

Abstract
The genetic basis of acute lung injury (ALI) is poorly understood. The myosin light chain kinase (MYLK) gene encodes the nonmuscle myosin light chain kinase isoform, a multifunctional protein involved in the inflammatory response (apoptosis, vascular permeability, leukocyte diapedesis). To examine MYLK as a novel candidate gene in sepsis-associated ALI, we sequenced exons, exon–intron boundaries, and 2 kb of 5′ UTR of the MYLK, which revealed 51 single-nucleotide polymorphisms (SNPs). Potential association of 28 MYLK SNPs with sepsis-associated ALI were evaluated in a case-control sample of 288 European American subjects (EAs) with sepsis alone, subjects with sepsis-associated ALI, or healthy control subjects, and a sample population of 158 African American subjects (AAs) with sepsis and ALI. Significant single locus associations in EAs were observed between four MYLK SNPs and the sepsis phenotype (P < 0.001), with an additional SNP associated with the ALI phenotype (P = 0.03). A significant association of a single SNP (identical to the SNP identified in EAs) was observed in AAs with sepsis (P = 0.002) and with ALI (P = 0.01). Three sepsis risk-conferring haplotypes in EAs were defined downstream of start codon of smooth muscle MYLK isoform, a region containing putative regulatory elements (P < 0.001). In contrast, multiple haplotypic analyses revealed an ALI-specific, risk-conferring haplotype at 5′ of the MYLK gene in both European and African Americans and an additional 3′ region haplotype only in African Americans. These data strongly implicate MYLK genetic variants to confer increased risk of sepsis and sepsis-associated ALI.
Keywords: MYLK/MLCK, genetic association, SNP, ALI, sepsis
Acute lung injury (ALI) is characterized by profound inflammation, increased vascular permeability, and alveolar flooding, a combination of events which frequently results in acute respiratory failure. The incidence of ALI in the United States (17–64 per 100,000 person-years) is higher than in other developed countries, with mortality rates for patients with acute respiratory distress syndrome, the more severe form of ALI, ranging from 34–58% (1, 2). The risk of ALI appears to be disproportionately higher in African Americans, an observation which cannot be explained by socioeconomic factors alone (3), suggesting a genetic influence on susceptibility and outcome. Studies on the genetic basis of sepsis, the most common predisposing condition leading to ALI, are limited but have elucidated several candidates, including a single-nucleotide polymorphism (SNP) in the TNF promoter (−308) (4) and a promoter polymorphism in the CD14 gene (−260) (5). A single variant (insertion/deletion) in the gene encoding angiotensin-converting enzyme (ACE) is associated with ALI with patients homozygous for the deletion (and therefore carriers of the ACE DD genotype) appearing to be at high risk (6). Similarly, an SNP in the surfactant protein-B gene (1,580 C/T) is a risk factor for acute respiratory distress syndrome (7–9). Although intriguing, these studies are limited by a focus on single variants within a candidate gene, the absence of adequate methodologies, and the complete lack of validation in replicate independent populations. Despite these caveats, it remains widely believed that the identification of genetic polymorphisms in candidate genes may provide new insight into the molecular pathogenesis of sepsis and ALI and lead to the development of new diagnostic and therapeutic targets.
The obvious absence of available families with a history of ALI has precluded linkage analysis approaches for examining the genetic basis of ALI. As a result, studies have used the candidate gene approach based on extensive expression profiling, the selection of putative candidate genes emanating from pathway analysis, or reports of similar association in related disorders. Based upon a systematic interrogation of endothelial barrier properties under conditions of lung inflammation, a defining feature of sepsis and ALI, we have speculated that the gene encoding human myosin light chain kinase (MLCK), spanning 217 kb on chromosome 3q21, may represent a viable candidate gene involved in ALI susceptibility and disease. The human MYLK gene encodes 3 proteins within a single gene, including the nonmuscle and smooth muscle MLCK isoforms (10–12). In addition, using a separate promoter in an intron in the 3′ region, MYLK encodes telokin, a small protein identical in sequence to the C-terminus of MYLK that is independently expressed in smooth muscle and functions to stabilize unphosphorylated myosin filaments (13). A pseudogene is located on the p arm of chromosome 3 (14). Seven transcript variants that produce seven isoforms of the calcium/calmodulin-dependent enzyme, as well as two transcripts that produce two isoforms of telokin, have been identified. We have previously demonstrated that the nonmuscle MLCK isoform encoded by MYLK is a multifunctional protein centrally involved in multiple aspects of the inflammatory response, including apoptosis, vascular barrier regulation and permeability, and leukocyte diapedesis (15–18). MLCK is a molecular target in ventilator-associated lung injury (19), and in vivo studies in MYLK mutant mice demonstrate an essential role for MYLK in murine sepsis, again implicating MYLK as a potential drug-discovery target (20). Despite this compelling rationale, common variants in MYLK have yet to be identified and directly implicated as a major causal allele.
To characterize the functional role of the MYLK gene as a potential ALI candidate, we performed direct sequencing of the MYLK gene, which contains 32 exons, exon–intron boundaries (including 100 bases of intronic sequence on either side), and 2 kb of the 5′ UTR in 36 subjects (European Americans [EAs] and African Americans [AAs]) with sepsis, sepsis-associated ALI, or healthy control subjects and identified 51 SNPs. We combined our SNP discovery and data available from public resources to construct two sets of biallelic markers with minor allelic frequency (MAF) ⩾ 10% among EAs and AAs, and tested for association in our case-control–designed study population stratified by ethnicity.
MATERIALS AND METHODS
Human MYLK Gene Sequencing and Polymorphism Analysis
Genomic DNA was extracted and purified from lymphocyte buffy coats removed from 20 ml of EDTA-treated blood using a commercial kit (PUREGENE; Gentra Systems, Inc., Minneapolis, MN). DNA was stored at −20°C in tris-EDTA buffer until further use. The full-length MYLK gene (GeneBank Accession no.: U48959; 217.6 kb containing 32 exons) (11) was assessed by direct sequencing of PCR amplicons using individual DNA samples from 36 subjects with either ALI (n = 12) or sepsis (n = 12) or who were healthy control subjects (n = 12) comprised equally of EAs and AAs. PCR primers were designed to amplify exons, exon–intron boundaries (including 100 bases of intronic sequence on either side), 3′ UTR, and 2 kb upstream of 5′ UTR. Primers were synthesized on the ABI 3948 DNA synthesizer (ABI, Foster City, CA). Sequencing was performed on an ABI 3700 sequencer, following standard protocols (21, 22), at the DNA Analysis Facility, Johns Hopkins University (Baltimore, MD). SNPs were identified by manual inspection using Sequencher 4.1 (Gene Codes Corp., Ann Arbor, MI). Representative sequence of gene encoding human EC MYLK with GenBank accession no. U48959.2 (GI:7239695) was used as reference sequence for the numbering of residues. Nucleotide numbering uses the A of the ATG translation initiation start site as nucleotide +1 for coding SNPs. Positions are given in the corresponding intron/exon based on U48959.2, and the reference intron sequence can be found in genomic contig NT_005543 for intronic variations. Positions for SNPs in 5′ UTR are counted upstream from the ATG, which is found in exon 2.
