemistry. 2006;281:5072–5083. doi: 10.1074/jbc.M508050200. [DOI] [PubMed] [Google Scholar]
- Erdman AR, Mangravite LM, Urban TJ, Lagpacan LL, Castro RA, de la Cruz M, Chan W, Huang CC, Johns SJ, Kawamoto M, et al. The human organic anion transporter 3 (OAT3; SLC22A8): Genetic variation and functional genomics. American Journal of Physiology: Renal Physiology. 2006;290:F905–F912. doi: 10.1152/ajprenal.00272.2005. [DOI] [PubMed] [Google Scholar]
- Feng B, Dresser MJ, Shu Y, Johns SJ, Giacomini KM. Arginine 454 and lysine 370 are essential for the anion specificity of the organic anion transporter, rOAT3. Biochemistry. 2001;40:5511–5520. doi: 10.1021/bi002841o. [DOI] [PubMed] [Google Scholar]
- Feng B, Shu Y, Giacomini KM. Role of aromatic transmembrane residues of the organic anion transporter, rOAT3, in substrate recognition. Biochemistry. 2002;41:8941–8947. doi: 10.1021/bi0200615. [DOI] [PubMed] [Google Scholar]
- Fielding CJ, Fielding PE. Relationship between cholesterol trafficking and signaling in rafts and caveolae. Biochimica et Biophysica Acta: Biomembranes. 2003;1610:219–228. doi: 10.1016/s0005-2736(03)00020-8. [DOI] [PubMed] [Google Scholar]
- Fritzsch G, Rumrich G, Ullrich KJ. Anion transport through the contraluminal cell membrane of renal proximal tubule. The influence of hydrophobicity and molecular charge distribution on the inhibitory activity of organic anions. Biochimica et Biophysica Acta. 1989;978:249–256. doi: 10.1016/0005-2736(89)90122-3. [DOI] [PubMed] [Google Scholar]
- Fujita T, Brown C, Carlson EJ, Taylor T, de la Cruz M, Johns SJ, Stryke D, Kawamoto M, Fujita K, Castro R, et al. Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1) Pharmacogenetics and Genomics. 2005;15:201–209. doi: 10.1097/01213011-200504000-00003. [DOI] [PubMed] [Google Scholar]
- Gekle M, Mildenberger S, Sauvant C, Bednarczyk D, Wright SH, Dantzler WH. Inhibition of initial transport rate of basolateral organic anion carrier in renal PT by BK and phenylephrine. American Journal of Physiology: Renal Physiology. 1999;277(2 Pt 2):F251–F256. doi: 10.1152/ajprenal.1999.277.2.F251. [DOI] [PubMed] [Google Scholar]
- Geng L, Kuze K, Satlin L, Healy D, Henderson S, Burrow C, Wilson PGY. Localization and developmental expression of the organic anion transporter protein mOAT; Paper presented at the American Society of Nephrology, 32nd Annual Meeting; 1999. [Google Scholar]
- George RL, Wu X, Huang W, Fei YJ, Leibach FH, Ganapathy V. Molecular cloning and functional characterization of a polyspecific organic anion transporter from Caenorhabditis elegans. Journal of Pharmacology and Experimental Therapeutics. 1999;291:596–603. [PubMed] [Google Scholar]
- Graessler J, Graessler A, Unger S, Kopprasch S, Tausche AK, Kuhlisch E, Schroeder HE. Association of the human urate transporter 1 with reduced renal uric acid excretion and hyperuricemia in a German Caucasian population. Arthritis and Rheumatism. 2006;54:292–300. doi: 10.1002/art.21499. [DOI] [PubMed] [Google Scholar]
- Groves CE, Suhre WB, Cherrington NJ, Wright SH. Sex differences in the mRNA, protein, and functional expression of organic anion transporter (Oat) 1, Oat3, and organic cation transporter (Oct) 2 in rabbit renal proximal tubules. Journal of Pharmacology and Experimental Therapeutics. 2006;316:743–752. doi: 10.1124/jpet.105.094979. [DOI] [PubMed] [Google Scholar]
- Grundemann D, Gorboulev V, Gambaryan S, Veyhi M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature. 1994;372:549–552. doi: 10.1038/372549a0. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Gao B, Meier PJ. Transport of xenobiotics across the blood-brain barrier. News in Physiological Sciences. 2002;17:231–234. doi: 10.1152/nips.01402.2002. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochimica et Biophysica Acta: Biomembranes. 2003;1609:1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflügers Archiv European Journal of Physiology. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- Hagos Y, Bahn A, Asif AR, Krick W, Sendler M, Burckhardt G. Cloning of the pig renal organic anion transporter 1 (pOAT1) Biochimie. 2002;84:1221–1224. doi: 10.1016/s0300-9084(02)00022-6. [DOI] [PubMed] [Google Scholar]
- Hagos Y, Braun IM, Krick W, Burckhardt G, Bahn A. Functional expression of pig renal organic anion transporter 3 (pOAT3) Biochimie. 2005;87:421–424. doi: 10.1016/j.biochi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Hasannejad H, Takeda M, Taki K, Shin HJ, Babu E, Jutabha P, Khamdang S, Aleboyeh M, Onozato ML, Tojo A, et al. Interactions of human organic anion transporters with diuretics. Journal of Pharmacology and Experimental Therapeutics. 2004;308:1021–1029. doi: 10.1124/jpet.103.059139. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kusuhara H, Sugiyama D, Ito K, Ueda S, Endou H, Sugiyama Y. Functional involvement of rat organic anion transporter 3 (rOat3; Slc22a8) in the renal uptake of organic anions. Journal of Pharmacology and Experimental Therapeutics. 2002;300:746–753. doi: 10.1124/jpet.300.3.746. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Narikawa S, Huang XL, Minematsu T, Usui T, Kamimura H, Endou H. Characterization of the renal tubular transport of zonampanel, a novel alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor antagonist, by human organic anion transporters. Drug Metabolism and Disposition. 2004;32:1096–1102. doi: 10.1124/dmd.32.10.. [DOI] [PubMed] [Google Scholar]
