

Abstract
We have expanded the field of “DNA computers” to RNA and present a general approach for the solution of satisfiability problems. As an example, we consider a variant of the “Knight problem,” which asks generally what configurations of knights can one place on an n × n chess board such that no knight is attacking any other knight on the board. Using specific ribonuclease digestion to manipulate strands of a 10-bit binary RNA library, we developed a molecular algorithm and applied it to a 3 × 3 chessboard as a 9-bit instance of this problem. Here, the nine spaces on the board correspond to nine “bits” or placeholders in a combinatorial RNA library. We recovered a set of “winning” molecules that describe solutions to this problem.
Keywords: DNA computing, satisfiability, RNA evolution, in vitro selection, SELEX
Adleman (1) introduced DNA-based computing as an approach to solving mathematical problems, which uses DNA as a data carrier and techniques of molecular biology to operate on DNA. Since then, there have been relatively few experimental demonstrations of DNA-based computations (2, 3), although the theoretical foundation is strong (4, 5). Here we introduce a method for the construction of binary nucleic acid libraries, and we introduce RNA as a molecule for computation to present a general approach for the solution of the famous satisfiability (SAT) problems of propositional logic.
Using a combination of a binary RNA library and ribonuclease (RNase) H digestion, we developed a destructive algorithm (6) that would hydrolyze RNA strands that did not fit the constraints of a chosen problem, instead of an algorithm that required efficient hybridization extraction (1, 2). The use of restriction endonucleases (3) would have also permitted a logical binary-style operation, as each restriction enzyme cleaves only in the presence of its recognition site, and cleaves the double-stranded DNA sufficiently to completion. However, by using RNase H in the context of different sets of oligonucleotides, one can go beyond the set of available restriction enzymes. Here, RNase H acts as a “universal restriction enzyme” because it allows selective marking of virtually any RNA strands for digestion in parallel, and use of a thermostable RNase H (7) ensures fidelity of hybridization between DNA and RNA strands, minimizing incorrect marking of noncognate strands.
Materials and Methods
Chemical Synthesis of DNA “Half Libraries.”
The DNA library was prepared as two halves by using mix and split phosphoramidite chemistry with the sequence codes shown in Table 1. In brief, bit n set to 0 and spacer n were synthesized on one column, and bit n set to 1 and spacer n were synthesized on the other column. The columns were decrimped and the two resins poured together and mixed, and then half of this mixture (containing approximately equal proportions of 0's and 1's at bit position n) was returned to each column for synthesis of the next variable position, and the entire process was repeated (Fig. 1). Each of the four combinatorial synthesis products was purified by polyacrylamide gel electrophoresis on a 6% denaturing gel (Sequagel, National Diagnostics) and was recovered by a “crush and soak” procedure (500 mM ammonium acetate/1 mM EDTA/0.1% SDS) and ethanol precipitation (8). Annealing of the four half-sized strands in a 50-μl PCR reaction containing 1 μM of each of four halves (9) and primer overlap extension during five thermocycles (94°C for 30 sec, 45°C for 30 sec, and 72°C for 30 sec per cycle) generated the DNA library of full sized strands with a total of 1,024 (210) unique strands. (Note that the 3 × 3 instance of this problem considers nine bits, or a total of 512 different chessboards. Bit 10 was unused in the actual selections.) The degeneracy of the DNA pool was verified by direct sequencing (10) using 5′ end-radiolabeled primers TXR (Prefix; [CAT]4CTCGAGAATT) and 32.41 (Suffix complement; [CTA]4CGGGATCCTAATGACCAAGG) in cycle sequencing reactions (SequiTherm Excel, Epicentre Tech).
Table 1.
