

Abstract
Pulmonary infection is the dominant clinical feature of cystic fibrosis (CF), but the basis for this susceptibility remains incompletely understood. One hypothesis is that CF airway surface liquid (ASL) is abnormal and interferes with neutrophil function. To study this possibility, we developed an in vitro system in which we collected ASL from primary cultures of normal and CF airway epithelial cells. Microbial killing was less efficient when bacteria were incubated with neutrophils in the presence of ASL from CF epithelia compared with normal ASL. Antimicrobial functions of human neutrophils were assessed in ASL from CF and normal epithelia using a combination of quantitative bacterial culture, flow cytometry, and microfluorescence imaging. The results of these assays of neutrophil function were indistinguishable in CF and normal ASL. In contrast, the direct bactericidal activity of ASL to Escherichia coli and to clinical isolates of Staphylococcus aureus and Pseudomonas aeruginosa was substantially less in CF than in normal ASL, even when highly diluted in media of identical ionic strength. Together, these observations indicate that the antimicrobial properties of ASL in CF are compromised in a manner independent of ionic strength of the ASL, and that this effect is not mediated through a direct effect of the ASL on phagocyte function.
Keywords: airway surface liquid, cystic fibrosis, inflammation, ionic strength, neutrophil
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-regulated Cl− channel (1, 2). CF manifests as a multiorgan disease involving the pancreas, salivary glands, genital tubes, and liver canaliculi. Involvement of the respiratory tract, however, is the dominant clinical feature, and progressive pulmonary disease accounts for most of the morbidity and mortality in patients with CF (1, 3). The life expectancy of affected individuals has increased remarkably in recent years, and those born today are expected to survive into their 40s, an astonishing improvement compared with the median survival of 10 years in the 1940s (3–5) Nonetheless, this improvement must be tempered with the reality that even now, the vast majority of patients with CF will die from pulmonary disease consequent to progressive inflammatory damage (3, 5–7).
Mutations in CFTR lead to unique alterations in the microenvironment of the lung that facilitate bacterial infection with a restricted group of microorganisms. Reported abnormalities in the CF lung include altered fluid and ionic fluxes across the respiratory epithelium, diminished glutathione levels, excessive and dehydrated luminal mucus, diminished mucociliary clearance, altered patterns of epithelial surface molecule glycosylation, and decreased activity of bactericidal factors such as lysozyme, lactoferrin, defensins, and cathelicidins in the airway surface liquid (ASL) (8–15). Notably, alterations in antibacterial factors may be consequent to the action of neutrophil-derived proteinases such as elastase (16, 17). Another possibility is that CFTR itself may serve as an epithelial receptor for Pseudomonas aeruginosa, triggering bacterial internalization and aiding in their clearance. Thus, according to this concept, mutations in CFTR result in diminished clearance of this common CF pathogen and may explain the frequency with which CF lungs are colonized with P. aeruginosa (18). However, despite intensive investigation, the relative importance of these multiple factors to the clinical manifestations of CF remains uncertain.
Neutrophil-predominant airway inflammation in CF begins early during the neonatal period, increases throughout childhood and adolescence, and is associated with persistent airway infection, but whether it is a cause or a consequence of infection remains conjectural (19–26). Paradoxically, despite the presence of increased numbers of these phagocytes, bacteria survive and often thrive in CF lungs, suggesting a defect in antimicrobial functions of phagocytes in the milieu of the CF airway. One possibility is that there is an intrinsic defect in phagocyte function. Indeed, there are reports of defects in CF neutrophils, including increased oxidant production (27) and alterations in Cl− flux and intracellular pH regulation (28), that may thwart antimicrobial functions and predispose to inflammatory tissue injury. However, because expression of CFTR mRNA and protein is normally exceedingly low in professional phagocytes such as neutrophils and macrophages (29), whether these reported defects are intrinsic to the phagocytes or secondary to extrinsic factors, such as abnormalities in the ASL (see above), or to the systemic effects of inflammation as a consequence of infection in CF, remains unresolved. Accordingly, the purpose of the current study was to determine if the ASL collected from CF epithelia inhibits antimicrobial functions of neutrophils. To address this issue without the confounding factor of systemic inflammation in CF, we developed an in vitro model system that allowed us to collect ASL from cultured CF and non-CF epithelial cells and study its effects on neutrophil antimicrobial functions ex vivo.
MATERIALS AND METHODS
Chemicals
Collagen (Human Placental type VI), calcium chloride, magnesium chloride, fibronectin, silicone oil, N-formyl-Met-Leu-Phe (fMLP), cytochalasin D, and phorbol 12-myristate 13-acetate (PMA) were from Sigma-Aldrich (St. Louis, MO). Sheep red blood cells (sRBCs) were from ICN/Cappel (Aurora, OH). Texas Red sulfonyl chloride, dihydrorhodamine (DHR), and fluorescein-5-isothiocyanate were from Molecular Probes, Inc. (Eugene, OR). Paraformaldehyde (16%) (PFA) and glutaraldehyde (EM-grade) were from Canemco Inc. (St. Laurent, PQ, Canada). Collagen (Bovine type I) was from BD Biosciences (Bedford, MA). Trypan blue stain was from Life Technologies (Grand Island, NY).
Antibodies
Rabbit anti-sheep IgG was from ICN/Cappel. Mouse monoclonal antibody to human CD63 antigen (LP9) and mouse monoclonal antibody to human CD66b antigen (80H3) were from Serotec Ltd (Kidlington, UK). Fluorescein isothiocyanate (FITC)-labeled mouse monoclonal antibody to the human CD11b (Mac-1) antigen (VIM12) was from Caltag Laboratories (Burlingame, CA). Alexa Fluor 488–labeled goat anti-mouse and Alexa Fluor 488–labeled goat anti-rabbit IgG secondary antibodies were from Molecular Probes.
Tissue Culture Reagents
UltroSerG powdered media was from BioSepra (Marlborough, MA). Bronchial Epithelial Cell Basal Medium (BEBM) and BEGM Singlequots were from Cambrex (Walkersville, MD). PBS was from Wisent Inc. (St. Bruno, PQ, Canada) or the University of Toronto Media Services (Toronto, ON, Canada). Plastic Transwell inserts were from Costar (Corning, NY). Plastic MilliCell inserts were purchased from Millipore Corporation (Bedford, MA). Metal Attofluor Cell Chambers were from Molecular Probes. Glass coverslips (25 mm) were from VWR Scientific, Inc. (West Chester, PA).
Airway Epithelial Culture
Human bronchial epithelial cells, harvested and processed according to methods described previously (30), were provided by Dr. J. Zabner and the “In Vitro Models and Cell Culture Core” of the University of Iowa. In brief, sections of major bronchi were dissected, rinsed, enzymatically digested, and differentially plated to yield a population of primary human lung epithelial cells. Cells were plated on collagen-coated matrices on either MilliCell or Transwell tissue culture inserts and grown to confluence in an air–liquid interface. Cells were grown to confluence in UltroSerG or BEGM media and, once confluent, the apical surfaces were washed multiple times with antibiotic-free media and aspirated creating an air–liquid interface in which cells were maintained. ASL was collected from these surfaces either directly, in the case of bovine collagen–coated Transwell inserts, or by the addition of a rinsing step with a small volume (20 μl) PBS plus 1 mM Ca2+ and 1 mM Mg2+ or UltroSerG media without antibiotics in the case of MilliCell inserts (see below). A brief centrifugation step removed unwanted cellular debris (see Figure 1 for an overview of the experimental design).