- Hediger MA, Johnson RJ, Miyazaki H, Endou H. Molecular physiology of urate transport. Physiology. 2005;20:125–133. doi: 10.1152/physiol.00039.2004. [DOI] [PubMed] [Google Scholar]
- Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell AL, Karlsson J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metabolism and Disposition. 2007;35:1333–1340. doi: 10.1124/dmd.107.014902. [DOI] [PubMed] [Google Scholar]
- Hirai T, Chida K. Protein kinase C zeta (PKC zeta): Activation mechanisms and cellular functions. Journal of Biochemistry. 2003;133:395–395. doi: 10.1093/jb/mvg017. [DOI] [PubMed] [Google Scholar]
- Hirai T, Heymann JAW, Shi D, Subramaniam S. Comparative structural analysis of the ‘symmetric’, substrate-bound state of the oxalate transporter OxlT with the ‘cytoplasmically-open’ state of the MFS transporters GlpT and LacY; Paper presented at the Biophysical Society 48th Annual Meeting; 2004. p. 3173. [Google Scholar]
- Hong M, Tanaka K, Pan Z, Ma J, You G. Determination of the external loops and the cellular orientation of the N- and the C-termini of the human organic anion transporter hOAT1. Biochemical Journal. 2007a;401:515–520. doi: 10.1042/BJ20061171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhou F, Lee K, You G. The putative transmembrane segment 7 of human organic anion transporter hOAT1 dictates transporter substrate binding and stability. Journal of Pharmacology and Experimental Therapeutics. 2007b;320:1209–1215. doi: 10.1124/jpet.106.117663. [DOI] [PubMed] [Google Scholar]
- Hong M, Zhou F, You G. Critical amino acid residues in transmembrane domain 1 of the human organic anion transporter hOAT1. Journal of Biological Chemistry. 2004;279:31478–31482. doi: 10.1074/jbc.M404686200. [DOI] [PubMed] [Google Scholar]
- Hosoyamada M, Ichida K, Enomoto A, Hosoya T, Endou H. Function and localization of urate transporter 1 in mouse kidney. Journal of the American Society of Nephrology. 2004;15:261–268. doi: 10.1097/01.asn.0000107560.80107.19. [DOI] [PubMed] [Google Scholar]
- Hosoyamada M, Sekine T, Kanai Y, Endou H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. American Journal of Physiology: Renal Physiology. 1999;276:F122–F128. doi: 10.1152/ajprenal.1999.276.1.F122. [DOI] [PubMed] [Google Scholar]
- Huang YF, Lemieux MJ, Song JM, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- Ichida K, Hosoyamada M, Hisatome I, Enomoto A, Hikita M, Endou H, Hosoya T. Clinical and molecular analysis of patients with renal hypouricemia in Japan — influence of URAT1 gene on urinary urate excretion. Journal of the American Society of Nephrology. 2004;15:164–173. doi: 10.1097/01.asn.0000105320.04395.d0. [DOI] [PubMed] [Google Scholar]
- Ichida K, Hosoyamada M, Kimura H, Takeda M, Utsunomiya Y, Hosoya T, Endou H. Urate transport via human PAH transporter hOAT1 and its gene structure. Kidney International. 2003;63:143–155. doi: 10.1046/j.1523-1755.2003.00710.x. [DOI] [PubMed] [Google Scholar]
- Iida A, Saito S, Sekine A, Mishima C, Kondo K, Kitamura Y, Harigae S, Osawa S, Nakamura Y. Catalog of 258 single-nucleotide polymorphisms (SNPs) in genes encoding three organic anion transporters, three organic anion-transporting polypeptides, and three NADH:ubiquinone oxidoreductase flavoproteins. Journal of Human Genetics. 2001;46:668–683. doi: 10.1007/s100380170019. [DOI] [PubMed] [Google Scholar]
- Islinger F, Gekle M, Wright SH. Interaction of 2,3-dimercapto-1-propane sulfonate with the human organic anion transporter hOAT1. Journal of Pharmacology and Experimental Therapeutics. 2001;299:741–747. [PubMed] [Google Scholar]
- Iwai M, Suzuki H, Ieiri I, Otsubo K, Sugiyama Y. Functional analysis of single nucleotide polymorphisms of hepatic organic anion transporter OATP1B1 (OATP-C) Pharmacogenetics. 2004;14:749–757. doi: 10.1097/00008571-200411000-00006. [DOI] [PubMed] [Google Scholar]
- Jacobsson JA, Haitina T, Lindblom J, Fredriksson R. Identification of six putative human transporters with structural similarity to the drug transporter SLC22 family. Genomics. 2007;90:595–609. doi: 10.1016/j.ygeno.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Jariyawat S, Sekine T, Takeda M, Apiwattanakul N, Kanai Y, Sophasan S, Endou H. The interaction and transport of beta-lactam antibiotics with the cloned rat renal organic anion transporter 1. Journal of Pharmacology and Experimental Therapeutics. 1999;290:672–677. [PubMed] [Google Scholar]
- Jenny M, Wrulich OA, Schwaiger W, Ueberall F. Relevance of atypical protein kinase C isotypes to the drug discovery process. Chembiochemistry. 2005;6:491–499. doi: 10.1002/cbic.200400186. [DOI] [PubMed] [Google Scholar]
- Kaler G, Truong DM, Khandelwal A, Nagle M, Eraly SA, Swaan PW, Nigam SK. Structural variation governs substrate specificity for organic anion transporters (oat) homologs: Potential remote sensing by oat family members. Journal of Biology and Chemistry. 2007;282:23841–23853. doi: 10.1074/jbc.M703467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler G, Truong DM, Sweeney DE, Logan DW, Nagle M, Wu W, Eraly SA, Nigam SK. Olfactory mucosa-expressed organic anion transporter, Oat6, manifests high affinity interactions with odorant organic anions. Biochemical and Biophysical Research Communications. 2006;351:872–876. doi: 10.1016/j.bbrc.2006.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamdang S, Takeda M, Noshiro R, Narikawa S, Enomoto A, Anzai N, Piyachaturawat P, Endou H. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. Journal of Pharmacology and Experimental Therapeutics. 2002;303:534–539. doi: 10.1124/jpet.102.037580. [DOI] [PubMed] [Google Scholar]