Nucleotide sequences for each bit and spacer of the combinatorial library
| Bit | Knight absent code, bit set to 0 | Knight present code, bit set to 1 | Spacer code |
|---|---|---|---|
| a | CTCTTACTCAATTCT | TCCTCACATTACTTA | TCTAC |
| b | CATATCAACATCTTA | ACTTCCTTTATATCC | ATAAC |
| c | ATCCTCCACTTCACA | TTATAACAAACATCC | CTTAA |
| d | TTAAAATCTTCCCTC | ACATAACCCTCTTCA | TTTAC |
| e | CTATTTATCCACACC | ACCTTACTTTCCATA | TACAA |
| f | GCTTCAAACAATTCC | GTACATTCTCCCTAC | TCCTT |
| g | AACTCTCAAATTCAA | CATAATCTTATATTC | TCAAT |
| h | CTAACCTTTACTTCA | ATAATCACATACTTC | TCCAA |
| i | CATTCCTTATCCCAC | TCCACCAACTACCTA | ACACA |
| j | CACCCTTTCTCCTCT | TTTTAAATTTCACAA | SUFFIX |
Figure 1.

Modular construction of the combinatorial DNA library from two half-libraries. Linear mixing and splitting steps yield exponential increases in pool complexity. Each half is synthesized on two columns beginning with bit f or its reverse complement. Synthesis of one half continues through bit a and the prefix; the other half contains the reverse complement of bits f through j and the suffix. Because the halves are complementary at position f, primer extension of the two halves creates the full-length 10-bit library. Black and white boxes represent 1 and 0, respectively; shaded boxes represent prefix, suffix, and spacer sequences.
The 271-bp DNA pool was amplified by PCR with primers T7TXR (TTCTAATACGACTCACTATAGG[CAT]4CTCGAGAATT) and 32.41 (1 μM each; 15 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec). The 251-bp RNA library was then synthesized by in vitro T7-transcription from the synthetic DNA pool and was purified on a 4% denaturing polyacrylamide gel.
DNA Bit Oligonucleotides.
The set of DNA bit oligonucleotides used both to operate on the RNA library and as the downstream primers in readout PCR were from Operon Technologies (Alameda, CA) and are shown in Table 2.
Table 2.
Nucleotide sequences for each DNA bit oligonucleotide used in the computation
| Hybridizes to bit set to 0 | Hybridizes to bit set to 1 | ||
|---|---|---|---|
| a0 | AGAATTGAGTAAGAG | a1 | TAAGTAATGTGAGGA |
| b0 | gttatTAAGAGGTTGATATG | b1 | gttatGGATATAAAGGAAGT |
| c0 | TGTGAAGTGGAGGAT | c1 | GGATGTTTGTTATAA |
| d0 | gtaaaGAGGGAAGATTTTAA | d1 | gtaaaTGAAGAGGGTTATGT |
| e0 | GGTGTGGATAAATAG | e1 | TATGGAAAGTAAGGT |
| f0 | GGAATTGTTTGAAGC | f1 | GTAGGGAGAATGTAC |
| g0 | attgaTTGAATTTGAGAGTT | g1 | attgaGAATATAAGATTATG |
| h0 | TGAAGTAAAGGTTAG | h1 | GAAGTATGTGATTAT |
| i0 | GTGGGATAAGGAATG | i1 | TAGGTAGTTGGTGGA |
Twenty-base oligonucleotides are complementary to both the 15-nt bit sequence (uppercase) and adjacent 5-nt spacer (lowercase) to increase the melting temperatures of weaker hybridizing oligonucleotides.
Implementing the RNA Algorithm.