Figure 1.

Preparation of ASL from both MilliCell and Transwell culture wells. This figure illustrates how various neutrophil functions were tested in ASL. Top aqueous ASL layers of both MilliCell and Transwell cultures were collected by pipette and placed in Eppendorf tubes. PBS (20 μl) was added to facilitate the collection of ASL from the MilliCell cultures. ASL was then centrifuged to remove any debris. ASL was used for functional assays as described in Materials and Methods.
MilliCell inserts have been the standard in our laboratory for years and support epithelial growth well. These inserts were used in initial experiments. However, the volume of ASL generated is very small and retrieval requires washing of the monolayer with small volumes of buffer. As an alternative method we used Transwell inserts. These are transparent, thus allowing for microscopic visualization of the epithelial monolayer and the possibility of direct imaging of live neutrophils added directly to the epithelial monolayers in situ. Epithelia grown on these inserts tended to produce larger volumes of ASL, thus facilitating collection. MilliCell inserts were used exclusively for bacterial killing (neutrophil-mediated) and secretion assays, since these could be done using very small volumes of ASL or on diluted ASL. ASL collected from both insert types was used for bacterial killing (ASL alone) and chemokinesis experiments. The results did not differ significantly between the two systems. ASL from cells grown on Transwell inserts was used for phagocytosis and respiratory burst assays, as these required larger volumes of ASL that was not diluted.
Human Peripheral Blood Neutrophil Isolation
Human neutrophils were isolated from anticoagulated peripheral blood in a mixture of sodium metrizoate and dextran 500 (1-Step Polymorphs; Accurate Chemical and Scientific Corp., Westbury, NY) according to the manufacturer's instructions. Blood was drawn from healthy human volunteers without CF. Approval was obtained from the University of Toronto Research Ethics Board (ref # 14262). Isolated neutrophils were > 98% pure as determined by modified Wright-Giemsa staining and > 99% viable as determined by Trypan blue staining. Isolated cells were kept in suspension by gentle rotation in Hank's Balanced Salt Solution (HBSS; University of Toronto Media Services, Toronto, ON, Canada) containing 2.5% (vol/vol) heat-inactivated FBS (Cansera, Etobicoke, ON, Canada). PBS with 1 mM Ca2+, 1 mM Mg2+, and 10 mM glucose was the buffer used for the chemokinesis, phagocytosis, and respiratory burst assays. Cell numbers were measured by hemocytometer (Reichert Scientific instruments, Buffalo, NY) or Coulter counter (Coulter Electronics, Inc., Hialeah, FL). Cellular morphology was evaluated by staining the cells with Hemacolor Stain Set (EM Science, Gibbstown, NJ) and examination under the light microscope at a magnification of ×400.
Bacterial Culture
Escherichia coli DH5α and clinical isolates of Staphylococcus aureus and P. aeruginosa obtained from the Clinical Microbiology Laboratory, Hospital for Sick Children, Toronto, were used in this study. Bacterial strains were maintained at −80°C as glycerol stocks, and streaked onto LB agar plates to obtain single colonies before use in these studies. One colony of the indicated bacterial strain was selected and grown overnight in LB medium and then washed once with sterile PBS. After washing, cells were sedimented at 6,000 × g for 10 min at 22°C and then resuspended in PBS. The final density of the cells was 1 × 109 cfu/ml. Bacterial opsonization was accomplished by incubating the cells in heat-inactivated FBS for 30 min at 37°C and then washing the cells once in sterile PBS. Viable bacterial counts were measured after plating 10-fold serial dilutions of bacteria on LB agar plates.
Bacterial Killing
Bacterial killing was assessed by quantitative agar plate culture. ASL samples were used either undiluted or diluted with sterile Milli-Q water or PBS to a final dilution factor of between 1:50 and 1:250 depending on the type of bacteria studied as indicated. Bacteria (500–2,000 cfu) were added to each sample and incubated for times between 0 and 120 min at 37°C as indicated. Viable bacteria were counted by plating serial dilutions of bacteria on LB agar plates. In experiments examining bacterial phagocytosis and killing by neutrophils, 0.5 × 106 neutrophils were placed in ASL samples, either undiluted or diluted as indicated. Bacteria (1.5 × 106 cfu; multiplicity of infection of 3:1), were added to the ASL and incubated for 2, 10, or 120 min at 37°C as indicated. Bacterial recovery was performed by centrifugation at 16,000 × g for 7 min, followed by resuspension in ice-cold sterile 0.2% (vol/vol) Triton X-100. Viable bacterial counts were determined as described above and compared to cfu counts at time 0 in order to determine percent bacterial survival.
Chemokinesis
Glass coverslips were coated with 50 μg/ml fibronectin for 45 min at room temperature then thoroughly rinsed with PBS. Coated coverslips were then mounted in cell chambers and overlaid with water-saturated silicon oil. One microliter of normal ASL, CF ASL, PBS, or tissue culture media (BEGM) was injected under the oil onto the surface of the coverslip. Although this is more technically challenging, injecting ASL under oil eliminated evaporative losses, allowing us to study the small ASL volumes generated in our in vitro systems. Neutrophils were then injected into this droplet with a Nanoliter 2000 injection apparatus (World Precision Instruments Inc., Sarasota, FL) and allowed to settle and attach for 5 min at 37°C. Images were collected with an Orca ER camera (Hamamatsu Photonics K.K., Hamamatsu, Japan) using Metafluor software (Universal Imaging Corporation, Molecular Devices Corporation, Sunnyvale, CA) once every 15 s for 25 min (100 images). fMLP (final concentration ∼ 10−7 M) was then injected into the droplet and cells imaged as before. The images acquired using Metafluor were imported into Metamorph (Universal Imaging Corporation, UK) and five individual cells from each experiment were randomly picked and tracked both with and without fMLP. Cell movement was expressed in μm/min.