- Kikuchi R, Kusuhara H, Hattori N, Kim I, Shiota K, Gonzalez FJ, Sugiyama Y. Regulation of tissue-specific expression of the human and mouse urate transporter 1 gene by hepatocyte nuclear factor 1 alpha/beta and DNA methylation. Molecular Pharmacology. 2007;72:1619–1625. doi: 10.1124/mol.107.039701. [DOI] [PubMed] [Google Scholar]
- Kikuchi R, Kusuhara H, Hattori N, Shiota K, Kim I, Gonzalez FJ, Sugiyama Y. Regulation of the expression of human organic anion transporter 3 by hepatocyte nuclear factor 1alpha/beta and DNA methylation. Molecular Pharmacology. 2006;70:887–896. doi: 10.1124/mol.106.025494. [DOI] [PubMed] [Google Scholar]
- Kikuchi R, Kusuhara H, Sugiyama D, Sugiyama Y. Contribution of organic anion transporter 3 (Slc22a8) to the elimination of p-aminohippuric acid and benzylpenicillin across the blood–brain barrier. Journal of Pharmacology and Experimental Therapeutics. 2003;306:51–58. doi: 10.1124/jpet.103.049197. [DOI] [PubMed] [Google Scholar]
- Kimura H, Takeda M, Narikawa S, Enomoto A, Ichida K, Endou H. Human organic anion transporters and human organic cation transporters mediate renal transport of prostaglandins. Journal of Pharmacology and Experimental Therapeutics. 2002;301:293–298. doi: 10.1124/jpet.301.1.293. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Slitt AL. Regulation of hepatic transporters by xenobiotic receptors. Current Drug Metabolism. 2005;6:309–328. doi: 10.2174/1389200054633826. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Ohbayashi M, Kohyama N, Yamamoto T. Mouse organic anion transporter 2 and 3 (mOAT2/3[Slc22a7/8]) mediates the renal transport of bumetanide. European Journal of Pharmacology. 2005a;524:44–48. doi: 10.1016/j.ejphar.2005.09.054. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Ohshiro N, Sakai R, Ohbayashi M, Kohyama N, Yamamoto T. Transport mechanism and substrate specificity of human organic anion transporter 2 (hOat2 [SLC22A7]) Journal of Pharmacy and Pharmacology. 2005b;57:573–578. doi: 10.1211/0022357055966. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Ohshiro N, Shibusawa A, Sasaki T, Tokuyama S, Sekine T, Endou H. Isolation, characterization and differential gene expression of multispecific organic anion transporter 2 in mice. Molecular Pharmacology. 2002;62:7–14. doi: 10.1124/mol.62.1.7. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Ohshiro N, Tsuchiya A, Kohyama N, Ohbayashi M, Yamamoto T. Renal transport of organic compounds mediated by mouse organic anion transporter 3 (moat3): Further substrate specificity of moat3. Drug Metabolism and Disposition. 2004;32:479–483. doi: 10.1124/dmd.32.5.479. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Endou H. The SLC22 drug transporter family. Pflügers Archiv European Journal of Physiology. 2004;447:666–676. doi: 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- Komoda F, Sekine T, Inatomi J, Enomoto A, Endou H, Ota T, Matsuyama T, Ogata T, Ikeda M, Awazu M, et al. The W258X mutation in SLC22A12 is the predominant cause of Japanese renal hypouricemia. Pediatric Nephrology. 2004;19:728–733. doi: 10.1007/s00467-004-1424-1. [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Sekine T, Utsunomiya-Tate N, Tsuda M, Kojima R, Cha SH, Sugiyama Y, Kanai Y, Endou H. Molecular cloning and characterization of a new multispecific organic anion transporter from rat brain. Journal of Biological Chemistry. 1999;274:13675–13680. doi: 10.1074/jbc.274.19.13675. [DOI] [PubMed] [Google Scholar]
- Kuze K, Graves P, Leahy A, Wilson P, Stuhlmann H, You G. Heterologous expression and functional characterization of a mouse renal organic anion transporter in mammalian cells. Journal of Biological Chemistry. 1999;274:1519–1524. doi: 10.1074/jbc.274.3.1519. [DOI] [PubMed] [Google Scholar]
- Kwak JO, Kim HW, Oh KJ, Kim DS, Han KO, Cha SH. Co-localization and interaction of organic anion transporter 1 with caveolin-2 in rat kidney. Experimental and Molecular Medicine. 2005a;37:204–212. doi: 10.1038/emm.2005.28. [DOI] [PubMed] [Google Scholar]
- Kwak JO, Kim HW, Oh KJ, Ko CB, Park H, Cha SH. Characterization of mouse organic anion transporter 5 as a renal steroid sulfate transporter. Journal of Steroid Biochemistry and Molecular Biology. 2005b;97:369–375. doi: 10.1016/j.jsbmb.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Kwak JO, Kim HW, Song JH, Kim MJ, Park HS, Hyun DK, Kim DS, Cha SH. Evidence for rat organic anion transporter 3 association with caveolin-1 in rat kidney. International Union of Biochemistry and Molecular Biology Life. 2005c;57:109–117. doi: 10.1080/15216540500104750. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Usmani KA, Chanas B, Ghanayem B, Xi T, Hodgson E, Mohrenweiser HW, Goldstein JA. Genetic findings and functional studies of human CYP3A5 single nucleotide polymorphisms in different ethnic groups. Pharmacogenetics. 2003a;13:461–472. doi: 10.1097/00008571-200308000-00004. [DOI] [PubMed] [Google Scholar]
- Lee W, Ingram AD, Leake BF, Kim RB. Identification of nonsynonomous polymorphisms of human organic anion transporter 1 (OAT1) among African-, European- and Chinese-American subjects. Drug Metabolism Reviews. 2003b;35:75. [Google Scholar]