RNase H digestions destroyed the RNA strand of RNA⋅DNA hybrids marked by hybridization of the pool RNA to the complementary bit oligonucleotides shown in Table 2. One hundred-microliter RNase H reactions contained 20 μg of gel-purified RNA (250 pmol), 1 nmol of each bit oligonucleotide, 5 units of Hybridase Thermostable RNase H (Epicentre Tech), 50 mM Tris⋅HCl (pH 7.5), 100 mM NaCl, and 10 mM MgCl2. After preincubation at 72°C for 3 min to denature strands, enzyme and MgCl2 were added at 45°C. The reaction was held at 45°C for 60 min and then was stopped by addition of 10 mM EDTA. Spin column chromatography (CLONTECH Chroma Spin-200, DEPC-H2O) was used to remove DNA bit oligonucleotides and short RNA digestion products. The eluted RNA was ethanol-precipitated, and remaining full-length (undigested) RNA was purified on a 5% denaturing polyacrylamide gel. Recovered RNA was reverse transcribed in 80 μl with 1.25 μM suffix complement (Superscript II, BRL). One-tenth of this reaction was amplified by PCR (200 μl, 1 μM primers T7TXR and 32.41, 2–5 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec). After ethanol precipitation, the two halves of each pool were combined, and half of the DNA was transcribed in vitro for the next step in the algorithm.
Multiplex Colony PCR Readout.
PCR products were directly cloned by using the TOPO-TA cloning kit (Invitrogen). Cells were grown on LB-agar plates containing 50 μg/ml ampicillin (8). Positive (white) colonies were randomly picked, were transferred to 50 μl of LB media containing 50 μg/ml ampicillin, and were grown for 4–6 hours at 37°C. Colony PCR (11) of 2 μl of media (95°C for 5 min, followed by 25 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec with 0.2 μM primers TXR and 32.41) allowed rapid screening and recovery of individual DNA library strands. To readout the bit settings of cloned strands, 1 μl of a 1:100 dilution of the colony PCR product was first amplified by linear PCR (9) (10 μl, 0.2 μM primer T7TXR; 10 cycles as above) in the presence of prefix only to generate an excess of single-stranded DNA; then equimolar mixtures of 5′ end-radiolabeled “0”- or “1”-complementary DNA oligonucleotides (≤1 nM each) were added to the same tube, and the reaction was continued for an additional five cycles. Reaction products were separated on a 6% denaturing polyacrylamide gel (Long Ranger gel solution, FMC Bioproducts). The dried gel was analyzed by autoradiography on a PhosphorImager (Molecular Dynamics).
Error Analysis.
To determine potential sources of error, we measured the efficiency and recovery of each operation.
Digestion of the RNA library by thermostable RNase H.
5′ end radiolabeled RNA library (5 pmol) was subjected to RNase H digestion in the presence of combinations of DNA bit oligonucleotides (a0, f1, h1; b0, g1, i1; c0, d1, h1; d0, c1, i1; f0, a1, g1; g0, b1, f1; h0, a1, c1; i0, b1, d1; 75 pmol each; Table 2) under conditions as described above in 15 μl and 0.5 units RNase H. The digestion products were quantified on a 10% denaturing polyacrylamide gel by using a PhosphorImager. The amount of digestion products were in the expected range [12.5% undigested pool RNA, 12.5% first (longest), 25% second, and 50% third (shortest) digestion product]. Digestion of single RNA clones under the same conditions was almost complete (≥98% in all cases but 96% for i0, b1, d1).
Spin-column chromatography and gel purification.
Column chromatography removed 5′ end radiolabeled DNA bit oligonucleotides (100 pmol) with >99% efficiency and recovery of 40–50% of 50 pmol 5′ end radiolabeled RNA library. Recovery of 50 and 30 pmol of cloned RNA molecules (the expected range after a digestion step) from a 6% denaturing polyacrylamide gel was 52% and 75%, respectively.
Reverse transcription and PCR.
Thirty and fifteen picomoles of three cloned RNA molecules were reverse transcribed in the presence of 5′ end radiolabeled primer 32.41. Efficiencies were 50% in one of the clones and 25% in two other clones. One-tenth of these reactions was subjected to PCR (200 μl, 1 μM primers T7TXR and 5′ end radiolabeled 32.41, 1–5 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec). Samples were removed after each cycle and were resolved on a 12% denaturing polyacrylamide gel. Amplification increased exponentially after cycles 1–4, but plateaued after the fifth cycle of PCR.
Results and Discussion
The “Knight Problem.”