Exocytosis
Neutrophils suspended in CF or normal (wild-type [WT]) ASL or PBS were pretreated with 1 μM cytochalasin D for 5 min at 37°C before stimulation with 10−6 M fMLP for an additional 5 min. Cells were washed once and incubated with monoclonal antibodies against CD63, CD66b, or FITC-CD11b in HBSS with 0.2% (wt/vol) sodium azide for 45 min at 4°C. Cells were then fixed with 2% (vol/vol) paraformaldehyde. Cells that had been exposed to unlabeled antibodies were washed once and incubated with Alexa Fluor 488–labeled secondary antibodies in HBSS solution for 45 min at 4°C. Cells were then washed and analysed by flow cytometry using a FACScan flow cytometer equipped with CELLQuest software (Becton Dickinson, San Jose, CA). The relative fluorescence intensity (RFI) represents the geometric mean of 10,000 cells gated on light scattering properties. The ratio between stimulated and unstimulated cells for CD63 and CD66b was calculated by dividing the RFI difference between stimulated cells stained with both primary and secondary antibodies and cells stained with only secondary antibodies by the RFI difference between unstimulated cells stained with both antibodies and cells stained with secondary antibodies only:
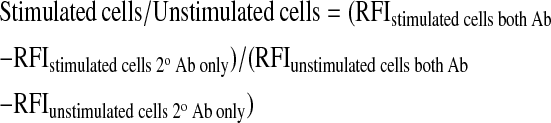 |
Phagocytosis
Two assays were used to assess the phagocytic ability of neutrophils. First, sheep erythrocytes were opsonized with rabbit anti-sheep IgG for 1 hr at 37°C with shaking, stained with Texas Red for 20 min at 4°C, and washed with PBS. Neutrophils and erythrocytes were combined (1:4 to 1:5) and kept on ice to allow binding but prevent ingestion. One microliter of the combined neutrophil and erythrocyte mixture was added to 10 μl of WT ASL, CF ASL, or control (PBS++), mixed well, sedimented in a microcentrifuge, and incubated for 5 min at 37°C. The cells were then gently resuspended and incubated a further 20 min, after which they were placed on ice and incubated with 1:10,000 goat anti-rabbit Alexa 488 for 15 min to label nonphagocytosed erythrocytes. Cells were then mounted (Dako mounting medium; Dako Corp., Carpinteria, CA) on a glass slide, visualized with a Leica DMIRB inverted fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany) and scored in terms of the number of phagocytosed erythrocytes per 100 neutrophils.
In a second assay, 1–2 × 108 P. aeruginosa were labeled with 1 mg/ml FITC in PBS, and placed on a rotator at room temperature for 30 min. After washing with PBS, bacteria were fixed with 2% EM-grade glutaraldehyde and placed on a rotator at room temperature for 30 min. After rinsing, bacteria and neutrophils were combined at a multiplicity of infection of 20:1, sedimented briefly, and incubated in 10 μl of normal ASL, CF ASL, or control buffer (PBS++ or USG-antibiotics) for 5 min 37°C. The cells were then gently resuspended and incubated a further 20 min, after which they were removed to ice. Five microliters was added to 5 μl 0.5% trypan blue (to quench extracellular fluorescence) on a glass slide with a glass coverslip, visualized with a Leica DMIRB inverted fluorescence microscope and scored for phagocytic index.
Oxidant Production (Respiratory Burst)
Neutrophils (0.5–1 × 106) were sedimented (10 s at 75% speed in a microcentrifuge) and resuspended in tissue culture medium, normal or CF ASL. One microliter of 10−4 M dihydrorhodamine was added to each tube followed by 1 μl of 10−6 M PMA or DMSO control, mixed, and incubated for 15 min at 37°C in the dark. Cells were sedimented and resuspended in 10 μl PBS and added to 400 μl PBS containing 4% paraformaldehyde and analyzed by flow cytometry on a FACScan flow cytometer equipped with CELLQuest software (Becton Dickinson, Palo Alto, CA).
Statistical Analysis
ANOVA was applied to evaluate the significance of differences using SPSS for Windows (version 12; SPSS Inc., Chicago, IL). If significant by ANOVA, correction for multiple comparisons was made using least squared differences. A P value of < 0.05 was considered to be significant. Data that were normally distributed are expressed as mean values ± 1 SEM.
RESULTS
Bacterial Killing Is Defective When Neutrophils Are Incubated in CF ASL
To determine if the ASL from CF epithelia modulates neutrophil-mediated bacterial killing, we compared the efficiency of bacterial killing by neutrophils incubated in CF or normal (WT) ASL. For these studies, 1.5 × 106 E. coli (used here as a well-characterized model for the study of bacterial phagocytosis) were incubated with 0.5 × 106 neutrophils in CF or normal ASL. The ASL was diluted 250-fold based on preliminary experiments that demonstrated extremely rapid (< 1 min) killing of bacteria in undiluted ASL from both normal (control) and CF epithelia. Under the conditions of this assay, bacteria may continue to proliferate and thus greater than 100% “survival” may be noted. After 2 min of incubation at 37°C, significantly fewer bacteria were killed in the CF ASL group compared with the normal ASL group (data not shown). This difference was also evident at 10 min (Figure 2), at which time 101 ± 9.2% of the initial bacteria remained viable in CF ASL as compared to 67 ± 6.4% in normal ASL. These data reflect the results of a minimum of six independent experiments with ASL from three different normal (control) and CF epithelial cell isolates. From these observations we conclude that bacterial killing is deficient when neutrophils are incubated in ASL from CF as compared to normal epithelial cells.
Figure 2.

Neutrophil-associated bacterial killing is reduced in CF ASL. Quantitative bacterial survival after incubating bacteria and neutrophils (ratio of 3:1) for 10 min at 37°C in normal (WT) ASL and cystic fibrosis (CF) ASL at 1/250 dilution. Data shown as mean ± SEM are from a minimum of six independent experiments. For each experiment, the colony counts were averaged from three plates. *P < 0.05.
It is noteworthy that the experimental protocol was designed to minimize the amount of residual antibiotics present in the ASL, as they could clearly influence bacterial survival. First, before the air–liquid interface culture, epithelial cells were washed multiple times with antibiotic-free culture medium and the apical surface aspirated to near dryness, allowing fresh ASL to accumulate. Second, the epithelial monolayers were cultured for a minimum of 1 wk in antibiotic-free medium, during which time the apical surface of the epithelial cells was washed with antibiotic-free medium and aspirated to near dryness every other day. The basal culture medium was also changed every second day and replaced with antibiotic-free medium. To control for the possible presence of residual antibiotics, we sampled the medium from the basolateral side of the wells from CF and normal epithelial cell cultures and determined that this medium had no detectable antimicrobial activity (data not shown). This argues strongly against the possibility that residual antibiotics were responsible for the differential bacterial killing observed between CF and normal ASL.
Assessment of Neutrophil Antimicrobial Functions in ASL
To examine further the mechanism by which ASL influences bacterial killing, we assessed a variety of neutrophil properties relevant to their antimicrobial function including motility, oxidant production, exocytosis of granule contents, and phagocytosis. This necessitated the development of miniaturized assays (see Materials and Methods and Figure 1) to allow the comparison of neutrophil functions in very small amounts of CF and normal ASL. An effort was made in all cases to use minimally diluted or undiluted ASL, to reproduce the conditions prevailing in situ as closely as possible.