Patient Cohort Recruitment and Demographics
A case-control designed study of sepsis and ALI was reviewed and approved by the Johns Hopkins Institutional Review Board. DNA was obtained from four clinical sites by members of the Consortium to Evaluate Lung Edema Genetics (CELEG), a collaborative enrollment network spearheaded by investigators from Johns Hopkins University and Medical College of Wisconsin. The primary population was a European-American dataset, which included (1) patients with sepsis-associated ALI (n = 92), (2) patients with sepsis alone (n = 114), and (3) healthy control subjects (n = 85). An African-American dataset served as the replicate population (46 with ALI, 51 with sepsis, and 61 control subjects) (Table 1). Definitions of sepsis and ALI were in accordance with the American College of Chest Physicians (23) and Society of Critical Care Medicine Consensus statements (24). Admission to the intensive care units was a requirement for enrollment, and virtually all patients experienced severe sepsis or septic shock. The definition of ALI required a PaO2/FiO2 ratio of < 300 and bilateral pulmonary infiltrates on a chest radiograph, with sepsis as the predisposing illness (patients with a non-sepsis cause of ALI were not enrolled). Exclusion criteria were allogeneic bone marrow transplant and severe leukopenia (WBC < 1,000/μl). APACHE II scores were recorded to ensure comparability of the severity of illness between ALI and sepsis groups (25). Healthy control subjects were defined as individuals without any recent acute illness or any chronic illness requiring a physician's care.
TABLE 1.
DEMOGRAPHIC AND CLINICAL CHARACTERISTICS OF THE STUDY POPULATION (CELEG)
| Patients with ALI
|
Patients with Sepsis
|
Healthy Control Subjects
|
||||
|---|---|---|---|---|---|---|
| EA | AA | EA | AA | EA | AA | |
| N | 92 | 43 | 99 | 51 | 85 | 61 |
| Sex (M/F) | 55/37 | 24/19 | 59/40 | 33/18 | 41/44 | 25/36 |
| Age | 53.1 ± 16.6 | 47.9 ± 17.0 | 60.7 ± 19.2 | 58.1 ± 15.8 | 32.8 ± 10.5† | 35.0 ± 10.8† |
| APACHE II | 24.8 ± 7.7 | 25.2 ± 7.8 | 22.1 ± 8.6 | 25.7 ± 7.3 | N.A. | N.A. |
| Survival, % | 58.3† | 59.1* | 74.7 | 73.7 | 100 | 100 |
| Cancer, % | 19.7 | 4.5 | 28.0 | 15.8 | N.A. | N.A. |
| CLD, % | 18.0 | 4.8 | 10.7 | 0 | N.A. | N.A. |
| ESRF, % | 13.1 | 4.5 | 12.0 | 15.8 | N.A. | N.A. |
| COPD, % | 8.2 | 9.1 | 16.2 | 10.5 | N.A. | N.A. |
| Alcoholism, % | 14.8 | 28.6 | 9.3 | 16.7 | N.A. | N.A. |
| Diabetes, % | 21.3 | 13.6 | 16 | 36.8 | N.A. | N.A. |
| CHF, % | 6.6 | 4.5 | 14.9 | 21.1 | N.A. | N.A. |
| HIV, % | 0 | 27.3 | 1.3 | 5.3 | N.A. | N.A. |
| AIDS, % | 0 | 18.2 | 0 | 5.3 | N.A. | N.A. |
| Anemia, % | 19.7 | 22.7 | 23.0 | 52.6 | N.A. | N.A. |
| Insult | ||||||
| Lung, % | 61 | 78.3 | 43.8 | 50 | N.A. | N.A. |
| Abdomen, % | 8.5 | 0 | 15.1 | 16.7 | N.A. | N.A. |
| UTI, % | 5.1 | 0 | 16.4 | 22.2 | N.A. | N.A. |
| Bloodstream, % | 5.1 | 4.3 | 8.2 | 5.6 | N.A. | N.A. |
| Other, % | 20.3 | 17.4 | 16.4 | 5.6 | N.A. | N.A. |
Definition of abbreviations: ALI, acute lung injury; APACHE II, Acute Physiology and Chronic Health Evaluation; CELEG, Consortium to Evaluate Lung Edema Genetics; CHF, congestive heart failure; CLD, chronic liver disease; COPD, chronic obstructive pulmonary disease; ESRF, end stage renal failure; N.A., not applicable; UTI = urinary tract infection.
Total cohort = 431 subjects, all sepsis subjects enrolled are either severe sepsis or septic shock. Age and APACHE II data expressed as mean ± SD. Survival is defined as survival to hospital discharge, measured up to 60 d. Ages and APACHE II are not significantly different between racial groups within a diagnosis.
P = 0.044 for survival difference ALI versus sepsis group.
Age of controls different versus all other groups.
SNP Genotyping
Genotyping was performed using a 5′ nuclease Taqman allelic discrimination assay (Applied Biosystems, Foster City, CA) on the 7900HT Sequence Detection System, which can detect different forms of the same gene that differ by nucleotide substitution. To account for differences in minor allele frequencies (MAF) according to ethnicity, two sets of markers with a MAF ⩾ 10% (the exception being four SNPs in the EA panel used for LD testing with MAF between 6 and 8%) were selected to screen the entire MYLK gene. EAs and AAs were analyzed separately. First, we selected SNPs available from the ABI Taqman SNP Genotyping Assay list (Assays-on-Demand, AOD) according to MAF and relative location between SNP to build the frame of the dense SNP map of the gene. Novel SNPs identified by direct sequencing were next selected to reduce the gaps using the ABI Customized Taqman SNP Genotyping Assay (Assays-by-Design) service or dbSNP (NCBI) for one additional SNP (rs9829784, MYLK_P1). Primers and probe sets were designed on the basis of types of base change and flanking sequences of each SNP. The EA set contained 28 SNP markers (15 Taqman assays and 13 novel SNPs) spanning a total of 214.7 kb of sequence on human chromosome 3 with an average inter-SNP distance of 8 kb. The AA set contained 25 SNP markers (17 Taqman assays and 8 novel SNPs) spanning a total of 204 kb with an average distance of 8.5 kb. A total of 17 SNPs were common markers, with an MAF > 10% in both ethnic groups. Genotyping of ∼ 7% of samples was repeated as the quality control procedure.