Chess problems create a class of SAT problems that are flexible in both scale and complexity. A “true” or “1” variable represents the presence of a knight at positions a–i whereas a “false” or “0” variable represents the absence of a knight at that position. This is a particular instance of a class of NP-complete SAT problems. For a 3 × 3 board
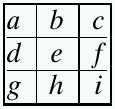 |
we represent the problem as follows (∧, “and”; ∨, “or”):
((¬h ∧ ¬f) ∨ ¬a) ∧ ((¬g ∧ ¬i) ∨¬b) ∧ ((¬d ∧ ¬h) ∨ ¬c) ∧ ((¬c ∧ ¬i) ∨ ¬d) ∧ ((¬a ∧ ¬g) ∨ ¬f) ∧ ((¬b ∧ ¬f) ∨ ¬g) ∧ ((¬a ∧ ¬c) ∨ ¬h) ∧((¬d ∧ ¬b) ∨ ¬i).
In this particular example, this simplifies to
((¬h ∧ ¬f) ∨¬a) ∧ ((¬g ∧ ¬i) ∨¬b) ∧((¬d ∧ ¬h) ∨¬c)∧ ((¬c ∧ ¬i) ∨¬d) ∧((¬a ∧ ¬g) ∨¬f).
This reduces the number of operations that one has to perform.
Design and Synthesis of a 10-Bit RNA Library.
A 10-bit pool size was chosen because this represents a pool of significant complexity, containing 1,024 different strands. Each strand of RNA follows the template below (4):
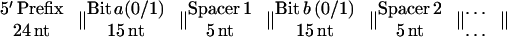 |
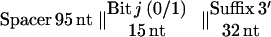 |
Each bit can be set to an “on” (= 1) or “off” (= 0) sequence. Using computer simulations, we incorporated three important criteria into the design of the combinatorial pool: (i) Each of the bit encodings must be fundamentally different. Hence, sequences were chosen to maximize the Hamming Distances between different library strands and between the different parts of individual strands (no more than five matches over a 20-nt window, both within and between all 210 possible strands). Bit sequences were also selected to have an average melting temperature of 45°C. (ii) The strands were biased to avoid secondary structure so that each bit position would be equally accessible to the enzymes and oligonucleotides that operate on the pool molecules. This was accomplished by using a three-letter alphabet for the bits and spacers in the library, A, C, and U, eliminating the potential for both G-C pairs and G:U pairs (which form in RNA) as well as G stacking. (iii) The strands were controlled to avoid hybridization to themselves or to any other library strands by more than seven consecutive base pairs (12) because such interactions would interfere with our ability to operate on the RNA strands by making these regions inaccessible to reagents.
To satisfy all of these criteria, a simple computer program (permute, published as supplemental material on the PNAS web site, www.pnas.org) generated random nucleotides for bit and spacer sequences to satisfy the second criterion and then permuted the sequences until they fulfilled all three criteria. The prefix and suffix provide PCR primer-binding sites, with the prefix containing the T7 RNA polymerase promoter for transcription. The resulting sequences are shown in Table 1.
We synthesized a 10-bit DNA library as two halves by a recursive mix and split strategy (Fig. 1). The first half contained bits a through f, and the second half contained the complement of bits f through j. Bit n set to 0 and spacer n were synthesized on one column, and bit n set to 1 and spacer n were synthesized on the other column. The contents of both columns were mixed, and half of the mixture was returned to each column. The partially synthesized molecules were further extended by synthesis of the next bit set to 0 and corresponding spacer on one column, and the next bit set to 1 and corresponding spacer on the other column, and the entire process was repeated. The first half (130 nt) contained the prefix and bits a through f, and the second half (110 nt) contained the complements of bits f through j and the suffix. Primer extension from overlapping bit f by Taq DNA polymerase created full-length library strands, and in vitro transcription produced the RNA library. Equal two-fold degeneracy of each bit position was verified by directly sequencing the library (data not shown). Note that use of a 10-bit library to solve an instance of a 9-bit SAT problem allows one to ignore any position that computes less reliably, analogous to the problem of avoiding “bad blocks” on computer disks.