- Ljubojevic M, Balen D, Breljak D, Kusan M, Anzai N, Bahn A, Burckhardt G, Sabolic I. Renal expression of organic anion transporter OAT2 in rats and mice is regulated by sex hormones. American Journal of Physiology: Renal Physiology. 2007;292:F361–F372. doi: 10.1152/ajprenal.00207.2006. [DOI] [PubMed] [Google Scholar]
- Ljubojevic M, Herak-Kramberger CM, Hagos Y, Bahn A, Endou H, Burckhardt G, Sabolic I. Rat renal cortical OAT1 and OAT3 exhibit gender differences determined by both androgen stimulation and estrogen inhibition. American Journal of Physiology: Renal Physiology. 2004;287:F124–F138. doi: 10.1152/ajprenal.00029.2004. [DOI] [PubMed] [Google Scholar]
- Lopez-Nieto CE, You G, Bush KT, Barros EJ, Beier DR, Nigam SK. Molecular cloning and characterization of NKT, a gene product related to the organic cation transporter family that is almost exclusively expressed in the kidney. Journal of Biological Chemistry. 1997;272:6471–6478. doi: 10.1074/jbc.272.10.6471. [DOI] [PubMed] [Google Scholar]
- Lu R, Chan BS, Schuster VL. Cloning of the human kidney PAH transporter: Narrow substrate specificity and regulation by protein kinase c. American Journal of Physiology: Renal Physiology. 1999;276:F295–F303. doi: 10.1152/ajprenal.1999.276.2.F295. [DOI] [PubMed] [Google Scholar]
- Maesaka JK, Fishbane S. Regulation of renal urate excretion: A critical review. American Journal of Kidney Diseases. 1998;32:917–933. doi: 10.1016/s0272-6386(98)70067-8. [DOI] [PubMed] [Google Scholar]
- Marshall EKJ, Grafflin AL. The structure and function of the kidney of Lophius piscatorius. Bulletin of the Johns Hopkins Hospital. 1928;43:205–235. [Google Scholar]
- Marshall EKJ, Grafflin AL. The function of the proximal convoluted segment of the renal tubules. Journal of Cellular and Comparative Physiology. 1932;1:161–176. [Google Scholar]
- Marshall EKJ, Vickers JL. The mechanism of the elimination of phenolsulphonephthalein by the kidney: A proof of secretion by the convoluted tubules. Bulletin of the John Hopkins Hospital. 1923;34:1–7. [Google Scholar]
- Marzolini C, Tirona RG, Kim RB. Pharmacogenomics of the OATP and OAT families. Pharmacogenomics. 2004;5:273–282. doi: 10.1517/phgs.5.3.273.29831. [DOI] [PubMed] [Google Scholar]
- Miller D, Lowes S, Pritchard JB. The molecular basis of xenobiotic transport and metabolism in the choroid plexus. In: Chodobski ZW, editor. The blood–cerebrospinal fluid barrier. Taylor & Francis; New York, NY: 2005. pp. 147–173. [Google Scholar]
- Miller DS. Confocal imaging of xenobiotic transport across the blood–brain barrier. Journal of Experimental Zoology Part A: Comparative experimental biology. 2003;300:84–90. doi: 10.1002/jez.a.10313. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Anzai N, Ekaratanawong S, Sakata T, Shin HJ, Jutabha P, Hirata T, He X, Nonoguchi H, Tomita K, et al. Modulation of renal apical organic anion transporter 4 function by two PDZ domain-containing proteins. Journal of the American Society of Nephrology. 2005;16:3498–3506. doi: 10.1681/ASN.2005030306. [DOI] [PubMed] [Google Scholar]
- Monte JC, Nagle MA, Eraly SA, Nigam SK. Identification of a novel murine organic anion transporter family member, OAT6, expressed in olfactory mucosa. Biochemical and Biophysical Research Communications. 2004;323:429–436. doi: 10.1016/j.bbrc.2004.08.112. [DOI] [PubMed] [Google Scholar]
- Mori K, Ogawa Y, Ebihara K, Aoki T, Tamura N, Sugawara A, Kuwahara T, Ozaki S, Mukoyama M, Tashiro K, et al. Kidney-specific expression of a novel mouse organic cation transporter-like protein. Federation of European Biochemical Societies Letters. 1997;417:371–374. doi: 10.1016/s0014-5793(97)01325-2. [DOI] [PubMed] [Google Scholar]
- Morita N, Kusuhara H, Sekine T, Endou H, Sugiyama Y. Functional characterization of rat organic anion transporter 2 in LLC-PK1 cells. Journal of Pharmacology and Experimental Therapeutics. 2001;298:1179–1184. [PubMed] [Google Scholar]
- Nishizato Y, Ieiri I, Suzuki H, Kimura M, Kawabata K, Hirota T, Takane H, Irie S, Kusuhara H, Urasaki Y, et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: Consequences for pravastatin pharmacokinetics. Clinical Pharmacology and Therapeutics. 2003;73:554–565. doi: 10.1016/S0009-9236(03)00060-2. [DOI] [PubMed] [Google Scholar]
- Ogasawara K, Terada T, Asaka J, Katsura T, Inui K. Human organic anion transporter 3 gene is regulated constitutively and inducibly via a cAMP-response element. Journal of Pharmacology and Experimental Therapeutics. 2006;319:317–322. doi: 10.1124/jpet.106.108233. [DOI] [PubMed] [Google Scholar]
- Ogasawara K, Terada T, Asaka J, Katsura T, Inui K. Hepatocyte nuclear factor-4{alpha} regulates the human organic anion transporter 1 gene in the kidney. American Journal of Physiology: Renal Physiology. 2007;292:F1819–F1826. doi: 10.1152/ajprenal.00017.2007. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S. New aspects of the blood–brain barrier transporters; its physiological roles in the central nervous system. Biological and Pharmaceutical Bulletin. 2004;27:1489–1496. doi: 10.1248/bpb.27.1489. [DOI] [PubMed] [Google Scholar]
- Ohtsuki S, Kikkawa T, Mori S, Hori S, Takanaga H, Otagiri M, Terasaki T. Mouse reduced in osteosclerosis transporter functions as an organic anion transporter 3 and is localized at abluminal membrane of blood–brain barrier. Journal of Pharmacology and Experimental Therapeutics. 2004;309:1273–1281. doi: 10.1124/jpet.103.063370. [DOI] [PubMed] [Google Scholar]