Test for Population Structure
Ancestry Informative Markers (AIMs, a total of 33) were typed in the African American cohort, with only 19 AIMs typed for 178 individuals and 14 typed only for 83 individuals (see Table E5 in the online supplement). All genotyping data was used together in the following analysis. Tests of equality of proportions were conducted for minor allele frequencies, testing for differences in ALI versus control subjects, sepsis versus control subjects, and ALI versus sepsis pairwise comparisons. Individual assignments of proportions of the three source populations (West African, European, and Native American) were obtained using maximum likelihood estimation (MLE) method. To get an overall assessment of stratification, we used a chi-square test on contingency tables for all markers of allele frequencies and numbers of alleles for the two disease groups and control subjects. The statistic was calculated as 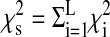 , where
, where 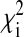 is the χ2 statistic computed at the ith marker locus and L is the set of unlinked marker loci typed on data from our 33 AIMs (26).
is the χ2 statistic computed at the ith marker locus and L is the set of unlinked marker loci typed on data from our 33 AIMs (26).
Statistical Analysis
Statistic comparisons were performed for age, Apache II, and survival variables in the patients' characteristics table. Racial groups within the three diagnosis groups were compared by two-tailed Mann-Whitney testing, and all groups were compared across diagnosis groups by one-way ANOVA; survival comparisons were by two-tailed Fisher testing in AAs and chi-square testing in EAs using STATA (Version 8.0; Stata Corp., College Station, TX). Departures from HWE proportions at each locus were tested among cases and control subjects separately, using the genhw procedure available in STATA 8.0. Pairwise linkage disequilibrium was evaluated using HaploView software (http://www.broad.mit.edu/personal/jcbarret/haploview) (27). LD blocks were defined using the default algorithm (Confidence Intervals), and allelic frequencies were estimated by gene counting. Risk estimation on the case-control dataset for each marker was conducted using direct calculation of odds ratios (STATA 8.0). Genotypic model was used if not otherwise stated. Sliding window haplotypic analyses were performed to screen the entire gene region for the haplotype with the most significant association with the disease phenotype using the SNPem software which estimates the haplotype frequencies via the method of maximum likelihood from genotype data through the use of the E-M algorithm under the assumption of HWE (28). The “omnibus” likelihood ratio test P value from SNPem was used as the “global P value” for 2- to 4-SNP windowing. Statistical significance of the P value was defined at the 5% level for all the analysis.
RESULTS
Patient Characteristics
The primary population was a European American dataset, which included (1) patients with sepsis-associated ALI (n = 92), (2) patients with sepsis alone (n = 114), and (3) healthy control subjects (n = 85). A secondary population consisted of a cohort of 158 African-American patients also comprised of patients with sepsis (n = 51), patients with sepsis-induced ALI (n = 46), and control subjects (n = 61) (Table 1). All sepsis subjects enrolled had either severe sepsis or septic shock. There were no significant differences in co-morbid factors between the case patients and the control subjects. Age and APACHE II scores were not significantly different between the two populations (European and African American) within a diagnosis, although the age of the control subjects in both groups was significantly different compared with the other groups (P < 0.01). Predictably, survival rates were significantly reduced in ALI cohorts versus sepsis subjects in both ethnic groups (P = 0.04).
Identification of Novel Polymorphisms in the MYLK Gene
Direct sequencing of MYLK to systematically characterize linkage disequilibrium and haplotype structure yielded 57 genetic variations with 51 polymorphic base substitutions (10 exonic, 31 intronic, 9 in the 5′ UTR, and 1 located in the noncoding exon 1) and 6 insertion/deletion variants (Table E1). Five of the 10 coding MYLK SNPs conferred an amino acid change including: Pro21His, Pro147Ser, Val261Ala, Ser1341Pro, and Arg1450Gln. Comparison of MYLK SNPs (identified by direct sequencing) with public information (NCBI dbSNP, ABI SNP Genotyping list), confirmed four novel nonsynonymous cSNPs (MYLK_002, 007, 033, 044). Matches were found for three cSNPs, such as MYLK_034 (rs1254392), MYLK_036 (rs820463), and MYLK_003 (rs3796164), by their locations on chromosome 3. The genomic organization of MYLK on chromosome 3, as well as the positions of 17 selected common MYLK SNPs (Table 2) with minor allelic frequencies, are displayed using original computational programs (Figure 1A).
TABLE 2.
CHROMOSOME 3 LOCATION, MINOR ALLELE FREQUENCY, AND TYPE OF SELECTED MYLK SNPS IN EUROPEAN AMERICAN SUBJECTS
| dbSNP or Celera ID | Chr.3 Location (Genome Build 34) | Inter-SNP Distance (bp) | MAF (chromosomes) | Type | Nucleotide/Amino Acid Change | |
|---|---|---|---|---|---|---|
| MYLK_016* | 124,873,039 | 0 | 0.17 (164) | c.-1747 | A/T | |
| MYLK_017* | 124,872,673 | 366 | 0.20 (164) | c.-1381 | T/C | |
| MYLK_021* | 124,872,210 | 463 | 0.19 (166) | 5′UTR (c.-918) | A/G | |
| MYLK_022* | 124,872,078 | 132 | 0.20 (162) | 5′ UTR (c.-786) | T/G | |
| MYLK_011* | 124,871,364 | 714 | 0.17 (166) | Ex1+48 | C/T | |
| AOD29† | hcv1602689 | 124,862,305 | 9,059 | 0.11 (160) | Intron | G/C |
| MYLK_P1 | rs9829784 | 124,861,037 | 1,268 | 0.22 (158) | Intron | C/T |
| AOD28 | rs10934651 | 124,854,110 | 8,195 | 0.06 (166) | Intron | C/G |
| AOD27 | rs4678062 | 124,850,009 | 4,101 | 0.21 (164) | Intron | C/T |
| AOD26 | rs11714297 | 124,840,480 | 9,529 | 0.23 (170) | Intron | C/T |
| MYLK_007*† | 124,833,528 | 6,952 | 0.10 (168) | C.62 | C/A (Pro > His) | |
| AOD24† | rs11707609 | 124,824,325 | 9,203 | 0.36 (166) | Intron | G/A |
| AOD22† | hcv1602709 | 124,800,686 | 23,639 | 0.58 (166) | Intron | A/G |
| AOD20† | rs11718105 | 124,784,609 | 16,077 | 0.33 (164) | Intron | C/T |
| MYLK_004* | 124,773,739 | 10,870 | 0.38 (168) | C.1005 | T/C | |
| AOD15† | rs820336 | 124,736,682 | 37,057 | 0.21 (168) | Intron | A/G |
| AOD14† | rs33262 | 124,734,125 | 2,557 | 0.21 (164) | Intron | A/T |
| MYLK_024*† | 124,732,490 | 1,635 | 0.23 (168) | C.3558 | T/C | |
| MYLK_025*† | 124,732,006 | 484 | 0.10 (168) | IVS17+477 | C/T | |
| AOD12 | rs33264 | 124,730,689 | 1,317 | 0.18 (168) | Intron | G/C |
| AOD11† | rs11717814 | 124,729,452 | 1,237 | 0.21 (166) | Intron | C/G |
| AOD9† | rs820325 | 124,706,578 | 22,874 | 0.10 (160) | Intron | A/G |
| AOD7† | rs702032 | 124,692,874 | 13,704 | 0.07 (168) | Intron | C/T |
| MYLK_034*† | rs1254392 | 124,688,914 | 3,960 | 0.08 (166) | C.4317 | T/C |
| MYLK_036*† | 124,677,938 | 10,976 | 0.08 (166) | C.4842 | T/C | |
| AOD3† | rs820447 | 124,669,080 | 8,858 | 0.13 (158) | Intron | A/G |
| AOD2† | rs3845915 | 124,660,735 | 8,345 | 0.13 (168) | Intron | T/C |
| MYLK_037*† | 124,658,305 | 2,430 | 0.17 (162) | IVS31+72 | G/C |
Definition of abbreviations: MAF, minor allele frequency; MYLK, myosin light chain kinase; SNP, single-nucleotide polymorphism.