Algorithm for Solving the Knight Problem Using the RNA Library.
We executed the algorithm for solving a variant of the knight problem with our RNA combinatorial library as follows (Fig. 2): Initially we have a test-tube that encodes all 1,024 possible 10-bit strings. Each string has the general form x1… xn, where a variable xi is either 1 or 0, representing a bit set to on or off. (In our experiment, x1 = a, x2 = b,… , x9 = i.) Then we systematically perform the first operation to destroy strings that fail to satisfy the first clause ((¬h ∧ ¬f) ∨ ¬a) as follows: (i) One executes an OR clause by dividing the library into two “equal” collections. In one test-tube, we select those strands that contain a 1 at position a by annealing DNA bit oligonucleotide a0 to the library, hence digesting those strands with position a set to 0 (thereby setting bit a to 1). Simultaneously, we destroy any 1's at those bit positions that must be set to 0 to fulfill the clause (bits f and h in this case). In the other test-tube, we anneal DNA bit oligonucleotide a1 to perform the mirror operation setting bit position a to 0 (¬a). (ii) Undigested molecules are recovered and reverse transcribed. After not more than five PCR cycles, the contents of both tubes are mixed and amplified to high copy number during T7 RNA polymerization. The library is split again to execute the next OR clause, and this process is repeated until all clauses have been processed. The combinations of bit oligonucleotides in each step were as follows: a →1: a0, f1, h1; →0: a1; b →1: b0, g1, i1; →0: b1; c →1: c0, d1, h1; →0: c1; d →1: d0, c1, i1; →0: d1; f →1: f0, a1, g1; →0: f1.
Figure 2.

Outline of the RNA algorithm. A value of 1 or 0 is assigned to a specific bit position by destroying all strands in the RNA library which do not have the value at this position (the step shown sets bits a, f, and h). The library is divided into two equal ensembles. In one test-tube, a specific bit position is set to 1 along with its accompanying constraints; in the other set, this bit is set to 0, using targeted digestion by RNase H. Continuing with this series of operations on the combined ensemble of strands leads to execution of the algorithm, which ends with readout of a randomly chosen subset of the surviving strands.
“Bit Shuffling.”
Recombination events are likely to occur during PCR amplification of heterogeneous target sequences (13), especially because the strands in our 10-bit library share several stretches of sequence identity constrained by the bit structure of the library. To measure this effect, the PCR products derived from two clones (wt-I 011 111 001 and wt-II 010 110 010, a, … ,i) were combined and amplified for 25 cycles of PCR (0.2 μM primers TXR and 32.41; conditions as above). The resulting PCR products were cloned, and 20 clones were randomly chosen and analyzed by multiplex colony PCR readout at positions c, f, and h. Eight of twenty resulting clones (40%) were the product of bit shuffling. This indicates that bit shuffling poses a serious threat to successful execution of the algorithm; however, when only 15 instead of 25 PCR cycles were used, no bit shuffling was detected in 20 randomly chosen clones. This suggests that bit shuffling in our system is mostly due to a high proportion of incompletely extended strands annealing to heterologous target sequences, and it suggests that we can sufficiently reduce this problem by limiting the number of PCR cycles.
Consequently, no more than five cycles of PCR were performed between any two digestion steps and before cloning. Instead, we exploited T7 RNA polymerase as an amplifier, because this enzyme is more processive than Taq DNA polymerase.
Methods of “Readout.”
Readout of a randomly sampled set of clones was accomplished by colony PCR (10) followed by multiplex linear PCR. This creates a “bar code” for each strand (Fig. 3). If one wanted to solve a stricter version of the knight problem, one could select the board containing the maximal number of knights by choosing sequences of unequal length for 1's and 0's and purifying either the longest or shortest strands (3), but the use of bit sequences of unequal length would preclude the quick and streamlined readout in Fig. 3 because it relies on equal length for 1's and 0's (Table 1) to generate a bar code pattern. Furthermore, unlike that of Ouyang et al. (3), our approach requires neither the isolation of plasmid DNA nor actual sequencing of any clones for readout.