Chemokinesis
We first assessed neutrophil motility (chemokinesis) in response to the bacterially derived formyl peptide, fMLP, using a microscope-based live cell imaging system where cells were incubated in a minute droplet (1 μL) of CF or normal ASL trapped under a layer of silicone oil on a coverslip, maintained at physiologic temperature in a thermoregulated imaging chamber (Figure 3). The ASL was injected underneath the layer of silicone oil to minimize evaporative loss on the microscope stage. Subsequently, neutrophils were introduced into the aqueous phase using a micropipette and allowed to settle onto the glass coverslip, which had been previously coated with fibronectin (Figures 3A–3C). Differential interference contrast images were acquired every 15 s immediately before and for 10 min after addition of the chemoattractant. The migration velocity of individual cells was determined using image analysis software (Metamorph; Universal Imaging Corporation). The rates of spontaneous migration of unstimulated neutrophils in either normal or CF ASL were similar in all groups (Figures 3D and 3E). After stimulation with fMLP, neutrophil velocities increased markedly, but were not different between cells bathed in CF or normal ASL. The velocities of neutrophils in ASL collected from Transwell cultures tended to be somewhat higher than in ASL from MilliCell inserts, although this difference did not achieve statistical significance.
Figure 3.

Neutrophil chemokinesis is not altered by CF ASL. (A) Low-power view of neutrophils injected into a droplet of ASL. (B) Isolated neutrophils have settled down after being injected into the ASL droplet and are captured at the start (T = 0) of the experiment. (C) Positions of the same neutrophils after 25 min (T = 25 min). Movements of the five labeled neutrophils are illustrated with dotted white lines. (D) Quantitative velocities of neutrophils when stimulated (+) or not stimulated (−) with fMLP in PBS, normal (WT) ASL or cystic fibrosis (CF) ASL from MilliCell culture wells. (E) Quantitative velocities of unstimulated (−) or fMLP-stimulated (+) neutrophils in PBS, normal (WT) ASL, or CF ASL from Transwell culture wells. Data shown as mean ± SEM are from four independent experiments conducted on different days with cells from different donors.
Exocytosis of Granules
Secretion (exocytosis) of granule-associated proteolytic enzymes and antimicrobial peptides and proteins is essential for efficient microbial killing (31). We used flow cytometry to assess exocytosis by quantifying cell surface expression of markers of defined secretory granule types. As before, a miniaturized assay had to be devised to assess secretion in the very small volumes of ASL collected from the cultures (see Materials and Methods for details). CD63 was used as a marker of primary (azurophilic) granules, CD66b as a marker of secondary or specific granules, and CD11b as a marker of both secondary granules and secretory vesicles (32). As illustrated in Figure 4, there was a trend towards decreased agonist-induced secretion in neutrophils incubated in CF ASL compared with normal ASL, but these differences did not attain statistical significance. The mean ratio of stimulated to unstimulated cell fluorescence intensities (± SEM) for CF and normal ASL, respectively, were as follows: for CD11b 1.7 (± 0.31) and 2.3 (± 0.36), for CD63 1.7 (± 0.24) and 2.0 (± 0.41), for CD66b 3.5 (± 0.60) and 4.9 (± 1.88).
Figure 4.

Neutrophil exocytosis is not altered by CF ASL. (A) Flow cytometric assessment of exocytosis of neutrophil secondary granules using monoclonal antibodies against CD66b. Quiescent cells are indicated by the gray curve (filled underneath) and fMLP-stimulated cells are indicated by the black curve. The dot plot illustrated the light scattering properties of the neutrophil population. (B) Ratio of relative fluorescence intensity of unstimulated (−) and stimulated (+) neutrophil populations in media (BEGM), normal (WT) ASL, and CF ASL detected with antibodies to CD63 (primary granules), CD66b (secondary granules), and CD11b (secondary granules and secretory vesicles) antibodies. Data shown as mean ± SEM are from eight independent experiments for those labeled with CD66b antibodies and four independent experiments for those labeled with CD63 and CD11b antibodies.
Phagocytosis
Phagocytosis (ingestion) of microbial pathogens is essential for proficient bacterial killing (33). As an initial approach to examine the effects of ASL on neutrophil phagocytosis, IgG-opsonized sheep red blood cells were used as phagocytic prey. This system uses a combination of fluorescent tags and hypotonic lysis of extracellular erythrocytes to enable unequivocal differentiation of internalized (phagocytosed) prey from surface-bound prey (see Materials and Methods). To avoid dilution of the ASL, 1 μl of a concentrated mixture of neutrophils and phagocytic targets was added to 10 μl of freshly collected ASL. Internalization was quantified as the phagocytic index (number of particles ingested per 100 cells; Figures 5A and 5B). A minimum of 100 neutrophils were assessed for each condition, and the results represent experiments performed on six different days. As illustrated in Figure 5E, no differences were noted in the phagocytic ability of neutrophils incubated in ASL collected from CF or normal cultures or in PBS.
Figure 5.

Neutrophil phagocytosis is not altered by CF ASL. Two types of phagocytic prey were used to assess neutrophil phagocytosis: sheep red blood cells (sRBCs) opsonized with IgG (A, B, and E), and fixed fluorescent P. aeruginosa (C, D, and F). (A, B, and E) The number of neutrophils with 1, 2, 3 or > 3 red blood cells phagocytosed was enumerated and the mean percentage ± SEM of neutrophils in each group is displayed for neutrophils in PBS, normal (WT) ASL, and CF ASL (E). Approximately 100 neutrophils were counted for each condition on six different days. Arrowheads indicate internalized prey (A), small arrow indicates nonphagocytosed sRBCs (A and B). (C, D, and F) Phagocytosis of P. aeruginosa by neutrophils incubated in PBS, normal (WT) ASL, or CF ASL was also performed. Results are from four independent experiments on four different days and are displayed as mean phagocytic index ± SEM (E). Inserts show a DIC image (C) and corresponding fluorescent image (D) of engulfed P. aeruginosa (arrowheads).
We next assessed the ability of neutrophils to engulf P. aeruginosa, a pathogen that is important in CF. To maximize the relevance of these studies, we used a clinical isolate of P. aeruginosa cultured from a patient with CF. Preliminary experiments revealed that neutrophils ingested and degraded live bacteria very rapidly, making quantification of internalized bacteria difficult due to intracellular fragmentation. To circumvent this problem, we first briefly fixed these bacteria and then labeled them with FITC. Under these conditions, neutrophils readily internalized P. aeruginosa and single intracellular bacteria could be easily identified by fluorescence microscopy (Figures 5C and 5D). Under these conditions, there was no difference in the efficiency of phagocytosis between neutrophils incubated in normal or CF ASL (Figure 5F).
Respiratory Burst
Bacterial killing is also dependent on the ability of neutrophils to generate reactive oxygen species by a process termed the “oxidative burst” (34). To assess this important aspect of phagocyte function, flow cytometry was used to quantify the oxidant-mediated conversion of nonfluorescent DHR to its oxidized and fluorescent derivative, rhodamine 123 (35). For these studies, PMA was used as an effective agonist of the neutrophil NADPH oxidase. Each experiment was repeated on at least four separate days. Figure 6B illustrates that no differences were apparent in oxidant production in neutrophils incubated in ASL from CF or normal epithelial cultures or in PBS.
Figure 6.

Respiratory burst in neutrophils is not altered by CF ASL. (A) Fluorescence measurement of reactive oxygen species (ROS) production by neutrophils, when PMA-stimulated (black curve), or quiescent (gray curve filled underneath). The light-scattering properties of the neutrophil population are illustrated by the inserted dot plot. (B) Graphical quantification of DHR fluorescence of PMA-stimulated (+) or quiescent (−) neutrophils, in PBS, normal (WT) ASL, or CF ASL. Data are shown as mean ± SEM and are from five independent experiments.