Indicate SNPs selected by SNP discovery (as opposed to SNPs selected from the public database).
Common SNPs which have MAF ⩾ 10% in both European Americans and African Americans.
Figure 1.

Genomic organization of MYLK and position of selected common SNPs on chromosome 3 with minor allele frequencies and pairwise linkage disequilibrium from European American control subjects. A shows the MYLK gene structure based on the representative mRNA sequence (GenBank Accession no. U48959) on chromosome 3 (hg16; UCSC Genome Browser on Human July 2003 Assembly). Exons are represented by blue boxes connected by gray bars representing introns, and the black bars at both sides indicate 5′ and 3′ flanking regions (5′ to 3′ direction). The horizontal dashed lines below the gene structure represented the MAF cutoff lines of 25%, 50%, and 75%, respectively. Positions and MAFs of the 17 common SNPs analyzed are represented by vertical lines below the gene structure with green color stands for MAF in EAs and the pink color for AAs measured from the bottom edge of the gray bar. B shows the LD between 17 common SNPs in EAs (estimated from 170 chromosomes) and AAs (estimated from 120 chromosomes), respectively. The strength of LD between respective pairs of SNPs is depicted by progression of color: for all D′ with LOD of > 2, the color moves from red to light blue as D′ runs from 1 (represent perfect LD) to 0; for D′ with LOD of < 2, it is represented by white.
Fine Scale Mapping and Intragenic LD Patterns in the MYLK Gene
We selected a set of 36 SNPs (ABI SNP Genotyping list, 21 assays; SNP discovery effort, 15 customized assays) based on (1) gene location, (2) relative distance to each other, (3) minor allele frequency, and (4) compatibility with the genotyping method employed in two subgroups (EA and AA). Priorities were given to SNPs in coding regions causing amino acid changes and SNPs only found in cases by discovery effort. The selected markers did not demonstrate significant departure from Hardy-Weinberg equilibrium (HWE) in either ethnic group. The EA set contained 28 SNP markers (13 novel SNPs from SNP discovery) spanning a total of 214.7 kb on chromosome 3, resulting in an average 8-kb distance between SNPs. Four SNPs exhibited MAFs between 6 and 8%, and were not used for genetic association testing (Table 2). The AA set contained 25 SNP markers (8 novel SNPs) spanning 204 kb with an average inter-SNP distance of 8.5 kb (Table E2). The D′ measure of pairwise LD was estimated between SNPs separately for control subjects from the two ethnic groups, and LD blocks were constructed using confidence intervals (29). Six distinct LD blocks were seen in EAs (estimated from 170 normal control chromosomes) (Figure E2A). Block 3 and block 4 were merged to construct a 5-kb block that encompassed exon17 (the second exon encoding smooth muscle MLCK isoform) and surrounding intronic regions (containing SNPs rs820336, rs33262, MYLK_024, MYLK_025, rs33264, and rs11717814). Only two distinct LD blocks were apparent in AAs (120 chromosomes), with two additional blocks identified exhibiting MAF for key variants that were excessively reduced to meet HaploView power requirements (Figure E2B). The first distinct block in AAs also included exon17 and the upstream intron SNPs (rs820336, rs33262, MYLK_024). To compare LD patterns of MYLK between EAs and AAs, we examined pairwise LD between 17 SNP markers common to both sample subgroups with MAF ⩾ 10% (Figure 1B). The MYLK region displayed high levels of pairwise LD between neighboring SNPs and relatively low haplotype diversity in EAs (Figure 1B, left panel), with three distinct LD blocks (block1 = rs820336, rs33262, MYLK_024, MYLK_025; block2 = rs702032, rs1254392, MYLK_036; block3 = rs3845915, MYLK_037). In contrast, only two distinct LD blocks were seen in AAs (Figure 1B, right panel), but these overlapped with blocks found among EAs (block1-rs820336, rs33262, MYLK_024; block2- rs820447, rs3845915). Despite low levels of pairwise LD between neighboring SNPs, haplotype diversity is high compared with that seen in EAs. Analysis of 60 randomly selected EA control subjects (to approximate the number of AA subjects) failed to reveal any significant change in LD patterns described above, suggesting that the differences in sample size do not explain this difference (results not shown). Interestingly, the common allele among EAs was quite rare among AAs for eight MYLK SNPs (Table E2).
Single-Locus Tests for Association between MYLK Polymorphisms, Sepsis, and ALI
Significant single-locus associations were observed between four distinct SNPs (“G” allele for rs820336: odds ratio [OR], 1.97; 95% confidence interval [CI], 1.22–3.23, P = 0.004; “C” allele for rs33264: OR, 2.27; 95% CI, 1.37–3.82, P = 0.001; “G” allele for rs820325: OR, 2.04; 95% CI, 1.03–4.24, P = 0.02; “C” allele for MYLK_037: OR, 2.23; 95% CI, 1.31–3.85, P = 0.002) and the sepsis phenotype among EAs (Figure 2, vertical lines in black). There was evidence for association between one additional SNP and an increased risk of ALI (“T” allele for rs11718105: OR, 1.62; 95% CI, 1.02–2.58, P = 0.03). At the genotypic level, carriers of the CT genotype in EAs at rs11714297 (OR, 2.08; 95% CI, 1.09–3.99, P = 0.02) and carriers of the CT genotype at rs4678062 (OR, 2.04; 95% CI, 1.06–3.92, P = 0.03) showed a higher risk of ALI compared with noncarriers. Carriers of the “GG” genotype (3.6% in control subjects) have over 5-fold increased risk for sepsis and ALI at rs820336 (Table E3).
Figure 2.

Chromosome 3 distributions of −log10 (P value) for single MYLK SNP or haplotypes from SNP windowing across the MYLK gene in EAs. Results from the ALI versus control comparison (A), the sepsis versus control comparison (B), and the ALI versus sepsis comparison (C) are displayed. The y axis indicates the value of −log10 (P), the x axis indicates the relative position for each SNP marker locus at the MYLK gene region on human chromosome 3 in the 5′ to 3′ direction. The vertical line in black represents the position of each SNP marker on the x axis and its height on y axis, indicating the value of −log10 (P). The horizontal lines in red represent the 2-SNP windowing, and the lines in blue represent the 3-SNP windowing.