Figure 3.

Multiplex colony PCR readout. Colony PCR (10) allowed rapid screening and recovery of individual library strands. The boards represented by two clones are shown above their respective readouts on a 6% polyacrylamide gel. These clones represent the strings 010 011 010 and 001 011 000. Left lane, 10-bp ladder.
RNA Solutions.
After subjecting the RNA library to the algorithm outlined above, 43 clones were randomly chosen and interpreted by readout PCR. Forty-two of these represented solutions to our version of the Knight problem (Fig. 4). Ten solutions occurred more than once, even though they were selected at random. However, the overall distribution of these 42 clones was not significantly different from a random sampling of all 94 possible solutions (Table 3), with a very modest preference for higher numbers of knights. We did not attempt to recover the other 64 solutions, as they are all variants of the classes of solutions shown in Fig. 4, and it would require a much larger sampling to ensure representation of each unique solution at least once. One clone (represented as 010 111 011) contained two illegal interactions, resulting from incorrect placement of one knight. Hence, of 127 knights on 43 boards, only one knight had an unacceptable placement. This was effectively a 97.7% success rate in finding correct solution strands, which is indeed satisfactory for choosing a clone that is likely to encode a solution.
Figure 4.

Representations of the 31 unique boards analyzed by PCR readout. The last board contains one “illegal” white knight.
Table 3.
Expected and observed frequencies of boards with specific numbers of knights
| No. of knights per board | Expected frequency (percent of 94) | Observed frequency (percent of 42) |
|---|---|---|
| 5 | 2 (2.1) | 1 (2.3) |
| 4 | 18 (19.1) | 11 (26.2) |
| 3 | 36 (38.3) | 16 (38.1) |
| 2 | 28 (29.8) | 10 (23.8) |
| 1 | 9 (9.6) | 4 (9.5) |
| 0 | 1 (1.1) | 0 (0) |
DNA sequence analysis of four clones confirmed the results obtained by readout PCR and the robustness of our style of encoding that uses hybridization discrimination between “words” in DNA of 15 letters or more (14–16); each clone contained one to three point mutations or deletions, compared with the sequences in Table 1, but in general this did not affect either the algorithm or readout. However, the board with one incorrectly placed knight did contain an adjacent deletion and point mutation in bit 9 (TCCACTACTACCTA instead of TCCACCAACTACCTA), resulting in ineffective RNase H cleavage but correct readout with DNA bit oligonucleotide i1. Four additional clones that could not be interpreted by readout PCR contained 3–5 deletions in single bit positions, one of which would have led to incorrect placement of another knight. This suggests that the main source of error is clusters of mutations in the same bit positions, but one could reduce the prevalence of such deletions by very accurate size purification, essential for the scalability of this system. Algorithms that employ recursive cycles of selection allow enrichment of the number of solution strands in each round as a form of progressive error correction (17–20). Repeated rounds of digestion might therefore enhance recovery of solution strands over “non-solutions.” Thus, because 250 ≈ 1015 is approximately the number of RNA molecules that in vitro selection protocols can currently search, this projects an upper bound for the size of DNA or RNA computing experiments that could use exhaustive search algorithms. Fortunately, this is on the same order as many interesting problems in computer science, such as the Data Encryption Standard (DES) (20).
Because in vitro selection experiments (18) that search for binding or function within an enormous sequence space have captured molecules as unique as one in a quadrillion, and because these molecules may even encode the solution to a mathematical problem, like knights on a chessboard, nucleic acid-based computing may be viewed as a natural extension of simulated evolution experiments to searches of a defined mathematical space.