Direct Bactericidal Effects of ASL
The experiments detailed above indicated that, while bacterial killing was compromised under conditions in which bacteria were incubated with neutrophils in ASL collected from CF cultures compared to normal (WT) ASL, no significant differences were detected in the native antimicrobial functions of neutrophils in the two solutions. While there was a trend toward enhanced secretion in neutrophils incubated in normal ASL compared to CF ASL, it is unlikely that this alone accounts for the variation seen in bactericidal effects. This raised the possibility that the defect was attributable to differences in the intrinsic bactericidal properties of the ASL itself. To test this possibility, the survival of bacteria incubated in ASL derived from normal and CF cultures was compared, in the absence of neutrophils. Parallel measurements were performed using water as a control, since PBS was found to impair slightly bacterial survival and growth. For these studies, several types of bacteria were used. Initial studies used a laboratory strain of E. coli to facilitate development of the system. Using these bacteria, after 10 min of incubation in ASL, the number of remaining viable bacteria was quantified by serial dilution and quantitative agar plate culture, and is expressed as a percentage of the number present at time zero. A minimum of triplicate assays for each condition was performed and repeated on three separate days. At low dilution (1/125), 52 ± 2.3% of bacteria in the normal ASL survived as compared to 81% ± 2.5% in the CF ASL group and 100% ± 3% in the sterile water group. These differences were statistically significant (p < 0.05). At higher dilution (1/250), the differences were still apparent, with 76% ± 2.2%, 89% ± 2.2%, and 105% ± 2.2% bacterial survival in normal ASL, CF ASL, and sterile water, respectively (see Figure 7).
Figure 7.

More E. coli survive in CF ASL than in normal (WT) ASL. Survival of E. coli DH5α was quantified in sterile water (H2O), normal ASL (WT), and CF ASL at two different dilutions (1/125 and 1/250) after 10 min of incubation at 37°C. Data shown as mean percent survival (based on baseline colony counts at time 0) ± SEM are from 12 independent experiments for normal (WT) ASL, 15 independent experiments for CF ASL, and 6 independent experiments for H2O. For each experiment, the colony counts were averaged from three plates. *P < 0.05 compared with the CF ASL group.
To examine the antibacterial properties of ASL against bacteria more relevant to CF, we extended the studies to examine ASL-mediated bacterial killing of clinical isolates of S. aureus and P. aeruginosa. The latter was isolated from a patient with CF. Initial experiments revealed that the clinical isolates were much more resistant than E. coli to killing by ASL. Consequently, we used a higher concentration of ASL (1:50 dilution) and a longer period of incubation (2 h) for these experiments. Under these conditions, bacteria exposed to normal ASL had significantly less survival than bacteria grown in CF ASL or in control medium. In normal ASL, 31.5% of the initial number of S. aureus was present after 2 h, compared with 439% in the CF ASL and 596% in the control medium (see Figure 8A). These results represent four experiments performed on separate days. For P. aeruginosa, only 16% of bacteria survived in normal ASL, compared with 123% in CF ASL and 110% in the control medium (see Figure 8B). These data represent the mean of five independent experiments. These results indicate that there is a generalized bacterial killing defect intrinsic to CF ASL and that this defect is applicable to bacteria that are relevant to the pathogenesis of CF lung disease.
Figure 8.

More bacteria survive in CF ASL than normal (WT) ASL. Survival of (A) S. aureus and (B) P. aeruginosa was quantified in media, normal ASL (WT) and CF ASL (CF) at a 1:50 dilution after 2 h incubation at 37°C. Data shown as mean percent survival (based on baseline colony counts at time 0) ± SEM are from four independent experiments on four separate days for S. aureus and from five independent experiments on five separate days for P. aeruginosa. For each experiment, the colony counts were averaged from three plates. *P < 0.05 compared with the CF ASL group.
DISCUSSION
The current studies address the notion that abnormalities in the composition of the ASL in CF compromise the innate immune response and, specifically, bactericidal functions of neutrophils. To study this possibility, we implemented an in vitro system of primary cultures of human CF and normal human airway epithelial cells grown in an air–liquid interface on semipermeable tissue culture inserts. This model system enabled us to collect small (microliter) quantities of ASL from these cultures. In parallel, we developed techniques to assess the microbicidal functions of human peripheral blood neutrophils in very small volumes of ASL, using a combination of live microscopic imaging and flow cytometry. We observed that bacterial killing was deficient under conditions in which neutrophils were incubated in CF compared with normal ASL. However, further experiments delineated that neutrophil antimicrobial functions were not compromised by ASL from CF cultures, but rather that the primary defect was impaired bacterial killing by the ASL itself.
CF results from mutations in the CFTR gene, leading to the absence or deficiency of the membrane protein that is primarily responsible for chloride conductance in normal airway epithelia. This defect somehow predisposes the lungs to chronic bacterial infection, most notably with Pseudomonas species. One potential explanation for the selective susceptibility of the CF airway to infection with P. aeruginosa, proposed by Pier, is that CFTR itself is an epithelial cell receptor for the bacterium (18). According to this concept, subsequent to binding, the bacteria are engulfed by the epithelium and cleared through sloughing of cells from the lung. However, this paradigm does not explain why CF lungs are colonized with non-Pseudomonas species such as S. aureus, Haemophilus influenzae, and in some geographic locations Burkholderia cepacia, or why Pseudomonas and other bacteria are not killed by innate defenses found in the ASL. Data from our current study build on this foundation and suggest that S. aureus and P. aeruginosa are both relatively resistant to killing by ASL collected from CF epithelia, and thus may survive and proliferate in the milieu of the CF airway.
The defect in microbial killing observed in CF has been attributed to a number of factors in the ASL that can be considered in three categories: (1) a primary neutrophil defect, (2) alterations in ASL composition that secondarily lead to compromised neutrophil function (i.e., a secondary neutrophil defect), or (3) a primary defect in the antimicrobial properties of the ASL itself (i.e., a neutrophil-independent defect). It is recognized, however, that these are not mutually exclusive and that all three types of defect may coexist and contribute to the predisposition to lung inflammation/infection in CF. With respect to the first possibility, to the extent that neutrophils from patients with CF do express small amounts of CFTR, it is conceivable that they have intrinsic functional defects related to the dysfunction or absence of this protein. Indeed, one report suggests that the enhanced phagocyte oxidase activity observed in neutrophils from patients with CF, although harmful to homozygotes by contributing to lung damage, may confer a selective advantage to heterozygotes and may provide an explanation for the relatively high frequency of CFTR mutations in the general population (36). In addition to enhanced oxidase activity, stimulated CF neutrophils have augmented secretion of proinflammatory cytokines such as IL-8, lipoxygenase products, and granule enzymes (myeloperoxidase and elastase) compared with normal neutrophils (27, 37–40). Alternatively, CFTR dysfunction may alter intracellular ionic concentrations and/or the pH of the neutrophils and thus interfere with their ability to release enzymes normally (27, 28). Low cytosolic chloride concentrations can also independently modulate protein phosphorylation, actin polymerization, secretion of lysozyme, and the respiratory burst in neutrophils (41). Other neutrophil defects may be explained by in vivo priming due to exposure of neutrophils to increased levels of endogenous inflammatory mediators that are a consequence of extensive pulmonary infection and inflammation (37, 38). However, proinflammatory mediators can also paradoxically lead to depressed neutrophil function under some circumstances through receptor desensitization (42, 43), and this may explain in part the reduced bacterial killing noted in CF despite increased levels of inflammatory mediators. Further, L-selectin shedding, a normal event in the process of neutrophil adhesion and activation, is decreased in CF neutrophils, suggesting a defect in their inflammatory responsiveness (44). Thus, as there is evidence that CFTR may independently influence neutrophil function, and given that variable degrees of endogenous inflammation may unpredictably prime neutrophils isolated from patients with CF, we chose to study neutrophils from normal controls in order to focus on the potential effects of the ASL on neutrophil function.