We confirmed these associations in AAs for this particular SNP-rs820336 where there appeared to be increased risk for both sepsis (“A” allele for rs820336: OR, 2.46; 95% CI, 1.32–4.60, P = 0.002) and ALI (OR, 2.07; 95% CI, 1.09–3.95, P = 0.02) phenotypes. The “AA” genotype is rare in the African American control subjects (1.7%), compared to the “GG” phenotype (55.8%). However, the “AA” genotype frequency significantly increased in both the sepsis (19.6%) and ALI (23.9%) groups; carriers of the “AA” genotype have 18-fold increased risk for both diseases (recessive model) (Table E3). There was evidence for association between ALI and 1 additional SNP (hcv1602689: OR, 3.5; 95% CI, 1.12–12.90, P = 0.01) (Figure E2). Carriers of mutant “GG” genotype at MYLK_037 (OR, 2.40; 95% CI, 0.97–5.91, P = 0.05) also showed a trend for association with ALI only among AAs. No significant association was observed between MYLK variants studied and either severity or outcome of disease at the genotypic level.
Adjusting for Population Stratification in African Americans
Testing for population structure via ancestry informative markers (AIMs) in the AA sample population was performed, and the results we obtained indicated that there may be important population stratification for all three comparisons in the case-control groups: 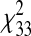 = 49.84853 (P < 0.05, sepsis versus control);
= 49.84853 (P < 0.05, sepsis versus control); 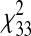 = 56.99234 (P < 0.01, ALI versus control);
= 56.99234 (P < 0.01, ALI versus control);  = 68.57489 (P < 0.001, ALI versus sepsis). Since admixture proportions can be considered significantly different across the case and control groups, the likelihood of confounding by differential admixture by case status is high. As a result, adjustment for population stratification was performed in the association analyses with MYLK variants, which showed positive association with the disease. All the observed positive associations were retained, with carriers of GG genotype for MYLK_037 demonstrating a greater association with the ALI phenotype after adjustment (OR, 4.163; P = 0.005).
= 68.57489 (P < 0.001, ALI versus sepsis). Since admixture proportions can be considered significantly different across the case and control groups, the likelihood of confounding by differential admixture by case status is high. As a result, adjustment for population stratification was performed in the association analyses with MYLK variants, which showed positive association with the disease. All the observed positive associations were retained, with carriers of GG genotype for MYLK_037 demonstrating a greater association with the ALI phenotype after adjustment (OR, 4.163; P = 0.005).
Haplotype Analysis using Sliding Window Approach
SNP windowing haplotype analysis was performed to define the strongest haplotype-phenotype associations as well as to further examine patterns of haplotype structure across the MYLK gene. Figure 2 displays the distributions of the global P value (displayed as –log10(P) on the y axis) derived from the two- and three-locus estimated haplotype frequency analysis for 24 SNPs on chromosome 3 in the MYLK gene region. Five different 2-SNP haplotypes showed significantly different frequencies between the sepsis and control groups (Figure 2, top panel) among EAs (markers MYLK_004, rs820336, P = 0.03; rs820336, rs33262, P = 0.001; MYLK_025, rs33264, P = 0.005; rs33264, rs11717814, P = 0.001; rs3845915, MYLK_037, P = 0.001), confirming significant single-locus associations observed with sepsis in this sample. Haplotype frequency comparisons between ALI and control groups (Figure 2, middle panel) yielded similar results. Table 3 displays haplotypes derived from two- and three-locus windowing indicating significant differences for both the individual haplotype frequency (estimated using permutation tests) and the overall haplotype frequency profile (i.e., the “omnibus” likelihood ratio test) comparisons between cases and control subjects (Table 3A) as well as similar values for ALI and sepsis (Table 3B). The GAT haplotype (over markers rs820336, rs33262, MYLK_024; 4.2 kb), the TCC haplotype (MYLK_024, MYLK_025, rs33264; 1.8 kb) and the CCC haplotype (MYLK_025, rs33264, rs11717814; 2.6 kb, observed only in EA cases) were significantly associated with increased risk (P < 0.0001, Table 3A). In contrast, EAs who carry the AAT haplotype were less likely to have sepsis (P = 0.01), and carriers of TCG and CGC haplotypes were less likely to have either sepsis or ALI (P = 0.001), suggesting the variant alleles of rs820336 and rs33264 have a major influence in determining susceptibility. Another AC haplotype involving markers rs3845915 and MYLK_037 (spanning 2.4 kb) was overrepresented among cases, with carriers exhibiting a 6.45-fold increased risk of sepsis (P < 0.001) and 5.13-fold increased risk of ALI (P = 0.001). More interestingly, the comparisons between ALI and sepsis groups (Figure 2, bottom panel) revealed an ALI-specific haplotype GGT (composed of markers MYLK_021, MYLK_022, MYLK_011; spanning a distance of 846 bp) located within the nonmuscle MLCK isoform-encoding region (between the 5′ UTR and the first exon) (Table 3B), suggesting this chromosome 3 segment may be important for ALI susceptibility among EAs. SNP windowing haplotype analyses of 25 MYLK SNP markers in AAs revealed two significantly different haplotype frequencies between sepsis and control groups (rs936170, rs820336, P = 0.01; rs820336, rs33262, P = 0.001; Figure E2). Comparisons between ALI and control groups (Figure E2, middle panel) also showed similar results, with two additional markers in the nonmuscle isoform-encoding region of MYLK (MYLK_002, MYLK_003, 4.8 kb; P < 0.01) as well as within the terminal 3′ region of the gene (rs820447, rs3845915, 8.3 kb; P = 0.03). AA carriers of the CC haplotype had an increased ALI risk, whereas carriers of the GA haplotype were less likely to have ALI (Table E4-A). The three-locus windowing revealed that both the GAT haplotype (using rs936170, rs820336, rs33262; a distance of 9.6 kb) and the ATC haplotype (based on rs820336, rs33262, MYLK_024; 4.2 kb) only existed in AA cases, with the ATC haplotype conferring the greatest risk of both sepsis and ALI (P = 0.00001 and P = 0.000001, respectively; Table E4-A). Similar to findings in EAs, comparisons between ALI and sepsis groups in AAs identified two significantly different haplotype frequencies at both the 5′ and the 3′ MYLK regions (Figure 2, bottom panel), suggesting that either these regions or neighboring regions are likely to harbor causal ALI variants. The CAG haplotype (composed of markers hcv1602689, MYLK_007, rs11707609; 28.8 kb) was overrepresented in the ALI group, with a frequency of 11.2% compared with 1% in the sepsis group (P = 0.01), whereas the CTA haplotype (MYLK_034, MYLK_036, rs820447; 19.8 kb) did not exist in the AA ALI group but had a frequency of 11.8% in the sepsis group (P = 0.001; Table E4-B).
TABLE 3.
HAPLOTYPE FREQUENCY ESTIMATES AND SIGNIFICANCE LEVELS OF CASE-CONTROL AND ALI VERSUS SEPSIS COMPARISONS FROM PERMUTATION TESTS IN 276 EUROPEAN AMERICAN SUBJECTS

Definition of abbreviations: ALI, acute lung injury; MYLK, myosin light chain kinase.