Supplementary Material
Acknowledgments
We thank Steve Lawrie, Vernadette Simon, and Saw Kyin for synthesis of the oligonucleotide portions of the combinatorial pool. We also thank Leonard Adleman, Paul Rothemund, and Erik Winfree for helpful discussions and two anonymous reviewers for comments. This research was supported in part by the National Science Foundation/Defense Advanced Research Planning Agency, the Air Force Office of Scientific Research, and a Deutsche Forschungsgemeinschaft fellowship to D.F.
Abbreviation
- SAT
satisfiability
Footnotes
See commentary on page 1328.
References
- 1.Adleman L M. Science. 1994;266:1021–1023. doi: 10.1126/science.7973651. [DOI] [PubMed] [Google Scholar]
- 2.Guarnieri F, Fliss M, Bancroft C. Science. 1996;273:220–223. doi: 10.1126/science.273.5272.220. [DOI] [PubMed] [Google Scholar]
- 3.Ouyang Q, Kaplan P D, Shumao L, Libchaber A. Science. 1997;278:446–449. doi: 10.1126/science.278.5337.446. [DOI] [PubMed] [Google Scholar]
- 4.Lipton R J. Science. 1995;268:542–545. doi: 10.1126/science.7725098. [DOI] [PubMed] [Google Scholar]
- 5.Landweber L F, Baum E B, editors. DNA Based Computers II. Providence, RI: Am. Math. Soc.; 1999. [Google Scholar]
- 6.Amos M, Gibbons A, Hodgson D. In: DNA Based Computers II. Landweber L F, Baum E B, editors. Providence, RI: Am. Math. Soc.; 1999. pp. 151–161. [Google Scholar]
- 7.Porter D, Curthoy N P. Anal Biochem. 1997;247:279–286. doi: 10.1006/abio.1997.2059. [DOI] [PubMed] [Google Scholar]
- 8.Sambrook J, Maniatis T, Fritsch E F. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 9.Landweber L F, Kreitman M. Methods Enzymol. 1993;218:17–26. doi: 10.1016/0076-6879(93)18004-v. [DOI] [PubMed] [Google Scholar]
- 10.Rao V B. PCR Methods Appl. 1994;4:S15–S23. doi: 10.1101/gr.4.1.s15. [DOI] [PubMed] [Google Scholar]
- 11.Trower M K. Methods Mol Biol. 1996;58:329–333. doi: 10.1385/0-89603-402-X:329. [DOI] [PubMed] [Google Scholar]
- 12.Zuker M. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 13.Meyerhans A, Vartanian J, Wain-Hobson S. Nucleic Acids Res. 1990;18:1687–1691. doi: 10.1093/nar/18.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frutos A G, Liu Q, Thiel A J, Sanner A M V, Condon A E, Smith L M, Corn R M. Nucleic Acids Res. 1997;25:4748–4757. doi: 10.1093/nar/25.23.4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith L M, Corn R M, Condon A E, Lagally M G, Frutos A G, Liu Q, Thiel A J. J Comp Biol. 1998;5:255–267. doi: 10.1089/cmb.1998.5.255. [DOI] [PubMed] [Google Scholar]
- 16.James K D, Boles A R, Henckel D, Ellington A D. Nucleic Acids Res. 1998;26:5203–5211. doi: 10.1093/nar/26.22.5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stemmer W P. Science. 1995;270:1510. doi: 10.1126/science.270.5241.1510. [DOI] [PubMed] [Google Scholar]
- 18.Landweber L F. In: DNA Based Computers II. Landweber L F, Baum E B, editors. Providence, RI: Am. Math. Soc.; 1999. pp. 181–189. [Google Scholar]
- 19.Boneh D, Dunworth C, Lipton R J, Sgall J. In: DNA Based Computers II. Landweber L F, Baum E B, editors. Providence, RI: Am. Math. Soc.; 1999. pp. 163–170. [Google Scholar]
- 20.Boneh D, Dunworth C, Lipton R J. In: DNA Based Computers. Lipton R J, Baum E B, editors. Providence, RI: Am. Math. Soc.; 1999. pp. 37–65. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.