- Pavlova A, Sakurai H, Leclercq B, Beier DR, Yu ASL, Nigam SK. Developmentally regulated expression of organic ion transporters NKT(OAT1), OCT1, NLT(OAT2), and Roct. American Journal of Physiology: Renal Physiology. 2000;278:F635–F643. doi: 10.1152/ajprenal.2000.278.4.F635. [DOI] [PubMed] [Google Scholar]
- Perry JL, Dembla-Rajpal N, Hall LA, Pritchard JB. A three-dimensional model of human organic anion transporter 1: Aromatic amino acids required for substrate transport. Journal of Biological Chemistry. 2006;281:38071–38079. doi: 10.1074/jbc.M608834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JL, Srimaroeng C, Dembla-Rajpal N, Hall LA, Pritchard JB. Substrate specificity in a model of the human organic anion transporter 3. Journal of the Federation of American Societies for Experimental Biology. 2007;21:758.7. [Google Scholar]
- Petrick JS, Klaassen CD. Importance of hepatic induction of constitutive androstane receptor and other transcription factors that regulate xenobiotic metabolism and transport. Drug Metabolism and Disposition. 2007;35:1806–1815. doi: 10.1124/dmd.107.015974. [DOI] [PubMed] [Google Scholar]
- Popowski K, Eloranta JJ, Saborowski M, Fried M, Meier PJ, Kullak-Ublick GA. The human organic anion transporter 2 gene is transactivated by hepatocyte nuclear factor-4{alpha} and suppressed by bile acids. Molecular Pharmacology. 2005;67:1629–1638. doi: 10.1124/mol.104.010223. [DOI] [PubMed] [Google Scholar]
- Popp C, Gorboulev V, Muller TD, Gorbunov D, Shatskaya N, Koepsell H. Amino acids critical for substrate affinity of rat organic cation transporter 1 line the substrate binding region in a model derived from the tertiary structure of lactose permease. Molecular Pharmacology. 2005;67:1600–1611. doi: 10.1124/mol.104.008839. [DOI] [PubMed] [Google Scholar]
- Price KL, Sautin YY, Long DA, Zhang L, Miyazaki H, Mu W, Endou H, Johnson RJ. Human vascular smooth muscle cells express a urate transporter. Journal of the American Society of Nephrology. 2006;17:1791–1795. doi: 10.1681/ASN.2006030264. [DOI] [PubMed] [Google Scholar]
- Pritchard JB. Luminal and peritubular steps in renal transport of p-aminohippurate. Biochimica et Biophysica Acta: Reviews on Biomembranes. 1987;906:295–308. doi: 10.1016/0304-4157(87)90015-3. [DOI] [PubMed] [Google Scholar]
- Pritchard JB. Coupled transport of p-aminohippurate by rat kidney basolateral membrane vesicles. American Journal of Physiology: Renal Physiology. 1988;255(4 Pt 2):F597–F604. doi: 10.1152/ajprenal.1988.255.4.F597. [DOI] [PubMed] [Google Scholar]
- Pritchard JB. Rat renal cortical slices demonstrate p-aminohippurate/glutarate exchange and sodium/glutarate coupled p-aminohippurate transport. Journal of Pharmacology and Experimental Therapeutics. 1990;255:969–975. [PubMed] [Google Scholar]
- Pritchard JB, Miller DS. Mechanisms mediating renal secretion of organic anions and cations. Physiological Reviews. 1993;73:765–796. doi: 10.1152/physrev.1993.73.4.765. [DOI] [PubMed] [Google Scholar]
- Pritchard JB, Miller DS. Expression systems for cloned xenobiotic transporters. Toxicology and Applied Pharmacology. 2005;204:256–262. doi: 10.1016/j.taap.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Race JE, Grassl SM, Williams WJ, Holtzman EJ. Molecular cloning and characterization of two novel human renal organic anion transporters (hOAT1 and hOAT3) Biochemical and Biophysical Research Communications. 1999;255:508–514. doi: 10.1006/bbrc.1998.9978. [DOI] [PubMed] [Google Scholar]
- Reid G, Wolff NA, Dautzenberg FM, Burckhardt G. Cloning of a human renal p-aminohippurate transporter, hROAT1. Kidney and Blood Pressure Research. 1998;21:233–237. doi: 10.1159/000025863. [DOI] [PubMed] [Google Scholar]
- Reyes JL, Melendez E, Alegria A, Jaramillo-Juarez F. Influence of sex differences on the renal secretion of organic anions. Endocrinology. 1998;139:1581–1587. doi: 10.1210/endo.139.4.5930. [DOI] [PubMed] [Google Scholar]
- Rizwan AN, Burckhardt G. Organic anion transporters of the SLC22 family: Biopharmaceutical, physiological, and pathological roles. Pharmaceutical Research. 2007;24:450–470. doi: 10.1007/s11095-006-9181-4. [DOI] [PubMed] [Google Scholar]
- Rizwan AN, Krick W, Burckhardt G. The chloride dependence of the human organic anion transporter 1 (hOAT1) is blunted by mutation of a single amino acid. Journal of Biological Chemistry. 2007;282:13402–13409. doi: 10.1074/jbc.M609849200. [DOI] [PubMed] [Google Scholar]
- Robertson EE, Rankin GO. Human renal organic anion transporters: Characteristics and contributions to drug and drug metabolite excretion. Pharmacology and Therapeutics. 2006;109:399–412. doi: 10.1016/j.pharmthera.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Rowntree LG, Geraghty JT. An experimental and clinical study of the functional activity of the kidneys by means of phenolsulphonephthalein. Journal of Pharmacology and Experimental Therapeutics. 1909;1:579–661. [Google Scholar]
- Sabolic I, Asif AR, Budach WE, Wanke C, Bahn A, Burckhardt G. Gender differences in kidney function. Pflügers Archiv European Journal of Physiology. 2007;455:397–429. doi: 10.1007/s00424-007-0308-1. [DOI] [PubMed] [Google Scholar]