(A) Haplotype frequency estimates and significance levels of ALI or sepsis versus control comparisons. Estimated haplotype frequencies are derived from the three-locus estimated haplotype frequency analyses for the three-SNP window across a 7.2-kb region and two-locus estimated haplotype frequency analyses for the two-SNP window across a 2.4kb region in the chromosome 3 MYLK gene region. (B) Haplotype frequency estimates and significance levels of the ALI versus sepsis comparison. Two estimated haplotype frequencies are derived from the three-locus estimated haplotype frequency analyses for three-SNP window across 0.846 kb at the 5′ end of the MYLK gene.
* P values based on 10,000 permutations.
† P(g) is referred as global P value which derived from “omnibus” likelihood ratio test for assessing the overall haplotype frequency profile differences between the cases and control subjects.
DISCUSSION
We first reported the full-length sequence of the MYLK gene spanning 217.6 kb on chromosome 3q21.1 (10, 11) and containing three putative promoter regions and 32 exons which encode the smooth muscle and nonmuscle MLCK isoforms that phosphorylate regulatory myosin light chains. Human nonmuscle cells, such as vascular endothelial cells (EC), only express the nonmuscle MLCK isoform, which contains a novel NH2-terminus stretch (amino acid 1–922) not present in the open reading frame of smooth muscle MLCK (11, 12). Both the nonmuscle and smooth muscle isoforms are post-translationally modified by phosphorylation (11, 30–32) with the novel N-terminal stretch of the nonmuscle isoform, a prominent site of phosphorylation by p60src (33). We have explored participation of the nonmuscle MYLK isoform in lung innate immunity and inflammatory responses and determined key involvement in regulating barrier function, fluid flow, inflammatory cell trafficking, creating new blood vessels, and in vascular cell apoptosis (15–18, 34–37). For example, the role of MLCK in inflammatory lung edema formation involves carefully orchestrated MLCK-dependent actomyosin-based cytoskeletal rearrangement (16). MLCK inhibition prevents the increased lung permeability produced by thrombin (15, 16), TGF-β1 (38), activated PMNs (39), ischemia/reperfusion injury (40), and by mechanical stress (19). Recently, selective nmMYLK knockout mice demonstrate an essential role for MYLK in susceptibility to sepsis-induced ALI and as a potential drug discovery target (20). Furthermore, the chromosome location of MYLK (3q21) is an active site for several inflammatory disorders including asthma, allergic rhinitis, COPD and atopic dermatitis (Genetic Linkage Map: http://www.grc.nia.nih.gov/branches/rrb/dna/chromosome3.htm). Despite the apparent clinical importance of this multifunctional enzyme, the role of MYLK as a candidate gene in ALI has remained largely unexplored. The goal of the current study was to evaluate MYLK as a potential candidate gene and drug target for sepsis and ALI and to identify genetic variants in MYLK conferring risk of sepsis and/or ALI using simple case/control samples.
A frequent systematic error in molecular epidemiologic studies involves imperfect sampling or classification procedures (41), a particular concern given the heterogeneous major risk factors in ALI (sepsis, multiple transfusions, trauma, pneumonia, burns, cardiopulmonary bypass, and pancreatitis) (42). We used clearly defined inclusion criteria for cases and control subjects, with all patients with ALI recruited developing severe physiologic derangements characteristic of ALI in the context of documented sepsis. We identified 51 SNPs among patients with ALI, patients with sepsis, or control subjects and assessed the importance of MYLK variations on the risk for sepsis and sepsis-induced ALI. Significant associations were observed between four MYLK SNPs and the sepsis phenotype in EAs by single-locus analyses. The associations in EAs of SNP rs820336 which located at the first intron of the smooth muscle isoform of the gene with increased risks of both sepsis and ALI were confirmed in a second AA population. We believe it likely that there are one or more functional MYLK variants at the neighboring region of this particular SNP responsible for susceptibility to both sepsis and ALI regardless of ethnicity. Studies to further characterize this region within MYLK are ongoing.
For the particular replicated SNP rs820336 (A > G), frequencies of the “AA” homozygote in EAs are higher (60.7% in control subjects, 47.3% in patients with sepsis, and 45.5% in patients with ALI) compared with the frequencies of mutant “GG” homozygote in the three subgroups (3.6%, 17.3%, and 13.6%, respectively). Carriers of the “GG” genotype have over 5-fold increased risk for sepsis and ALI. In contrast, the genotype frequencies were reversed in AAs: only 1.7% of the AA control subjects carried the “AA” genotype, as compared with 55.8% who carried the “GG” genotype. However, carriers of the “AA” genotype have 18-fold increased risk for both sepsis and ALI. It is not unusual for marker allele and haplotype frequencies to show considerable variability across populations, such that a “major” allele in one population is the “minor” allele in the other population. Given such frequency differences, the high-risk allele/haplotype can easily switch across populations. The low occurrence of “AA” in African Americans could be due to the European admixture, and more common interactions with other genetic or environmental risk factors in AAs are likely to account for the greater risk in this group (43).
Our haplotypic analysis results confirmed the findings from the single-locus analysis and further increased the power to discover regions in the N-terminal of the MYLK gene that specifically contribute to susceptibility to ALI in both ethnic groups. These results suggest the defined regions or nearby region may harbor causal variants that confer susceptibility to ALI. The CAC haplotype, which exists only in AAs (comprising SNPs hcv1602689-AOD29, MYLK_007, rs11707609-AOD24), is of particular interest. MYLK_007, which maps to exon 2 containing the transcription initiation site, is a nonsynonymous SNP conferring proline to histidine amino acid change. The other nonsynonymous coding SNPs—proline to serine (MYLK_002) and valine to alanine (MYLK_003)—were also associated with both phenotypes in AA, and, as with MYLK_007, require the functional consequence to be elucidated. These coding SNPs suggest the potential for major conformation changes in the enzyme either affecting enzymatic activity or interactions with other regulatory proteins such as p60Src (33) and macrophage migration inhibitory factor (MIF), a well accepted biomarker for ALI (44). We previously demonstrated that p60Src-mediated phosphorylation of two key tyrosine residues in the N-terminus results in 3-fold enhancement of MLC kinase activity (33). Mechanistic in vitro biochemical and cellular studies and in vivo knock-in transgenic animal studies will be needed to fully understand the ramifications of these SNPs in the context of ALI pathophysiology and the racial disparity which exists in disease morbidity and mortality.