In the present study, we observed initially that bacterial killing was reduced when neutrophils were incubated together with bacteria in ASL from CF cultures (Figure 2). In order to explore the possibility of a secondary neutrophil defect, we examined a variety of their antimicrobial functions including chemokinesis, granule secretion, phagocytosis, and oxidant production in CF and normal ASL. We observed that each of these critical antimicrobial functions was similar whether the milieu was CF or normal ASL (Figures 3–6). While we noted a trend toward reduced granule exocytosis in neutrophils incubated in CF ASL, this difference was not statistically significant and is unlikely by itself to explain the defective bacterial killing observed in our studies. Though this notion remains controversial, it has been suggested that salt concentrations may be altered in ASL from CF tissues, and investigators have proposed that high salt concentrations may directly influence neutrophil function (45, 46). There is also evidence that elevated chloride in particular can both enhance neutrophil release of IL-8 and reduce killing of Pseudomonas, presumably through disruption of phagocytosis (47). Alternatively, low sodium concentrations may alter some neutrophil antimicrobial functions, specifically killing of P. aeruginosa (48). However, these reports are not universally accepted, as other investigators have not observed defective neutrophil-mediated killing of P. aeruginosa or S. aureus in conditions of either high or low sodium concentrations (45). In the current study, no significant defect in neutrophil function was observed in CF ASL, and thus a secondary neutrophil defect due to abnormalities induced by CF ASL is not supported by our experiments and does not explain our findings.
With respect to the third possibility, we examined the intrinsic (direct) bactericidal activity of isolated ASL from normal and CF epithelial cells. These studies revealed that ASL from normal epithelial cells exhibited potent and rapid microbial killing activity toward both Gram-positive (S. aureus) and Gram-negative (E. coli and P. aeruginosa) bacteria even when substantially diluted (Figures 7 and 8). By contrast, the bacterial killing activity of CF ASL was deficient. These data indicate that normal ASL has potent microbial killing activity that is poorly sensitive to changes in the salt concentration, because it is present even when diluted up to 250 times with water. We specifically examined the ability of ASL to kill clinical isolates of S. aureus and P. aeruginosa to ensure the relevance of this system to CF pathogens. As these bacteria are more resilient than E. coli, we used more concentrated ASL (1:50) for a longer period of time (2 h). In this situation, CF ASL was again found to be less effective in bacterial killing when compared with normal ASL (Figure 8). Taken together, these observations suggest that the primary innate antimicrobial defect in CF is not one of altered phagocytic function, but rather is a consequence of a defect in the intrinsic antimicrobial properties of the ASL itself.
In developing this experimental system, we carefully considered the possibility that residual antibiotics might be a confounding variable in our system. To minimize this possibility, the epithelial cultures were washed multiple times and cultured for weeks in antibiotic-free medium. Moreover, CF epithelia require four antibiotics to prevent bacterial contamination in the initial phase of culture, whereas normal epithelia require only two. Despite the use of additional antibiotics in the CF epithelia, the ASL derived from these cultures was always much less effective in bacterial killing than that of control epithelia.
In the current studies, the fact that differences in bactericidal activity were preserved after dilution of the ASL argues against the possibility of a salt-sensitive mechanism for bacterial killing in ASL. Our observations, however, do not preclude the possibility that there are additional salt-dependent antimicrobial effects in the milieu of the ASL in vivo. In fact, the “high salt” hypothesis suggests that defective chloride ion absorption leads to increased ionic strength of the CF ASL (49). Further, such alterations in ionic strength may interfere with defensin function and thus compromise microbial killing (50–52). However, recent data indicate that the salt concentration of the CF ASL is normal and that the principal defect in CF lies in the regulation of the volume of the ASL (8, 53–55). The “low volume” ASL hypothesis asserts that while the ionic composition of CF ASL is normal, its volume is low as a result of enhanced sodium absorption (55). In this regard, mice overexpressing ENaC, a condition that results in increased sodium absorption in the airways, develop some of the clinical features of CF lung disease, including inflammation and poor bacterial clearance (46). These data strongly support the concept that the primary defect in the CF lung relates to altered ASL volume rather than altered ionic composition.
Our observation that there are salt-independent defects in the antimicrobial function of CF ASL is compatible with reports by other investigators. For example, Bals and coworkers, using a laboratory strain of P. aeruginosa (PAO1), previously described that CF ASL is lacking an antimicrobial factor that is independent of the salt concentration and can be restored by CFTR gene transfer (56). Our observations confirm and extend these findings by demonstrating that the impaired bactericidal activity in the CF microenvironment is not related to neutrophil functional defects and is applicable to clinically relevant bacterial pathogens. In addition, we report that the ASL-mediated bactericidal activity is preserved at up to 50-fold dilutions for P. aeruginosa and S. aureus and up to 250-fold for E. coli. The nature of the bactericidal activity in ASL remains unknown. In this regard, CFTR may regulate other properties of the ASL in addition to sodium and chloride conductance that may impact on microbial defense. For example, CFTR regulates ASL pH through exchange of bicarbonate for chloride. Though conflicting reports exist, it has been suggested that in CF the ASL is acidified (57). However, this potential hyperacidity cannot explain our in vitro findings, as the ASL retains its killing ability even when diluted in large volumes of well-buffered solution (cell culture medium or PBS).
CFTR mutations can also result in altered secretion by airway epithelial cells. There is evidence that CF cells have reduced mucin and protein secretion. It has been suggested that post-translational protein modifications such as sialylation may be influenced by CFTR (58), and thus a number of cell surface proteins may exhibit altered function in the presence of defective CFTR. This is consistent with work by Bals and colleagues, who suggest that the antimicrobial defect in CF ASL is protease-sensitive but is not due to alterations in the amount or function of lactoferrin, lysozyme, hBD-1, hBD-2, or LL-37 (56). In patients with CF, these and other innate immune molecules may be altered secondarily by leukocyte-derived products such as elastase that can cleave complement components (16) and cathepsins that inactivate lactoferrin (17). A key issue is that while decreased levels of innate immune molecules observed in vivo in CF (such as low lactoferrin) may be attributable to excessive proteinase action, in our simple in vitro system these neutrophil-derived factors (elastase or cathepsins) are not present. Thus any defect in innate immunity in our model is intrinsic to the epithelial cells and their secreted products. Identification of the defective factor(s) may provide an attractive target for pharmacotherapy, especially since this factor is bactericidal after extensive dilution.