- Saji T, Kikuchi R, Kusuhara H, Kim I, Gonzalez FJ, Sugiyama Y. Transcriptional regulation of human and mouse organic anion transporter 1 by hepatocyte nuclear factor 1 {alpha}/{beta} Journal of Pharmacology and Experimental Therapeutics. 2008;324(2):784–790. doi: 10.1124/jpet.107.128249. [DOI] [PubMed] [Google Scholar]
- Sauvant C, Holzinger H, Gekle M. Short-term regulation of basolateral organic anion uptake in proximal tubular OK cells: EGF acts via MAPK, PLA(2), and COX1. Journal of the American Society of Nephrology. 2002;13:1981–1991. doi: 10.1097/01.asn.0000024437.62046.af. [DOI] [PubMed] [Google Scholar]
- Sauvant C, Holzinger H, Gekle M. Short-term regulation of basolateral organic anion uptake in proximal tubular opossum kidney cells: Prostaglandin E2 acts via receptor-mediated activation of protein kinase A. Journal of the American Society of Nephrology. 2003;14:3017–3026. doi: 10.1097/01.asn.0000099376.87890.71. [DOI] [PubMed] [Google Scholar]
- Sauvant C, Holzinger H, Gekle M. Prostaglandin E2 inhibits its own renal transport by downregulation of organic anion transporters rOAT1 and rOAT3. Journal of the American Society of Nephrology. 2006;17:46–53. doi: 10.1681/ASN.2005070727. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Advanced Drug Delivery Reviews. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- Schmitt C, Burckhardt G. p-Aminohippurate/2-oxoglutarate exchange in bovine renal brush-border and basolateral membrane vesicles. Pflügers Archiv European Journal of Physiology. 1993;423:280–290. doi: 10.1007/BF00374407. [DOI] [PubMed] [Google Scholar]
- Schnabolk GW, Youngblood GL, Sweet DH. Transport of estrone sulfate by the novel organic anion transporter Oat6 (Slc22a20) American Journal of Physiology: Renal Physiology. 2006;291:F314–F321. doi: 10.1152/ajprenal.00497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T, Cha SH, Tsuda M, Apiwattanakul N, Nakajima N, Kanai Y, Endou H. Identification of multispecific organic anion transporter 2 expressed predominantly in the liver. Federation of European Biochemical Societies Letters. 1998;429:179–182. doi: 10.1016/s0014-5793(98)00585-7. [DOI] [PubMed] [Google Scholar]
- Sekine T, Watanabe N, Hosoyamada M, Kanai Y, Endou H. Expression cloning and characterization of a novel multispecific organic anion transporter. Journal of Biological Chemistry. 1997;272:18526–18529. doi: 10.1074/jbc.272.30.18526. [DOI] [PubMed] [Google Scholar]
- Shimada H, Moewes B, Burckhardt G. Indirect coupling to Na+ of p-aminohippuric acid uptake into rat renal basolateral membrane vesicles. American Journal of Physiology: Renal Physiology. 1987;253(5 Pt 2):F795–F801. doi: 10.1152/ajprenal.1987.253.5.F795. [DOI] [PubMed] [Google Scholar]
- Shin HJ, Anzai N, Enomoto A, He X, Kim do K, Endou H, Kanai Y. Novel liver-specific organic anion transporter OAT7 that operates the exchange of sulfate conjugates for short chain fatty acid butyrate. Hepatology. 2007;45:1046–1055. doi: 10.1002/hep.21596. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. European Journal of Pharmaceutical Sciences. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug–drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacology and Therapeutics. 2006;112:71–105. doi: 10.1016/j.pharmthera.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Shuprisha A, Lynch RM, Wright SH, Dantzler WH. PKC regulation of organic anion secretion in perfused S2 segments of rabbit proximal tubules. American Journal of Physiology: Renal Physiology. 2000;278:F104–F109. doi: 10.1152/ajprenal.2000.278.1.F104. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simonson GD, Vincent AC, Roberg KJ, Huang Y, Iwanij V. Molecular cloning and characterization of a novel liver-specific transport protein. Journal of Cell Science. 1994;107(Pt 4):1065–1072. doi: 10.1242/jcs.107.4.1065. [DOI] [PubMed] [Google Scholar]
- Smith HW, Goldring W, Chassis H. The measurement of the tubular excretory mass, effective blood flow, and filtration rate in the normal human kidney. Journal of Clinical Investigation. 1938;17:263–278. doi: 10.1172/JCI100950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soodvilai S, Chatsudthipong V, Evans KK, Wright SH, Dantzler WH. Acute regulation of OAT3-mediated estrone sulfate transport in isolated rabbit renal proximal tubules. American Journal of Physiology: Renal Physiology. 2004;287:F1021–F1029. doi: 10.1152/ajprenal.00080.2004. [DOI] [PubMed] [Google Scholar]
- Sugiyama D, Kusuhara H, Shitara Y, Abe T, Meier PJ, Sekine T, Endou H, Suzuki H, Sugiyama Y. Characterization of the efflux transport of 17beta-estradiol-D-17beta-glucuronide from the brain across the blood–brain barrier. Journal of Pharmacology and Experimental Therapeutics. 2001;298:316–322. [PubMed] [Google Scholar]
- Sun W, Wu RR, Van Poelje PD, Erion MD. Isolation of a family of organic anion transporters from human liver and kidney. Biochemical and Biophysical Research Communications. 2001;283:417–422. doi: 10.1006/bbrc.2001.4774. [DOI] [PubMed] [Google Scholar]