Linkage disequilibrium is a complex function of a number of genetic and evolutionary factors (mutation, recombination, gene conversion rates), demographic and selective events, and the age of the mutation itself (45). Both the boundaries of haplotype blocks and the specific haplotype observed are shared to a remarkable extent across populations. It has been suggested (29) that initial haplotype mapping in populations with longer-range LD might serve to make initial localization more efficient. The “tag” SNPs can be further selected and can be used in other studies, substantially reduce the time and effort for genotyping without losing significant haplotype information. In EAs, the MYLK gene region exhibits high levels of pairwise LD between neighboring SNPs and relatively low haplotype diversity, with 5- to 6-LD blocks accounting for the vast majority of EA chromosomes. The AA population is the result of relatively recent admixture between the European American and African populations, which is the potential resource of extended LD (46). Correlation between extended intervals of LD and functional genomic elements was observed (47). Both the CAG haplotype (28.8 kb) and the CTA haplotypes (19.8 kb) that associated with ALI in our AA samples are quite extended compared with findings in the EAs.
A small set of AAs replicated and supported our findings in EAs and added additional information that may contribute to the racial disparity in disease susceptibility and severity. We do not believe our findings can be explained by an admixture resulting in population stratification. However, it is also important to test and control for the genetic structure present in our AA population to avoid false positives. Since sepsis and sepsis-associated ALI predominantly affects middle-aged adults, recruitment of relatives is difficult and eliminated the possibility of using family-based design. An alternative approach involves using a set of unlinked genetic markers to infer details of population structure, and estimation of the ancestry of sampled individuals (48). Ancestry informative markers (AIMs) are genetic loci with alleles that have high frequency difference between populations defined for and specific to a particular admixture mapping application. AIMs can be used to estimate ancestry at the level of the population, subgroup (e.g., disease cases and control subjects), and individual (49). These studies confirmed the validity of not adjusting for population stratification.
In summary, our results, involving association testing in two case-control designed populations, are consistent with the notion that case-control association studies are a useful tool for shedding light on the genetic basis of disease predisposition and outcome. Our study used single-locus and haplotypic analyses using SNP markers across the entire MYLK gene to provide valuable information in terms of study design, haplotypic analysis approach, and the requirement for adjusting of stratification in AAs. Significant single-locus and haplotypic associations were observed in EA and AA populations between MYLK SNPs and both the sepsis and sepsis induced ALI phenotypes. In addition, results derived from multiple haplotypic analyses revealed a ALI-specific, risk-conferring haplotype at both the 5′ region of the nonmuscle MYLK gene in EAs and AAs and an additional haplotype within the 3′ region of the gene only in AAs. Consistent with the role of MYLK as a key modulator of inflammatory responses and a potential drug target, these data strongly implicate genetic variants in MYLK that confer increased risk of both sepsis and sepsis-associated ALI. The MYLK gene resides in 3q21, a genomic locale significantly associated with several inflammatory disorders, including asthma, allergic rhinitis, chronic obstructive pulmonary disease, and atopic dermatitis, suggesting that MYLK may represent a viable candidate gene in other inflammatory disorders. While a potential weakness of our study is the relatively small sample size and confirmatory studies (using greater populations) with fine scale mapping in defined MYLK regions are needed, these results provide needed validation of the candidate gene approach in complex disorders where family-based studies are not feasible.
Supplementary Material
Acknowledgments
The authors are grateful to all the study coordinators for recruitment of subjects into Consortium to Evaluate Lung Edema Genetics (CELEG), and thank Dr. Daniele Fallin for statistical input, William Shao and Mohan Parigi for information technology support, and Nancy Cox, Ph.D. (University of Chicago) for helpful discussions.
This work was supported by a NHLBI Program Project grant (HL 58064), the HopGene Program in Genomic Applications (U01 HL66583), and a Specialized Center for Clinically-Oriented Research (SCCOR) award (HL 73994). L.G. is supported in part by NIH T32 training grant.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2005-0404OC on January 6, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Frutos-Vivar F, Nin N, Esteban A. Epidemiology of acute lung injury and acute respiratory distress syndrome. Curr Opin Crit Care 2004;10:1–6. [DOI] [PubMed] [Google Scholar]
- 2.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care 2005;11:43–49. [DOI] [PubMed] [Google Scholar]
- 3.Moss M, Mannino DM. Race and gender differences in acute respiratory distress syndrome deaths in the United States: an analysis of multiple-cause mortality data (1979–1996). Crit Care Med 2002;30:1679–1685. [DOI] [PubMed] [Google Scholar]
- 4.O'Keefe GE, Hybki DL, Munford RS. The G→A single nucleotide polymorphism at the -308 position in the tumor necrosis factor-alpha promoter increases the risk for severe sepsis after trauma. J Trauma 2002;52:817–825. [DOI] [PubMed] [Google Scholar]
- 5.Gibot S, Cariou A, Drouet L, Rossignol M, Ripoll L. Association between a genomic polymorphism within the CD14 locus and septic shock susceptibility and mortality rate. Crit Care Med 2002;30:969–973. [DOI] [PubMed] [Google Scholar]
- 6.Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ, Humphries SE, Hill MR, Laurent GJ. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med 2002;166:646–650. [DOI] [PubMed] [Google Scholar]
- 7.Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, Floros J. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet 2000;58:181–191. [DOI] [PubMed] [Google Scholar]
- 8.Wang G, Christensen ND, Wigdahl B, Guttentag SH, Floros J. Differences in N-linked glycosylation between human surfactant protein-B variants of the C or T allele at the single-nucleotide polymorphism at position 1580: implications for disease. Biochem J 2003;369:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong MN, Wei Z, Xu LL, Miller DP, Thompson BT, Christiani DC. Polymorphism in the surfactant protein-B gene, gender, and the risk of direct pulmonary injury and ARDS. Chest 2004;125:203–211. [DOI] [PubMed] [Google Scholar]
- 10.Potier MC, Chelot E, Pekarsky Y, Gardiner K, Rossier J, Turnell WG. The human myosin light chain kinase (MLCK) from hippocampus: cloning, sequencing, expression, and localization to 3qcen-q21. Genomics 1995;29:562–570. [DOI] [PubMed] [Google Scholar]
- 11.Garcia JG, Lazar V, Patterson CE, Gallagher PJ, Verin AD. Endothelial cell myosin light chain kinase: cloning and regulation. Am J Respir Cell Mol Biol 1997;16:487–491. [DOI] [PubMed] [Google Scholar]