The bacterial killing defect in ASL may also be related to the ability of CFTR to modulate the redox state of ASL through secretion of the antioxidant glutathione (GSH). This theory is based on several key observations. First, CFTR can function as a “permeation pathway” allowing GSH transport into the ASL (59). Second, although ASL normally contains a high concentration of GSH (60), in CF it is greatly reduced (61). Finally, a reduction of GSH in the ASL leads to oxidative stress and can result in conditions very similar to those present in CF, including impaired mucolysis, reduced nitric oxide production and importantly, altered leukocyte function manifested as reduced bactericidal effectiveness (62). In addition to deleterious effects on neutrophils, innate immune molecules such as surfactant proteins can be oxidized, leading to their dysfunction (63). Jointly, these observations suggest that the altered redox state of CF ASL may modulate an antimicrobial factor(s) leading to a defective bacterial killing. Not surprisingly, antioxidant therapy and GSH replacement have been advocated and attempted in patients with CF (64, 65).
In summary, the current experiments demonstrate that CF ASL is intrinsically deficient in one or more antimicrobial factors that are present in normal ASL. These factors remain active against clinically relevant bacteria, S. aureus and P. aeruginosa, despite dilution, and their activity is therefore independent of salt concentration. Further, ASL collected from CF epithelia does not compromise neutrophil antimicrobial functions when studied in vitro, supporting the notion that there are neutrophil-independent defects in bacterial killing in CF. Additional studies will be required to delineate the nature of the CF-related antimicrobial defect.
This work was supported by operating grants from the Canadian Cystic Fibrosis Foundation to G.P.D. and S.G., and from the National Institutes of Health (P50 DK49096-06 SCOR) to G.P.D. and S.G. T.J.M. is supported by a Duncan Gordon Fellowship from the Hospital for Sick Children Foundation and a Fellowship from the Canadian Institutes for Health Research. G.P.D. is the recipient of a Tier 1 Canada Research Chair. S.G. holds the Pitblado Chair in Cell Biology from the Hospital for Sick Children. L.L.B. is a CIHR New Investigator and work in her laboratory is funded by the CCFF's BREATHE program. J.V.K. holds a joint Studentship from the CCFF and the CIHR Training Program in Cell Signaling in Mucosal Inflammation and Pain.
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Originally Published in Press as DOI: 10.1165/rcmb.2005-0146OC on November 15, 2005
References
- 1.Davis PB. Cystic fibrosis. Pediatr Rev 2001;22:257–264. [DOI] [PubMed] [Google Scholar]
- 2.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science 1989;245:1073–1080. [DOI] [PubMed] [Google Scholar]
- 3.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1996;154:1229–1256. [DOI] [PubMed] [Google Scholar]
- 4.Elborn JS, Shale DJ, Britton JR. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax 1991;46:881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003;168:918–951. [DOI] [PubMed] [Google Scholar]
- 6.Corey M, Farewell V. Determinants of mortality from cystic fibrosis in Canada, 1970–1989. Am J Epidemiol 1996;143:1007–1017. [DOI] [PubMed] [Google Scholar]
- 7.Schwiebert EM, Erik M, Dale J, Fuller C. Cystic fibrosis: a multiple exocrinopathy caused by dysfunctions in a multifunctional transport protein. Am J Med 1998;104:576–590. [DOI] [PubMed] [Google Scholar]
- 8.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998;95:1005–1015. [DOI] [PubMed] [Google Scholar]
- 9.Davis PB, editor. Pathophysiology of the lung disease in cystic fibrosis: lung biology in health and disease. New York: Marcel Dekker; 1993.
- 10.Cheng PW, Boat TF, Cranfill K, Yankaskas JR, Boucher RC. Increased sulfation of glycoconjugates by cultured nasal epithelial cells from patients with cystic fibrosis. J Clin Invest 1989;84:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelial fail to kill bacteria because of abnormal airway surface fluid. Cell 1996;85:229–236. [DOI] [PubMed] [Google Scholar]
- 12.Travis SM, Conway BA, Zabner J, Smith JJ, Anderson NN, Singh PK, Greenberg EP, Welsh MJ. Activity of abundant antimicrobials of the human airway. Am J Respir Cell Mol Biol 1999;20:872–879. [DOI] [PubMed] [Google Scholar]
- 13.Guggino WB. Cystic fibrosis and the salt controversy. Cell 1999;96:607–610. [DOI] [PubMed] [Google Scholar]
- 14.Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev 1999;79:S215–S255. [DOI] [PubMed] [Google Scholar]
- 15.Velsor LW, van Heeckeren A, Day BJ. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Physiol Lung Cell Mol Physiol 2001;281:L31–L38. [DOI] [PubMed] [Google Scholar]
- 16.McCormick LL, Karulin AY, Schreiber JR, Greenspan NS. Bispecific antibodies overcome the opsonin-receptor mismatch of cystic fibrosis in vitro: restoration of neutrophil-mediated phagocytosis and killing of Pseudomonas aeruginosa. J Immunol 1997;158:3474–3482. [PubMed] [Google Scholar]
- 17.Rogan MP, Taggart CC, Greene CM, Murphy PG, O'Neill SJ, McElvaney NG. Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J Infect Dis 2004;190:1245–1253. [DOI] [PubMed] [Google Scholar]
- 18.Pier GB. Role of the cystic fibrosis transmembrane conductance regulator in innate immunity to Pseudomonas aeruginosa infections. Proc Natl Acad Sci USA 2000;97:8822–8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med 1994;150:448–454. [DOI] [PubMed] [Google Scholar]
- 20.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995;151:1075–1082. [DOI] [PubMed] [Google Scholar]
- 21.Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R. The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr Pulmonol 1995;20:63–70. [DOI] [PubMed] [Google Scholar]
- 22.Noah TL, Black HR, Cheng PW, Wood RE, Leigh MW. Nasal and bronchoalveolar lavage fluid cytokines in early cystic fibrosis. J Infect Dis 1997;175:638–647. [DOI] [PubMed] [Google Scholar]
- 23.Kirchner KK, Wagener JS, Khan TZ, Copenhaver SC, Accurso FJ. Increased DNA levels in bronchoalveolar lavage fluid obtained from infants with cystic fibrosis. Am J Respir Crit Care Med 1996;154:1426–1429. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, Olinsky A, Phelan EM, Robertson CF, Phelan PD. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med 1997;156:1197–1204. [DOI] [PubMed] [Google Scholar]
- 25.Cantin A. Cystic fibrosis lung inflammation: early, sustained, and severe. Am J Respir Crit Care Med 1995;151:939–941. [DOI] [PubMed] [Google Scholar]
- 26.Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, Hiatt P, McCoy K, Wilson CB, Inglis A, et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol 2001;32:356–366. [DOI] [PubMed] [Google Scholar]