- Sweet DH. Organic anion transporter (Slc22a) family members as mediators of toxicity. Toxicology and Applied Pharmacology. 2005;204:198–215. doi: 10.1016/j.taap.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Chan LM, Walden R, Yang XP, Miller DS, Pritchard JB. Organic anion transporter 3 (Slc22a8) is a dicarboxylate exchanger indirectly coupled to the Na+ gradient. American Journal of Physiology: Renal Physiology. 2003;284:F763–F769. doi: 10.1152/ajprenal.00405.2002. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Miller DS, Pritchard JB, Fujiwara Y, Beier DR, Nigam SK. Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (Oat3 (Slc22a8)) knockout mice. Journal of Biological Chemistry. 2002;277:26934–26943. doi: 10.1074/jbc.M203803200. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Miller DS, Pritchard JB. Localization of an organic anion transporter-GFP fusion construct (rROAT1-GFP) in intact proximal tubules. American Journal of Physiology: Renal Physiology. 1999;276:F864–F873. doi: 10.1152/ajprenal.1999.276.6.F864. [DOI] [PubMed] [Google Scholar]
- Sweet DH, Wolff NA, Pritchard JB. Expression cloning and characterization of ROAT1: The basolateral organic anion transporter in rat kidney. Journal of Biological Chemistry. 1997;272:30088–30095. doi: 10.1074/jbc.272.48.30088. [DOI] [PubMed] [Google Scholar]
- Sykes D, Sweet DH, Lowes S, Nigam SK, Pritchard JB, Miller DS. Organic anion transport in choroid plexus from wild-type and organic anion transporter 3 (Slc22a8)-null mice. American Journal of Physiology: Renal Physiology. 2004;286:F972–F978. doi: 10.1152/ajprenal.00356.2003. [DOI] [PubMed] [Google Scholar]
- Tahara H, Shono M, Kusuhara H, Kinoshita H, Fuse E, Takadate A, Otagiri M, Sugiyama Y. Molecular cloning and functional analyses of OAT1 and OAT3 from cynomolgus monkey kidney. Pharmaceutical Research. 2005;22:647–660. doi: 10.1007/s11095-005-2503-0. [DOI] [PubMed] [Google Scholar]
- Takeda M, Babu E, Narikawa S, Endou H. Interaction of human organic anion transporters with various cephalosporin antibiotics. European Journal of Pharmacology. 2002a;438:137–142. doi: 10.1016/s0014-2999(02)01306-7. [DOI] [PubMed] [Google Scholar]
- Takeda M, Khamdang S, Narikawa S, Kimura H, Hosoyamada M, Cha SH, Sekine T, Endou H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. Journal of Pharmacology and Experimental Therapeutics. 2002b;302:666–671. doi: 10.1124/jpet.102.034330. [DOI] [PubMed] [Google Scholar]
- Takeda M, Khamdang S, Narikawa S, Kimura H, Kobayashi Y, Yamamoto T, Cha SH, Sekine T, Endou H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. Journal of Pharmacology and Experimental Therapeutics. 2002c;300:918–924. doi: 10.1124/jpet.300.3.918. [DOI] [PubMed] [Google Scholar]
- Takeda M, Sekine T, Endou H. Regulation by protein kinase C of organic anion transport driven by rat organic anion transporter 3 (rOAT3) Life Sciences. 2000;67:1087–1093. doi: 10.1016/s0024-3205(00)00694-9. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Zhou F, Kuze K, You G. Cysteine residues in the organic anion transporter mOAT1. Biochemical Journal. 2004b;380:283–287. doi: 10.1042/BJ20031724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Xu W, Zhou F, You GF. Role of glycosylation in the organic anion transporter OAT1. Journal of Biological Chemistry. 2004a;279:14961–14966. doi: 10.1074/jbc.M400197200. [DOI] [PubMed] [Google Scholar]
- Taniguchi A, Urano W, Yamanaka M, Yamanaka H, Hosoyamada M, Endou H, Kamatani N. A common mutation in an organic anion transporter gene, SLC22A12, is a suppressing factor for the development of gout. Arthritis and Rheumatism. 2005;52:2576–2577. doi: 10.1002/art.21242. [DOI] [PubMed] [Google Scholar]
- Thompson D, Srimaroeng C, Dallas S, Walden R, Miller D, Pritchard J. Functional characterization of human organic anion transporter 4 (hOAT4) in a Baculovirus expression system. Journal of the Federation of American Societies for Experimental Biology. 2007;21:758. 1. [Google Scholar]
- Ugele B, St-Pierre MV, Pihusch M, Bahn A, Hantschmann P. Characterization and identification of steroid sulfate transporters of human placenta. American Journal of Physiology: Endocrinology and Metabolism. 2003;284:E390–E398. doi: 10.1152/ajpendo.00257.2002. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ. Renal Transporters for organic anions and organic cations. Structural requirements for substrates. Journal of Membrane Biology. 1997;158:95–107. doi: 10.1007/s002329900247. [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Rumrich G. Contraluminal transport systems in the proximal renal tubule involved in secretion of organic anions. American Journal of Physiology: Renal Physiology. 1988;254:F453–F462. doi: 10.1152/ajprenal.1988.254.4.F453. [DOI] [PubMed] [Google Scholar]
- Uwai Y, Okuda M, Takami K, Hashimoto Y, Inui K. Functional characterization of the rat multispecific organic anion transporter OAT1 mediating basolateral uptake of anionic drugs in the kidney. Federation of European Biochemical Societies Letters. 1998;438:321–324. doi: 10.1016/s0014-5793(98)01328-3. [DOI] [PubMed] [Google Scholar]
- Wakida N, Tuyen DG, Adachi M, Miyoshi T, Nonoguchi H, Oka T, Ueda O, Tazawa M, Kurihara S, Yoneta Y, et al. Mutations in human urate transporter 1 gene in presecretory reabsorption defect type of familial renal hypouricemia. Journal of Clinical Endocrinology and Metabolism. 2005;90:2169–2174. doi: 10.1210/jc.2004-1111. [DOI] [PubMed] [Google Scholar]
- Weiner IM. Transport of weak acids and bases. In: Orloff J, Berliner RW, editors. Handbook of physiology, Section 8: Renal physiology. American Physiological Society; Washington, DC: 1973. pp. 521–554. [Google Scholar]
- Weiner IM, Blanchard KC, Mudge GH. Factors influencing renal excretion of foreign organic acids. American Journal of Physiology. 1964;207:953–963. doi: 10.1152/ajplegacy.1964.207.5.953. [DOI] [PubMed] [Google Scholar]