- 12.Lazar VL, Garcia JGN. A single human myosin light chain kinase gene transcribes multiple non-muscle isoforms. Genomics 1999;57:256–267. [DOI] [PubMed] [Google Scholar]
- 13.Watterson DM, Schavocky JP, Guo L, Weiss C, Chlenski A, Shirinsky VP, Van Eldik LJ, Haiech J. Analysis of the kinase-related protein gene found at human chromosome 3q21 in a multi-gene cluster: organization, expression, alternative splicing, and polymorphic marker J. Cell Biochem 1999;75:481–491. [PubMed] [Google Scholar]
- 14.Brand-Arpon V, Rouquier S, Massa H, de Jong PJ, Ferraz C, Ioannou PA, Demaille JG, Trask BJ, Giorgi D. A genomic region encompassing a cluster of olfactory receptor genes and a myosin light chain kinase (MLCK) gene is duplicated on human chromosome regions 3q13-q21 and 3p13. Genomics 1999;56:98–110. [DOI] [PubMed] [Google Scholar]
- 15.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell myosin light chain phosphorylation: role in thrombin-induced barrier dysfunction. J Cell Physiol 1995;163:510–522. [DOI] [PubMed] [Google Scholar]
- 16.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 2001;91:1487–1500. [DOI] [PubMed] [Google Scholar]
- 17.Garcia JGN, Herenyiova M, Cui Y, Verin AD, English D. Activation of endothelial cell myosin light chain kinase by adherent neutrophils: role in transendothelial migration. J Appl Physiol 1998;84:1817–1821. [DOI] [PubMed] [Google Scholar]
- 18.Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2001;280:L1168–L1178. [DOI] [PubMed] [Google Scholar]
- 19.Parker JC. Inhibitors of myosin light chain kinase and phosphodiesterase reduce ventilator-induced lung injury. J Appl Physiol 2000;89:2241–2248. [DOI] [PubMed] [Google Scholar]
- 20.Wainwright MS, Rossi J, Schavocky J, Crawford S, Steinhorn D, Velentza AV, Zasadzki M, Shirinsky V, Jia Y, Haiech J, et al. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc Natl Acad Sci USA 2003;100:6233–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, Heiner C, Kent SB, Hood LE. Fluorescence detection in automated DNA sequence analysis. Nature 1986;321:674–679. [DOI] [PubMed] [Google Scholar]
- 22.McCombie WR, Heiner C, Kelley JM, Fitzgerald MG, Gocayne JD. Rapid and reliable fluorescent cycle sequencing of double-stranded templates. DNA Seq 1992;2:289–296. [DOI] [PubMed] [Google Scholar]
- 23.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994;149:818–824. [DOI] [PubMed] [Google Scholar]
- 24.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992;20:864–874. [PubMed] [Google Scholar]
- 25.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med 1985;13:818–829. [PubMed] [Google Scholar]
- 26.Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 1999;65:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265. [DOI] [PubMed] [Google Scholar]
- 28.Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, Schork NJ. Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer's disease. Genome Res 2001;11:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al. The structure of haplotype blocks in the human genome. Science 2002;296:2225–2229. [DOI] [PubMed] [Google Scholar]
- 30.Verin AD, Gilbert-McClain LI, Patterson CE, Garcia JGN. Biochemical regulation of the nonmuscle myosin light chain kinase isoform in bovine endothelium. Am J Respir Cell Mol Biol 1998;19:767–776. [DOI] [PubMed] [Google Scholar]
- 31.Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science 1999;283:2083–2085. [DOI] [PubMed] [Google Scholar]
- 32.Goeckeler ZM, Masaracchia RA, Zeng Q, Chew TL, Gallagher P, Wysolmerski RB. Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. J Biol Chem 2000;275:18366–18374. [DOI] [PubMed] [Google Scholar]
- 33.Birukov KG, Csortos C, Ma SF, Dudek S, Bresnik A, Verin AD, Cotter R, Garcia JGN. Differential regulation of alternatively spliced endothelial cell MLCK isoforms by p60src. J Biol Chem 2001;276:8567–8573. [DOI] [PubMed] [Google Scholar]
- 34.Birukov KG, Birukova AA, Dudek SM, Verin AD, Crow MT, Zhan X, DePaola N, Garcia JG. Shear stress-mediated cytoskeletal remodeling and cortactin translocation in pulmonary endothelial cells. Am J Respir Cell Mol Biol 2002;26:453–464. [DOI] [PubMed] [Google Scholar]
- 35.Linz-McGillem LA, Moitra J, Garcia JG. Cytoskeletal rearrangement and caspase activation in sphingosine l-phosphate-induced lung capillary tube formation. Stem Cells Dev 2004;13:496–508. [DOI] [PubMed] [Google Scholar]
- 36.Petrache I, Birukov K, Zaiman AL, Crow MT, Deng H, Wadgaonkar R, Romer LH, Garcia JGN. Caspase-dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF-α-mediated bovine pulmonary endothelial cell apoptosis. FASEB J 2003;17:407–416. [DOI] [PubMed] [Google Scholar]
- 37.Wadgaonkar R, Linz-McGillem L, Zaiman AL, Garcia JG. Endothelial cell myosin light chain kinase (MLCK) regulates TNFalpha-induced NFkappaB activity. J Cell Biochem 2005;94:351–364. [DOI] [PubMed] [Google Scholar]
- 38.Hurst V, Goldberg PL, Minnear FL, Heimark RL, Vinvcent PA. Rearrangement of adherens junctions by transforming growth factor B1-role of contraction. Am J Physiol 1999;276:L582–L595. [DOI] [PubMed] [Google Scholar]
- 39.Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circ Res 2002;90:1214–1221. [DOI] [PubMed] [Google Scholar]
- 40.Khimenko PL, Moore TM, Wilson PS, Taylor AE. Role of calmodulin and myosin light-chain kinase in lung ischemia-reperfusion injury. Am J Physiol 1996;271:L121. [DOI] [PubMed] [Google Scholar]
- 41.Vineis P, McMichael AJ. Bias and confounding in molecular epidemiological studies: special considerations. Carcinogenesis 1998;19:2063–2067. [DOI] [PubMed] [Google Scholar]
- 42.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med 1995;151:293–301. [DOI] [PubMed] [Google Scholar]
- 43.Helgadottir A, Manolescu A, Helgason A, Thorleifsson G, Thorsteinsdottir U, Gudbjartsson DF, Gretarsdottir S, Magnusson KP, Gudmundsson G, Hicks A, et al. A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat Genet 2005; [Epub ahead of print]. [DOI] [PubMed]
- 44.Wadgaonkar R, Dudek SM, Zaiman AL, Linz-McGillem L, Verin AD, Nurmukhambetova S, Romer LH, Garcia JG. Intracellular interaction of myosin light chain kinase with macrophage migration inhibition factor (MIF) in endothelium. J Cell Biochem 2005;95:849–858. [DOI] [PubMed] [Google Scholar]
- 45.Shriver MD, Parra EJ, Dios S, Bonilla C, Norton H, Jovel C, Pfaff C, Jones C, Massac A, Cameron N, et al. Skin pigmentation, biogeographical ancestry and admixture mapping. Hum Genet 2003;112:387–399. [DOI] [PubMed] [Google Scholar]
- 46.Collins-Schramm HE, Chima B, Operario DJ, Criswell LA, Seldin MF. Markers informative for ancestry demonstrate consistent megabase-length linkage disequilibrium in the African American population. Hum Genet 2003;113:211–219. [DOI] [PubMed] [Google Scholar]
- 47.Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR. Whole-genome patterns of common DNA variation in three human populations. Science 2005;307:1072–1079. [DOI] [PubMed] [Google Scholar]
- 48.Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet 2000;67:170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kittles RA, Weiss KM. Race, ancestry, and genes: implications for defining disease risk. Annu Rev Genomics Hum Genet 2003;4:33–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.