- 27.Witko-Sarsat V, Allen RC, Paulais M, Nguyen AT, Bessou G, Lenoir G, Descamps-Latscha B. Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol 1996;157:2728–2735. [PubMed] [Google Scholar]
- 28.Coakley RJ, Taggart C, Canny G, Greally P, O'Neill SJ, McElvaney NG. Altered intracellular pH regulation in neutrophils from patients with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 2000;279:L66–L74. [DOI] [PubMed] [Google Scholar]
- 29.Yoshimura K, Nakamura H, Trapnell BC, Chu CS, Dalemans W, Pavirani A, Lecocq JP, Crystal RG. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res 1991;19:5417–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karp PH, Moninger TO, Weber SP, Nesselhauf TS, Launspach JL, Zabner J, Welsh MJ. An in vitro model of differentiated human airway epithelia: methods for establishing primary cultures. Methods Mol Biol 2002;188:115–137. [DOI] [PubMed] [Google Scholar]
- 31.Nathan C. Points of control in inflammation. Nature 2002;420:846–852. [DOI] [PubMed] [Google Scholar]
- 32.Borregaard N. Current concepts about neutrophil granule physiology. Curr Opin Hematol 1996;3:11–18. [DOI] [PubMed] [Google Scholar]
- 33.Greenberg S, Grinstein S. Phagocytosis and innate immunity. Curr Opin Immunol 2002;14:136–145. [DOI] [PubMed] [Google Scholar]
- 34.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys 2002;397:342–344. [DOI] [PubMed] [Google Scholar]
- 35.Waddell TK, Fialkow L, Chan CK, Kishimoto TK, Downey GP. Potentiation of the oxidative burst of human neutrophils: a signaling role for L-selectin. J Biol Chem 1994;269:18485–18491. [PubMed] [Google Scholar]
- 36.Regelmann WE, Skubitz KM, Herron JM. Increased monocyte oxidase activity in cystic fibrosis heterozygotes and homozygotes. Am J Respir Cell Mol Biol 1991;5:27–33. [DOI] [PubMed] [Google Scholar]
- 37.Koller DY, Urbanek R, Gotz M. Increased degranulation of eosinophil and neutrophil granulocytes in cystic fibrosis. Am J Respir Crit Care Med 1995;152:629–633. [DOI] [PubMed] [Google Scholar]
- 38.Corvol H, Fitting C, Chadelat K, Jacquot J, Tabary O, Boule M, Cavaillon JM, Clement A. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 2003;284:L997–1003. [DOI] [PubMed] [Google Scholar]
- 39.Taggart C, Coakley RJ, Greally P, Canny G, O'Neill SJ, McElvaney NG. Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-alpha and interleukin-8. Am J Physiol Lung Cell Mol Physiol 2000;278:L33–L41. [DOI] [PubMed] [Google Scholar]
- 40.Saak A, Schonfeld W, Knoller J, Steinkamp G, von der Hardt H, Konig W. Generation and metabolism of leukotrienes in granulocytes of patients with cystic fibrosis. Int Arch Allergy Appl Immunol 1990;93:227–236. [DOI] [PubMed] [Google Scholar]
- 41.Grinstein S, Furuya W, Downey GP. Activation of permeabilized neutrophils: role of anions. Am J Physiol 1992;263:C78–C85. [DOI] [PubMed] [Google Scholar]
- 42.Dai Y, Dean TP, Church MK, Warner JO, Shute JK. Desensitisation of neutrophil responses by systemic interleukin 8 in cystic fibrosis. Thorax 1994;49:867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawrence RH, Sorrelli TC. Decreased polymorphonuclear leucocyte chemotactic response to leukotriene B4 in cystic fibrosis. Clin Exp Immunol 1992;89:321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell KJ, McRedmond J, Mukherji N, Costello C, Keatings V, Linnane S, Henry M, Fitzgerald MX, O'Connor CM. Neutrophil adhesion molecule surface expression and responsiveness in cystic fibrosis. Am J Respir Crit Care Med 1998;157:756–761. [DOI] [PubMed] [Google Scholar]
- 45.Verghese MW, Boucher RC. Effects of ion composition and tonicity on human neutrophil antibacterial activity. Am J Respir Cell Mol Biol 1998;19:920–928. [DOI] [PubMed] [Google Scholar]
- 46.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 2004;10:487–493. [DOI] [PubMed] [Google Scholar]
- 47.Tager AM, Wu J, Vermeulen MW. The effect of chloride concentration on human neutrophil functions: potential relevance to cystic fibrosis. Am J Respir Cell Mol Biol 1998;19:643–652. [DOI] [PubMed] [Google Scholar]
- 48.Mizgerd JP, Kobzik L, Warner AE, Brain JD. Effects of sodium concentration on human neutrophil bactericidal functions. Am J Physiol 1995;269:L388–L393. [DOI] [PubMed] [Google Scholar]
- 49.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996;85:229–236. [DOI] [PubMed] [Google Scholar]
- 50.Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997;88:553–560. [DOI] [PubMed] [Google Scholar]
- 51.Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest 1998;102:874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bals R, Goldman MJ, Wilson JM. Mouse beta-defensin 1 is a salt-sensitive antimicrobial peptide present in epithelia of the lung and urogenital tract. Infect Immun 1998;66:1225–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verkman AS, Song Y, Thiagarajah JR. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am J Physiol Cell Physiol 2003;284:C2–15. [DOI] [PubMed] [Google Scholar]
- 54.Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS. Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc Natl Acad Sci USA 2001;98:8119–8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 2004;23:146–158. [DOI] [PubMed] [Google Scholar]
- 56.Bals R, Weiner DJ, Meegalla RL, Accurso F, Wilson JM. Salt-independent abnormality of antimicrobial activity in cystic fibrosis airway surface fluid. Am J Respir Cell Mol Biol 2001;25:21–25. [DOI] [PubMed] [Google Scholar]
- 57.Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, O'Neal WK, Boucher RC. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA 2003;100:16083–16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol 2003;285:C1–18. [DOI] [PubMed] [Google Scholar]
- 59.Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am J Physiol 1998;275:C323–C326. [DOI] [PubMed] [Google Scholar]
- 60.Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol 1987;63:152–157. [DOI] [PubMed] [Google Scholar]
- 61.Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol 1993;75:2419–2424. [DOI] [PubMed] [Google Scholar]
- 62.Hudson VM. Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic Biol Med 2001;30:1440–1461. [DOI] [PubMed] [Google Scholar]
- 63.Davis IC, Zhu S, Sampson JB, Crow JP, Matalon S. Inhibition of human surfactant protein A function by oxidation intermediates of nitrite. Free Radic Biol Med 2002;33:1703–1713. [DOI] [PubMed] [Google Scholar]
- 64.Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, Ratjen F, Mullinger B, Huber RM, Maier K, et al. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med 2004;169:822–828. [DOI] [PubMed] [Google Scholar]
- 65.Cantin AM. Potential for antioxidant therapy of cystic fibrosis. Curr Opin Pulm Med 2004;10:531–536. [DOI] [PubMed] [Google Scholar]