- Whitley AC, Sweet DH, Walle T. The dietary polyphenol ellagic acid is a potent inhibitor of hOAT1. Drug Metabolism and Disposition. 2005;33:1097–1100. doi: 10.1124/dmd.105.004275. [DOI] [PubMed] [Google Scholar]
- Wolff NA, Grunwald B, Friedrich B, Lang F, Godehardt S, Burckhardt G. Cationic amino acids involved in dicarboxylate binding of the flounder renal organic anion transporter. Journal of the American Society of Nephrology. 2001;12:2012–2018. doi: 10.1681/ASN.V12102012. [DOI] [PubMed] [Google Scholar]
- Wolff NA, Thies K, Kuhnke N, Reid G, Friedrich B, Lang F, Burckhardt G. Protein kinase C activation downregulates human organic anion transporter 1-mediated transport through carrier internalization. Journal of the American Society of Nephrology. 2003;14:1959–1968. doi: 10.1097/01.asn.0000079040.55124.25. [DOI] [PubMed] [Google Scholar]
- Wolff NA, Werner A, Burkhardt S, Burckhardt G. Expression cloning and characterization of a renal organic anion transporter from winter flounder. Federation of European Biochemical Societies Letters. 1997;417:287–291. doi: 10.1016/s0014-5793(97)01304-5. [DOI] [PubMed] [Google Scholar]
- Wright SH, Dantzler WH. Molecular and cellular physiology of renal organic cation and anion transport. Physiological Reviews. 2004;84:987–1049. doi: 10.1152/physrev.00040.2003. [DOI] [PubMed] [Google Scholar]
- Xu G, Bhatnagar V, Wen G, Hamilton BA, Eraly SA, Nigam SK. Analyses of coding region polymorphisms in apical and basolateral human organic anion transporter (OAT) genes [OAT1 (NKT), OAT2, OAT3, OAT4, URAT (RST)] Kidney International. 2005;68:1491–1499. doi: 10.1111/j.1523-1755.2005.00612.x. [DOI] [PubMed] [Google Scholar]
- Xu W, Tanaka K, Sun AQ, You G. Functional role of the C terminus of human organic anion transporter hOAT1. Journal of Biological Chemistry. 2006;281:31178–31183. doi: 10.1074/jbc.M605664200. [DOI] [PubMed] [Google Scholar]
- Yang XP, Bleasby K, Breen CM, Miller DS, Pritchard JB. Exploring the structure and function of the human organic anion transporter hOAT1. Journal of the Federation of American Societies for Experimental Biology. 2002;16:390. 1. [Google Scholar]
- You G. Towards an understanding of organic anion transporters:structure–function relationships. Medicinal Research Reviews. 2004;24:762–774. doi: 10.1002/med.20014. [DOI] [PubMed] [Google Scholar]
- You G, Kuze K, Kohanski RA, Amsler K, Henderson S. Regulation of mOAT-mediated organic anion transport by okadaic acid and protein kinase C in LLC-PK(1) cells. Journal of Biological Chemistry. 2000;275:10278–10284. doi: 10.1074/jbc.275.14.10278. [DOI] [PubMed] [Google Scholar]
- Youngblood GL, Sweet DH. Identification and functional assessment of the novel murine organic anion transporter Oat5 (Slc22a19) expressed in kidney. American Journal of Physiology: Renal Physiology. 2004;287:F236–F244. doi: 10.1152/ajprenal.00012.2004. [DOI] [PubMed] [Google Scholar]
- Zalups RK. Molecular interactions with mercury in the kidney. Pharmacological Reviews. 2000;52:113–143. [PubMed] [Google Scholar]
- Zalups RK, Ahmad S. Homocysteine and the renal epithelial transport and toxicity of inorganic mercury: Role of basolateral transporter organic anion transporter 1. Journal of the American Society of Nephrology. 2004;15:2023–2031. doi: 10.1097/01.ASN.0000135115.63412.A9. [DOI] [PubMed] [Google Scholar]
- Zalups RK, Ahmad S. Handling of cysteine S-conjugates of methylmercury in MDCK cells expressing human OAT1. Kidney International. 2005;68:1684–1699. doi: 10.1111/j.1523-1755.2005.00585.x. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Shirahatti NV, Mahadevan D, Wright SH. A conserved glutamate residue in transmembrane helix 10 influences substrate specificity of rabbit OCT2 (SLC22A2) Journal of Biological Chemistry. 2005;280:34813–34822. doi: 10.1074/jbc.M506342200. [DOI] [PubMed] [Google Scholar]
- Zhou F, Hong M, You G. Regulation of human organic anion transporter 4 by progesterone and protein kinase C in human placental BeWo cells. American Journal of Physiology: Endocrinology and Metabolism. 2007;293:E57–E61. doi: 10.1152/ajpendo.00696.2006. [DOI] [PubMed] [Google Scholar]
- Zhou F, Pan Z, Ma J, You G. Mutational analysis of histidine residues in human organic anion transporter 4 (hOAT4) Biochemical Journal. 2004a;384(Pt 1):87–92. doi: 10.1042/BJ20040751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Tanaka K, Pan Z, Ma J, You GF. The role of glycine residues in the function of human organic anion transporter 4. Molecular Pharmacology. 2004b;65:1141–1147. doi: 10.1124/mol.65.5.1141. [DOI] [PubMed] [Google Scholar]
- Zhou F, Xu W, Hong M, Pan Z, Sinko PJ, Ma J, You G. The role of N-linked glycosylation in protein folding, membrane targeting, and substrate binding of human organic anion transporter hOAT4. Molecular Pharmacology. 2005;67:868–876. doi: 10.1124/mol.104.007583. [DOI] [PubMed] [Google Scholar]
- Zhou F, Xu W, Tanaka K, You G. Comparison of the interaction of human organic anion transporter hOAT4 with PDZ proteins between kidney cells and placental cells. Pharmaceutical Research. 2008;25(2):475–480. doi: 10.1007/s11095-007-9359-4. [DOI] [PubMed] [Google Scholar]
- Zhou F, You G. Molecular insights into the structure–function relationship of organic anion transporters OATs. Pharmaceutical Research. 2007;24:28–36. doi: 10.1007/s11095-006-9144-9. [DOI] [PubMed] [Google Scholar]